Introduction
Macroautophagy, also known as autophagy, is a
catabolic process that operates by generating several benefits for
the cell, protecting it against multiple stress stimuli. In the
majority of cases, autophagy functions as a recycling and recovery
system for proteins, lipids and DNA damage; therefore, the cell
eliminates various compounds that may be toxic, and also generates
ATP. It has been reported that autophagy protects the cell against
DNA damage, chromosomal instability and metabolic stress (1). The exogenous expression of a great
variety of oncogenes leads to tumorigenesis which is often
accompanied by metabolic changes. In this aspect, one of the better
examples is aneuploid tumors, where the additional chromosomes bear
an increased amount of various proteins, affecting protein folding
and turnover. The overaccumulation of proteins induces the
activation of adaptive processes to alleviate this proteotoxic
effect, such as the increase in chaperone Hsp72, the activation of
AMPK or the degradation mediated by autophagy (1). These alterations of the metabolism in
this type of tumor offer the possibility of blocking autophagy or
generating metabolic stress as a therapeutic strategy due to the
differential sensitivity of cancer cells (2).
Numerous oncogenic processes favor adaptations of
signaling pathways to allow tumor transformation, although they
generate cellular stress. This affects the autophagic pathway since
in the majority of stress conditions the cells activate several
signaling pathways, such as mTOR, AMPK, p53, JNK, FoxO3 among
others, which regulate, positively or negatively, the induction of
autophagic vacuoles. Most of these pathways control the expression
of proteins related to vacuole generation, such as the Atgs. In
contrast, both p53 and FoxO3 have been found to induce genes such
as Sestrin 2 and DRAM or BNIP3 and GABARAP, respectively. However,
various functions of these genes have not been completely
elucidated since they are only associated with an increased
degradative phase and in some circumstances with cell death.
During the Ras-mediated process of oncogenesis,
there is an induction of autophagy through negative feedback in
which Ras-Raf activation shrinks the PI3K pathway, reducing AKT
phosphorylation (3). Therefore,
this decrease in Akt activity reduces FoxO repression and permits
the upregulation of protein, such as LC3, BNIP3 and ULK (4). In this manner, RAS-v12 overexpression
induces an increase in autophagy that is associated with
oncogene-induced senescence (OIS) hallmarks and also with the
formation of TOR-autophagy spatial coupling compartment (TASCC)
that mediates the secretion of interleukin-6/8, which are involved
in senescence (5,6). If once transformation is established
and the stress situation is maintained, this generates autophagy
dependence or an increase in the glucose uptake as previously
demonstrated (7,8).
In contrast, the involvement of AMPK in the
regulation of metabolism and autophagy has been widely
demonstrated. This kinase performs several functions that link
metabolism and proliferation. In addition to its function as a
sensor of the ATP/AMP ratio, this kinase is activated in response
to oxidative and proteolytic stress.
In this sense, it has been recently reported that
AMPK directly phosphorylates FoxO3 in its Ser413 and Ser588
residues, increasing the transcription of various FoxO3 regulated
genes, such as Gadd45α (9).
Thus, much of the metabolic adaptation in cells which undergo
oncogenic transformation may induce a differential response of AMPK
depending on ATP levels, oxidative stress and autophagy.
In the present study, we demonstrated that the
overexpression of c-Myc or RAS-v12 in untransformed mammary cells
activates AMPK and that the phosphorylation of FoxO3 increases the
transcription of genes such as BNIP3. Using a powerful system of
mitophagy induction, the linamarase/linamarin/glucose oxidase
system (lis/lin/GO) (10,11),
we demonstrated that c-Myc or RAS-v12 transformation sensitized
cells to autophagy, in vitro and in vivo.
Materials and methods
Reagents and antibodies
The antibody against FoxO3 P-ser413 was a kind gift
from Dr Anne Brunet (Department of Genetics, Stanford University,
Stanford, CA, USA). The commercial primary antibodies used were:
FoxO3 and phospho-AMPKα (Thr172) (Cell Signaling, Danvers, MA,
USA); pan-RAS (Calbiochem, Darmstadt, Germany); p62 (BD
Biosciences, Bedford, CA, USA); E-cadherin and c-Myc (sc-788)
(Santa Cruz Biotechnology, Santa Cruz, CA, USA); LC-3, BNiP3 and
actin (Sigma, St. Louis, MO, USA). The secondary antibodies for
western blot analysis were horseradish peroxidase-conjugated
anti-rabbit or anti-mouse IgGs (Santa Cruz Biotechnology). The
reagents used were: linamarin (lin; 500 µg/ml; Toronto
Research Chemicals, Toronto, Canada), glucose oxidase (GO; 5–5.5
mEU/ml), 3-methylad-enine (3MA; 10 mM) and thiazolyl blue
tetrazolium bromide (MTT; 200 µg/ml) (all from Sigma),
bafilomycin A1 and compound C (10 µM) (from Calbiochem).
Media and cell culture
The MCF7 cell line was cultured in Dulbecco's
modified Eagle's medium (DMEM) (Gibco, Life Technologies,
Barcelona, Spain) supplemented with 10% fetal calf serum (FCS) at
37°C in 7% CO2 and 97% relative humidity. The MCF10A
cell line was grown as recommended by Joan S. Brugge (Whitehead
Institute for Biomedical Research, Cambridge, MA, USA) in DMEM:F12
media (1:1) supplemented with 10 µg/ml insulin (Gibco), 20
ng/ml EGF (Tocris, Bristol, UK), 0.5 µg/ml hydrocortisone
(Calbiochem), 100 ng/ml cholera toxin and 5% horse serum at 37°C in
7% CO2 and 97% relative humidity.
Adenovirus, lentivirus and retrovirus
production
Adenolis vectors were obtained from Crucell
(Leiden, The Netherlands). Pseudotyped lentivectors were produced
using reagents and protocols from Didier Trono with the following
modifications: 293T cells were transiently co-transfected with 5
µg of the corresponding lentivector plasmid, 5 µg of
the packaging plasmid pCMVdR8.74 and 2 µg of the VSV-G
envelope protein plasmid pMD2G using Lipofectamine Plus reagent
following instructions of the supplier (Invitrogen, Life
Technologies, Barcelona, Spain). The lentivectors included
pMIG-h-c-Myc, that was kindly provided by Maria Soengas (CNIO,
Madrid, Spain) (12), and
pLenti-RAS-v12 provided by Judith Campisi (Lawrence Berkeley
National Laboratory, Berkeley, CA, USA) (13) which encode human c-Myc and
constitutively active RAS, respectively. To generate the different
MCF10A derivative cells, the retroviral vectors used were pBabe,
pBabe-dnFoxO3 and pBabe-FoxO3A (caFoxO3) that were kindly provided
by Dr Clemens Schmitt (Max Delbrück Center for Molecular Medicine,
Berlin, Germany). The retrovirus supernatant was prepared by
transfection of phoenix-Ampho cells (Garry Nolan; http://www.stanford.edu/group/nolan/)
with 5 µg of each plasmid with Lipofectamine Plus as
previously mentioned, and infected cells were selected with 0.5
µg/ml of puromycin.
Soft agar assay
To evaluate the tumorigenic potential,
2×104 viable cells/well were plated in soft agar in
6-well plates. Briefly, the base layer was constructed by mixing
equal agar and 2X medium, to obtain a final solution of 0.5% agar
in 1X DMEM:F12 medium and for the top layer, the agar was diluted
to 0.7% in distilled water with 2X DMEM medium. The cells were
immediately added to the mix to yield a final solution of 0.35%
agar in 1X DMEM medium, containing 30,000 cells/ml. The cells were
grown for 10 days at 37°C in a humidified atmosphere containing 5%
CO2, and viable colonies were then stained with 1
ml/well of 600 µg/ml MTT for 2 h and then were
photographed.
Immunoblot analysis
Cell lysates were prepared by extracting proteins
with lysis buffer (50 mM pH 7.5 Tris-HCl, 300 mM NaCl, 0.5% SDS and
1% Triton X-100). Proteins were separated by SDS-PAGE and
transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA,
USA). The blots were developed using peroxidase-conjugated
secondary antibody, and proteins were visualized using an enhanced
chemiluminescence-based detection kit (Pierce, Rockford, IL,
USA).
Mice and treatments
Nude mice, BALB/c nu/nu, were inoculated
subcutaneously in both flanks with 1×106
MCF7-Ras-lis cells (n=6/group). After 5 days, the animals
were intratumorally treated daily with lin (5 mg) and GO (2 EU) in
the right tumor while the left one was left untreated. All
treatments were performed under general anesthesia with isofluorane
(AP/DRUGS/220/96; Baxter, Valencia, Spain). Tumor size (length,
width and height) was measured when indicated with a caliper, and
the tumor volume (V) was calculated as: V = [π/6(length × width ×
height)]. For animal care and handling we followed the Spanish
legislation and guidelines (Spanish Royal Decree 1201/2005 BOE
published on October 21, 2005), and those from the European Union
(2003/65/CE from the European Parliament and Council July 2003) and
those of the CBMSO Institutional Biosafety Committee.
Statistical analysis
Statistical comparison of the data groups was
carried out using the Student's t-test. The differences are
expressed with their corresponding statistical significance or
p-value, which is the probability that the observation in a sample
occurred merely by chance under the null hypothesis.
Results
The purpose of the present study was to verify
whether oncogene-mediated transformation sensitizes cells to
autophagy. For this purpose we used two alternative approaches, the
transfection of c-Myc (14,15) or a mutant version of ras
(RAS-v12) (16), and we initially
used MCF10A, an untransformed mammary cell line, as a cell model.
Next, we transfected and generated cell lines expressing either
c-Myc or RASv12 (HrasVal12) or an empty vector containing GFP as an
internal control. The c-Myc- and RASv12-expressing cells were
characterized at three levels. First they were phenotypically
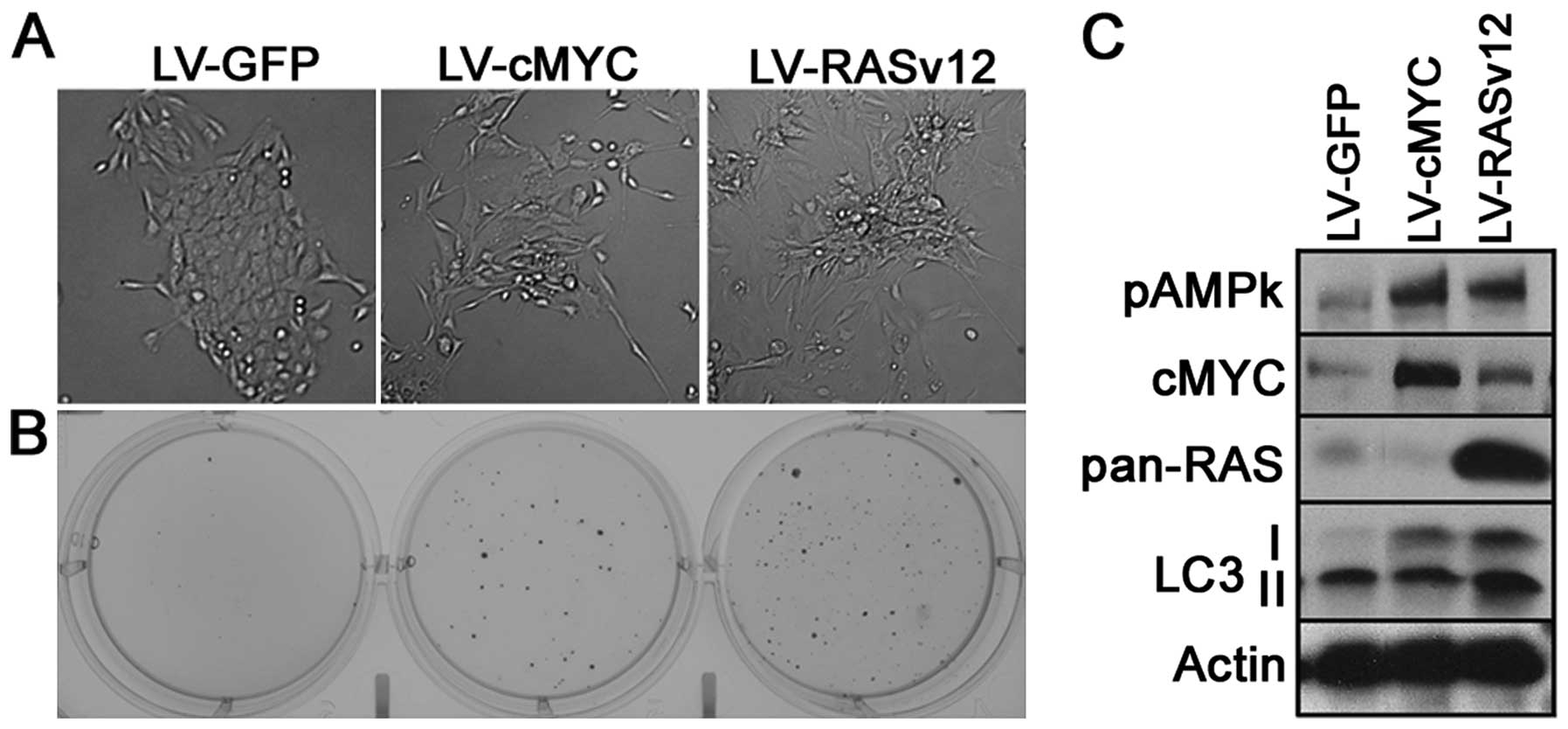
altered giving the appearance of a mesenchymal-like phenotype
(Fig. 1A). Second, both oncogenic
transformations conferred MCF10A cells the ability of
anchorage-independent growth forming colonies in soft agar when
compared with the mock-transfected cells (LV-GFP) (Fig. 1B). Third, they expressed and
maintained a high level of either c-Myc or RAS as confirmed by
western blotting (Fig. 1C).
As an initial indicator of autophagic vesicles, we
determined the level of LC3-II. We showed that c-Myc-transformed
cells, while more prominent in Ras-transformed cells, showed
an increase in the amount of total LC3B, as well as an increase in
the LC3-II form. Next, we inferred the activity of AMPK by
determining the phosphorylation levels of Thr172-AMPK. Notably,
both oncogenes were capable of increasing phosphorylation of this
residue (Fig. 1C). This kinase
activation permitted us to conclude that the amount of ATP/AMP and
NADP/NADPH were modified in response to the 'oncogenic stress'.
The increase in LC3-II indicated differences in
autophagy dynamics. Thus, we next analyzed how the
RASv12-transformed cells respond to a drug that induces
mitochondrial stress, when compared with the parental cell lines.
For this purpose, we used the MCF10A-GFP and MCF10A-RASv12 cells,
as well as a breast tumor cell line (MCF7), transfected with both
constructs (GFP and RASv12).
The cell lines containing GFP or RASv12, derived
from either MCF10A or MCF7 cells, were treated with
lis/lin/GO, that produced mitochondrial stress and
consequently induced potent autophagy as previously described
(10,11). The lis/lin/GO treatment
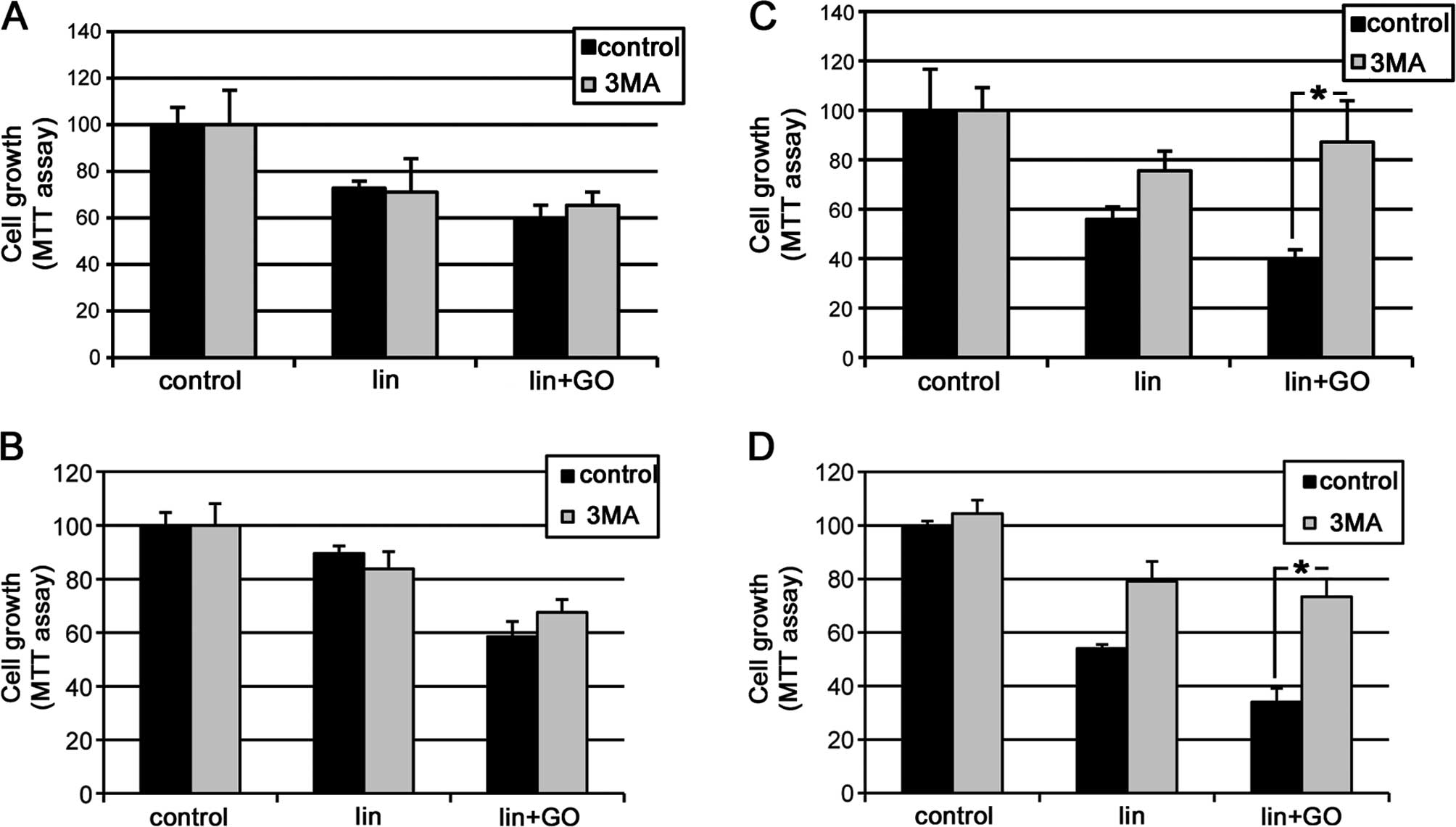
showed a marked growth inhibition in the MCF10A cells either
untransformed (LV-GFP) (Fig. 2A) or
transformed (LV-RASv12) (Fig. 2C)
as determined by the MTT assay. Similar results were observed in
the MCF7 and MCF7-RASv12 cells (Fig. 2B
and D).
Notably, when we used a wide inhibitor of class III
PI3K, 3-methyladenine (3MA), this effect was prevented or highly
diminished only in the RAS-transformed cells in both cell types
(Fig. 2C and D), whereas in the
non-transformed cells the toxic effect generated by the
lis/lin/GO system was not prevented (Fig. 2A and B).
Collectively, we conclude that the RAS-expressing
cells were more sensitive to the inhibition of autophagy. To
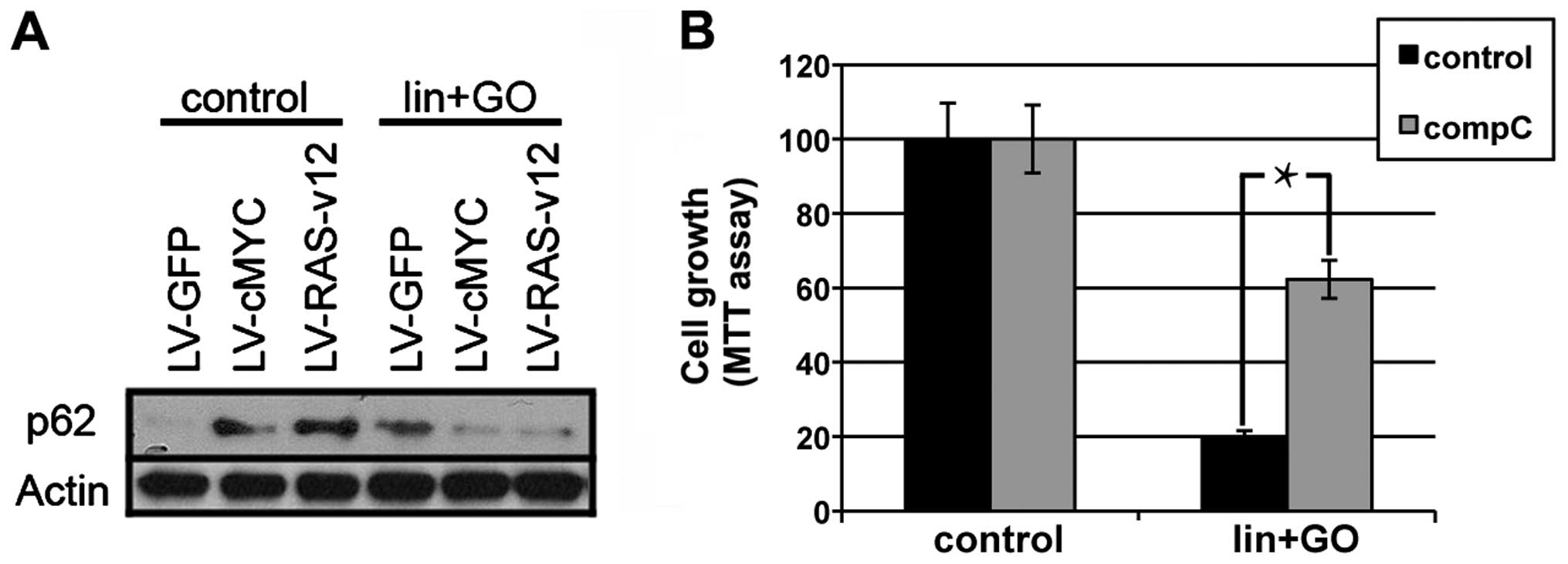
confirm this hypothesis, we determined whether the levels of p62,
one of the main partners of LC3B (17,18),
is modified by the lis/lin/GO treatment. Thus, we observed
that either c-Myc or RAS transformation increased the amount of
p62, and this protein level was reduced, probably by degradation,
when autophagy was triggered by lis/lin/GO treatment in
either the c-Myc- or Ras-expressing MCF10A cells (Fig. 3A).
There is a close relationship between metabolism and
cellular damage generated by stress. In addition the mitochondrial
toxic effect generated by the lis/lin/GO system permitted us
to hypothesize the participation of AMPK in AMP/ATP deregulation.
We showed that Myc or RAS transformation generated an increase in
AMPK activity (Fig. 1C), and
inhibition of AMPK by compound C (10 µM) partially recovered
the cell survival reduced by lis/lin/GO treatment in the
MCF10A-RASv12 cells (Fig. 3B).
It has been previously reported that AMPK mediates
FoxO3-positive regulation of energy balance and stress (9). This transcriptional factor controls
the expression of an important set of genes related to autophagy,
such as LC3B or BNIP3 among others, as reported in RAS-mediated
senescence or in muscular atrophy (4,19,20).
Therefore, to validate the activation of AMPK due to these tumor
transformations, we analyzed the expression of phosphorylated FoxO3
and the protein levels of LC3B and BNIP3.
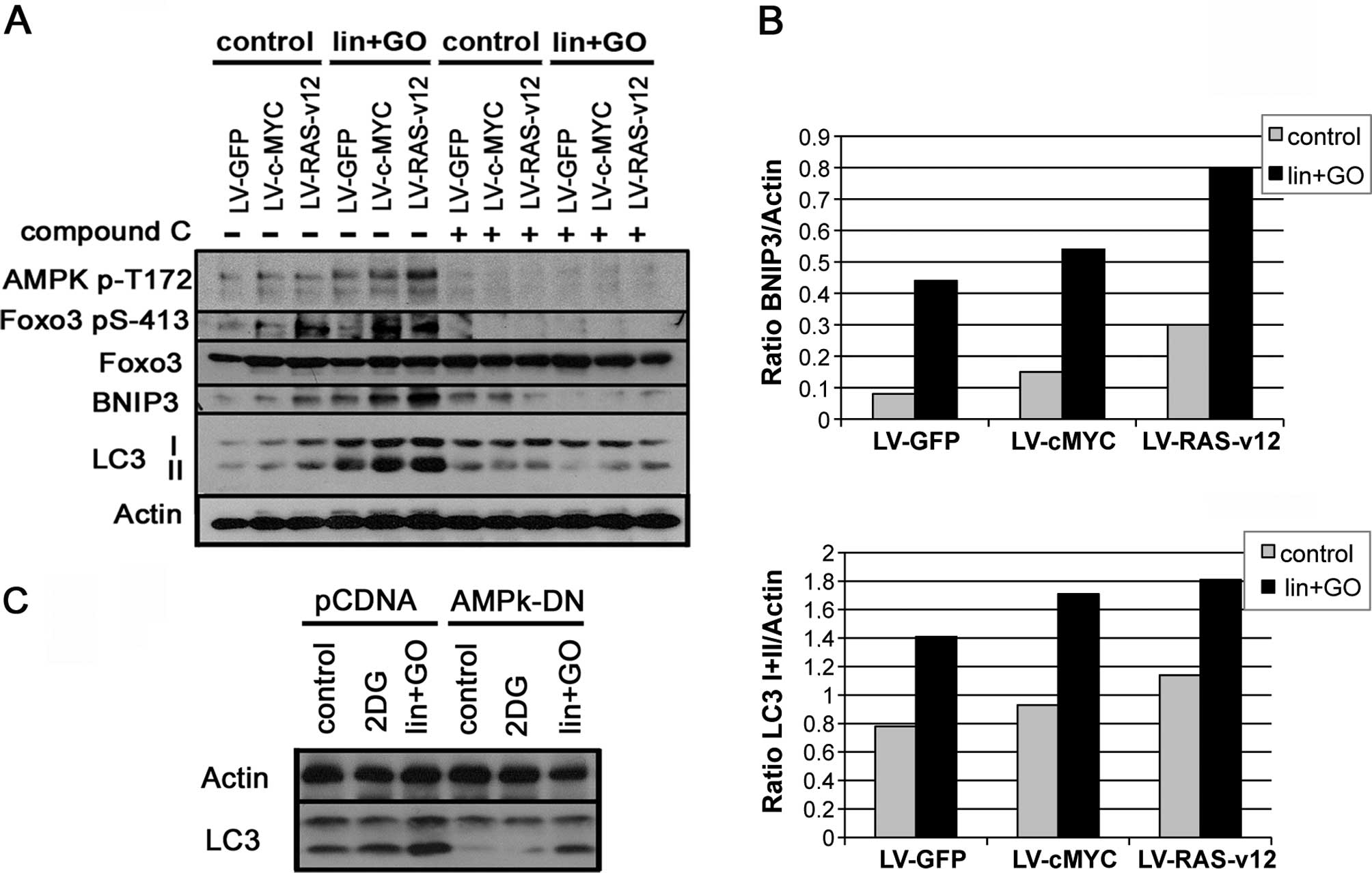
Our data indicated that both c-Myc and RAS activated
AMPK by increasing the level of phosphorylated AMPKα in Thr172
(pAMPK) (Fig. 4A); in parallel
LC3-II was increased (Fig. 4A).
This activation of AMPK was correlated with an increase in
p-FoxO3-Ser413, coinciding with an increase in BNIP3 and LC3B
(Fig. 4A). This AMPK kinase
activation was confirmed since following incubation with the
inhibitor compound C (10 µM) both FoxO3-Ser413
phosphorylation and BNIP3 and LC3B expression were severely reduced
(Fig. 4A). The additional stress
generated by the lis/lin/GO system increased p-AMPK-Thr172
as well as FoxO3-Ser413, which were correlated with an increase in
BNIP3 expression and LC3-II (Fig.
4B) more markedly in the c-Myc- and RAS-transformed cells.
Similarly the addition of AMPK inhibitor, compound C, completely
blocked FoxO3 phosphorylation, therefore preventing BNIP3 and
LC3-II induction (Fig. 4A).
Similarly to Compound C, the expression of dominant-negative
version of AMPK (AMPK-DN) reduced the enhancement of LC3-II
triggered by lin+GO (Fig. 4C).
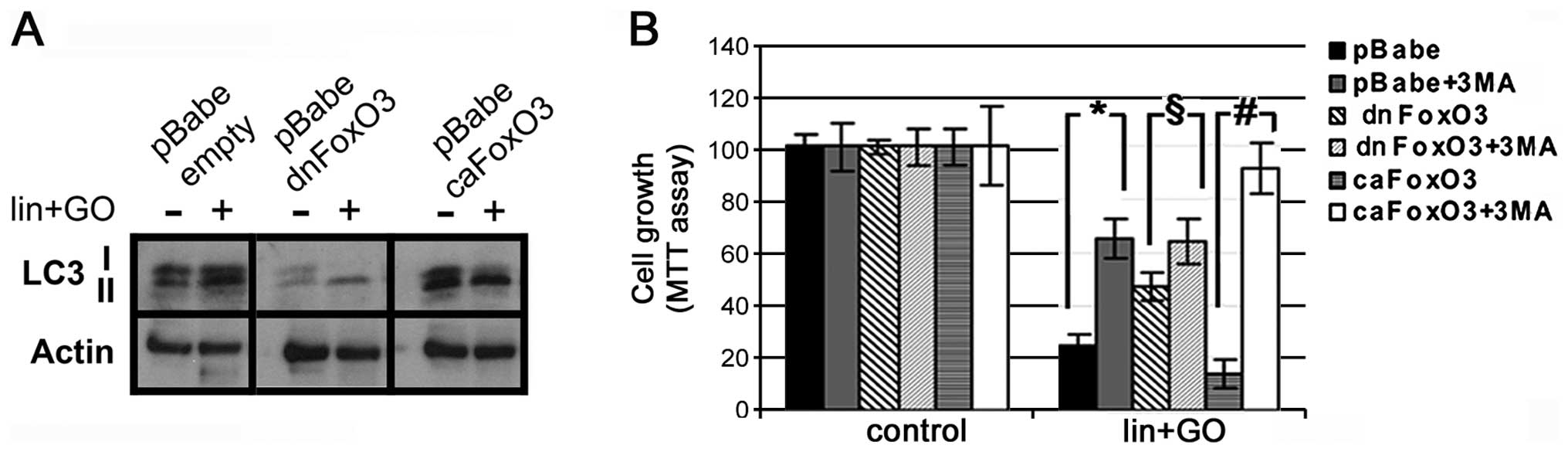
To assess the involvement of FoxO3 in the regulation
of LC3, we infected MCF10A-RASv12 cells with either a
dominant-negative (dnFoxO3) or a constitutively active version of
FoxO3 (caFoxO3), as well as an empty virus (pBabe) as a control. It
was observed that dnFoxO3 blocked the expression of LC3 as well as
the amount of LC3-II (Fig. 5A); in
contrast, the expression of caFoxO3 increased the level of LC3-II
(Fig. 5A).
Notably, the dnFoxO3 construct partially recovered
the cell survival rate following lis/lin/GO treatment, while
the caFoxO3 construct diminished the survival of the MCF10A-RASv12
cells (Fig. 5B). The subsequent
inhibition of class III PI3K by 3MA increased the survival of the
control cells (infected by LV-GFP). Notably, the presence of 3MA
increased the survival of the caFoxO3 cells even more than that in
the control cells (p=0.045 vs. p=0.0001) (Fig. 5B), whereas no additional increase
was observed in the dnFoxO3 3MA-treated cells (p=0.2) (Fig. 5B). All these data suggest that
autophagy induced by lis/lin/GO was mediated at least in
part by FoxO3 activity.
Based on the fact that the lis/lin/GO
treatment reduced the growth of MCF7-RASv12 cells, we aimed to
ascertain whether this affects the growth capacity of the
MCF7-RASv12 tumor cells in vivo. To address this issue, we
inoculated MCF7-RAS-lis cells into the flank of nude mice
and carried out treatment with vehicle or lis/lin/GO
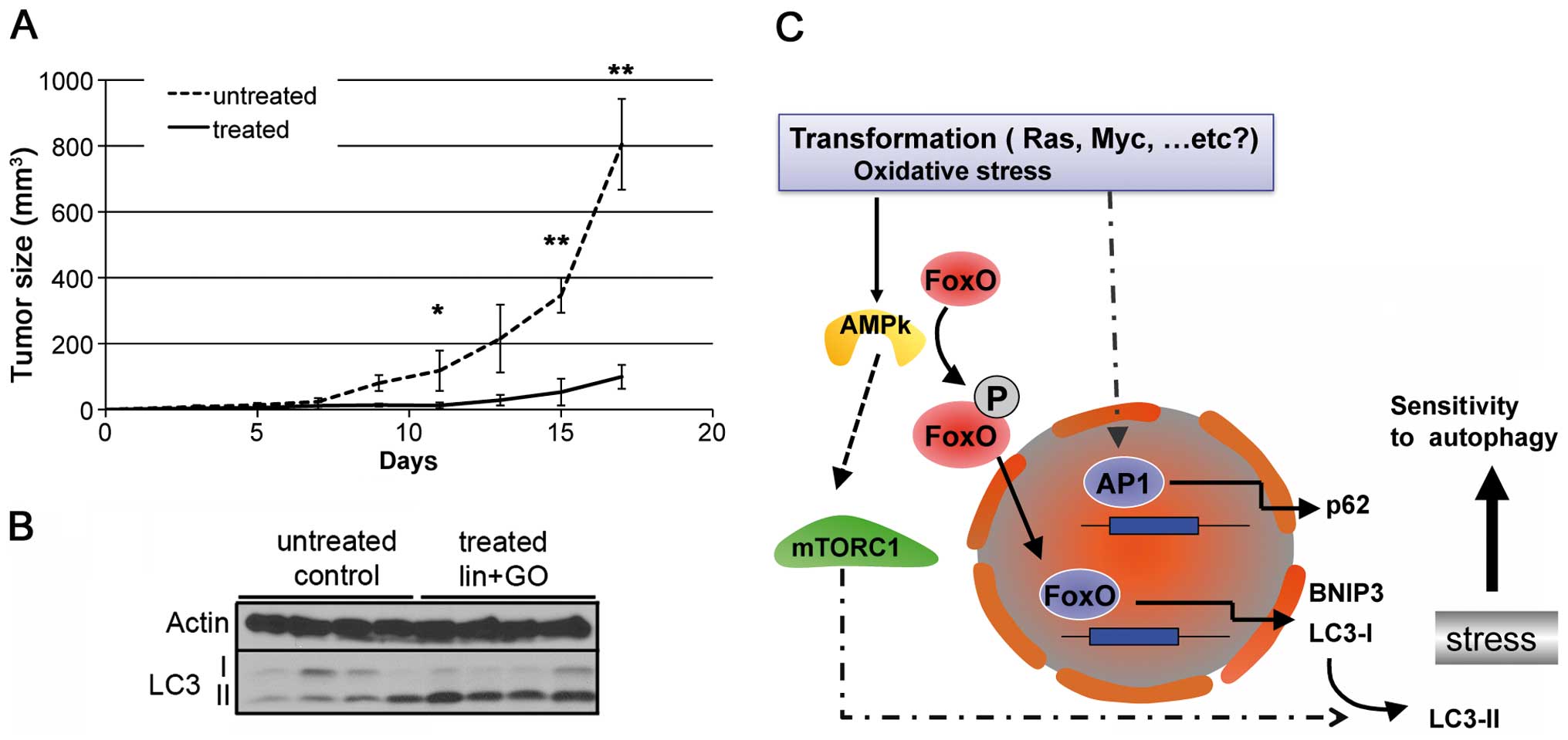
compounds as described in Materials and methods. As shown in
Fig. 6A, treatment with
lis/lin/GO significantly reduced in vivo tumor growth
from day 11 onwards. Additionally we analyzed the induction of
autophagy (LC3-II levels) in the untreated and treated tumors.
Western blot analyses of the samples (n=4) showed that tumor
extracts from the lis/lin/GO-treated tumors showed a
substantial increase in levels of LC3-II when compared with these
levels in the solvent-treated tumors (Fig. 6B).
In conclusion, transformation mediated by both
oncogenes enhanced the sensitivity of the transformed cells to
autophagy induction when compared with the untransformed cells.
This may have therapeutic interest. This increase in sensitivity
under a condition of potent mitochondrial stress is mediated, at
least in part, by the AMPK-FoxO3 axis as summarized in the scheme
in Fig. 6C.
Discussion
It is widely accepted that tumor cells modify
autophagy generating a dependency on this process for maintaining
survival and overcoming metabolic stress. Thus, tumor cells are
sensitive to the blockage of autophagy (14,21) as
well as to its stimulation (22–24).
It has been previously demonstrated that autophagy is necessary for
activated Ras-mediated transformation (7). In the present study, we demonstrated
that tumor processes driven by genes such as c-Myc or RAS (RAS-v12)
(14,16,25)
sensitized cells to autophagy induction by the lis/lin/GO
system. This system has been reported to generate mitochondrial
stress-triggering autophagy (10,11).
Mitochondrial targeting is an attractive antitumor strategy due to
the close relationship between oncogenic processes and the
modification of the mitochondrial metabolic status (26,27).
Additionally, mitophagy is a mechanism capable of recycling damaged
mitochondria, producing ATP (28)
and reducing cellular stress. Therefore, a disturbance in this
process can affect the proliferation and survival of tumor
cells.
The present data confirmed that the
lis/lin/GO system increased AMPK activity, as inferred from
phosphorylation of FoxO3 and an increase in genes that are
fundamental for autophagy induction such as BNIP3 and LC3B. These
data confirm previous reports that indicate that AMPK favors the
transcriptional activation of FoxO3 by the phosphorylation of its
Ser413 or Ser588 residues (9), and
this transcriptional factor controls the induction of BNIP3 and
LC3B genes depending on cell type and context (19,20).
In addition, our data demonstrated that the
oncogenes, c-Myc and RAS, also modified AMPK activity by generating
the upregulation of BNIP3 and LC3B. We demonstrated that the
lis/lin/GO-induced autophagy activated this pathway, which
was more evident in the RAS-transformed cells, and presented the
highest levels of BNIP3 and LC3B in a clear correlation with the
phosphorylation of FoxO3. Obviously, we cannot ignore the fact that
other regulatory elements such as p38 may contribute to the final
regulatory loop after transformation (29). In cases, transformed cells with or
without lis/lin/GO treatment, the inhibitor of AMPK or the
dominant-negative version of AMPK prevented the increase in LC3-II.
Thus, these data allow the conclusion that autophagy is enhanced in
c-Myc and RAS oncogenic models. A similar effect of enhanced
autophagy was reported in cancer cells transformed by various
tyrosine kinases such as EGFR or Abl (30–33).
In these cases, tumor progression was associated with an
enhancement of autophagy, dependent or independent, of tyrosine
kinase activity. Thus, it can be proposed that oncogenic
transformation which enhances autophagy is an important mechanism
for tumor growth and progression.
Thus, we propose that a high percentage of tumors
that bear oncogenes may have proliferative or metabolic stress
which makes cells more sensitive to autophagy affecting tumor
growth in vitro and in vivo. This susceptibility to
the induction of autophagy is regulated, at least in certain cases,
by the AMPK-FoxO pathway. Additionally, we hypothesize that
autophagy overstimulation by mitochondrial stress induction can be
considered as a therapeutic strategy against cancer.
Acknowledgments
We would like to thank Dr Clemens Schmitt, Dr Maria
Soengas, Dr Patricia Boya and Dr Anne Brunet for providing the
plasmids and antibodies. The laboratory of M.I. was supported by
funds from the Fondo de Investigación Sanitaria, Ministerio de
Sanidad y Consumo (PI06/0554), and from FINA Biotech. R.G. was
supported by the Fundación Mario Losantos del Campo and Juan de la
Cierva postdoctoral fellowship (Ministerio de Economía y
Competitividad). The laboratory of F.W. was supported by grants
from the Plan Nacional of the Dirección General de Ciencia y
Tecnología (SAF2012-39148-C03-01), from the European Union
(EU-FP7-2009-CT222887), and from CIBERNED (which is an initiative
of ISCIII). The Centro de Biología Molecular S.O. is also the
recipient of an institutional grant from the Ramón Areces
Foundation.
References
|
1
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tang YC, Williams BR, Siegel JJ and Amon
A: Identification of aneuploidy-selective antiproliferation
compounds. Cell. 144:499–512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Courtois-Cox S, Genther Williams SM,
Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein
PE, MacCollin M and Cichowski K: A negative feedback signaling
network underlies oncogene-induced senescence. Cancer Cell.
10:459–472. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Young AR, Narita M, Ferreira M, Kirschner
K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, et
al: Autophagy mediates the mitotic senescence transition. Genes
Dev. 23:798–803. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rong Y, McPhee CK, Deng S, Huang L, Chen
L, Liu M, Tracy K, Baehrecke EH, Yu L and Lenardo MJ: Spinster is
required for autophagic lysosome reformation and mTOR reactivation
following starvation. Proc Natl Acad Sci USA. 108:7826–7831. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Narita M, Young AR, Arakawa S, Samarajiwa
SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira
M, et al: Spatial coupling of mTOR and autophagy augments secretory
phenotypes. Science. 332:966–970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo JY, Chen HY, Mathew R, Fan J,
Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM,
Karantza V, et al: Activated Ras requires autophagy to maintain
oxidative metabolism and tumorigenesis. Genes Dev. 25:460–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Flier JS, Mueckler MM, Usher P and Lodish
HF: Elevated levels of glucose transport and transporter messenger
RNA are induced by ras or src oncogenes. Science. 235:1492–1495.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Greer EL, Oskoui PR, Banko MR, Maniar JM,
Gygi MP, Gygi SP and Brunet A: The energy sensor AMP-activated
protein kinase directly regulates the mammalian FOXO3 transcription
factor. J Biol Chem. 282:30107–30119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
García-Escudero V and Gargini R: Autophagy
induction as an efficient strategy to eradicate tumors. Autophagy.
4:923–925. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gargini R, García-Escudero V and Izquierdo
M: Therapy mediated by mitophagy abrogates tumor progression.
Autophagy. 7:466–476. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nikiforov MA, Riblett M, Tang WH,
Gratchouck V, Zhuang D, Fernandez Y, Verhaegen M, Varambally S,
Chinnaiyan AM, Jakubowiak AJ, et al: Tumor cell-selective
regulation of NOXA by c-MYC in response to proteasome inhibition.
Proc Natl Acad Sci USA. 104:19488–19493. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Beauséjour CM, Krtolica A, Galimi F,
Narita M, Lowe SW, Yaswen P and Campisi J: Reversal of human
cellular senescence: Roles of the p53 and p16 pathways. EMBO J.
22:4212–4222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thibodeaux CA, Liu X, Disbrow GL, Zhang Y,
Rone JD, Haddad BR and Schlegel R: Immortalization and
transformation of human mammary epithelial cells by a tumor-derived
Myc mutant. Breast Cancer Res Treat. 116:281–294. 2009. View Article : Google Scholar
|
|
16
|
Datta S, Hoenerhoff MJ, Bommi P, Sainger
R, Guo WJ, Dimri M, Band H, Band V, Green JE and Dimri GP: Bmi-1
cooperates with H-Ras to transform human mammary epithelial cells
via dysregulation of multiple growth-regulatory pathways. Cancer
Res. 67:10286–10295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duran A, Linares JF, Galvez AS,
Wikenheiser K, Flores JM, Diaz-Meco MT and Moscat J: The signaling
adaptor p62 is an important NF-kappaB mediator in tumorigenesis.
Cancer Cell. 13:343–354. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mammucari C, Milan G, Romanello V, Masiero
E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J,
et al: FoxO3 controls autophagy in skeletal muscle in vivo. Cell
Metab. 6:458–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao J, Brault JJ, Schild A, Cao P, Sandri
M, Schiaffino S, Lecker SH and Goldberg AL: FoxO3 coordinately
activates protein degradation by the autophagic/lysosomal and
proteasomal pathways in atrophying muscle cells. Cell Metab.
6:472–483. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maclean KH, Dorsey FC, Cleveland JL and
Kastan MB: Targeting lysosomal degradation induces p53-dependent
cell death and prevents cancer in mouse models of lymphomagenesis.
J Clin Invest. 118:79–88. 2008. View
Article : Google Scholar
|
|
22
|
Kanzawa T, Zhang L, Xiao L, Germano IM,
Kondo Y and Kondo S: Arsenic trioxide induces autophagic cell death
in malignant glioma cells by upregulation of mitochondrial cell
death protein BNIP3. Oncogene. 24:980–991. 2005. View Article : Google Scholar
|
|
23
|
Chang CP, Yang MC, Liu HS, Lin YS and Lei
HY: Concanavalin A induces autophagy in hepatoma cells and has a
therapeutic effect in a murine in situ hepatoma model. Hepatology.
45:286–296. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
García-Escudero V, Gargini R and Izquierdo
M: Glioma regression in vitro and in vivo by a suicide combined
treatment. Mol Cancer Res. 6:407–417. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim H, Farris J, Christman SA, Kong BW,
Foster LK, O'Grady SM and Foster DN: Events in the immortalizing
process of primary human mammary epithelial cells by the catalytic
subunit of human telomerase. Biochem J. 365:765–772. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levine AJ and Puzio-Kuter AM: The control
of the metabolic switch in cancers by oncogenes and tumor
suppressor genes. Science. 330:1340–1344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dang CV: Links between metabolism and
cancer. Genes Dev. 26:877–890. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin A, Yao J, Zhuang L, Wang D, Han J, Lam
EW and Gan B: The FoxO-BNIP3 axis exerts a unique regulation of
mTORC1 and cell survival under energy stress. Oncogene.
33:3183–3194. 2014. View Article : Google Scholar
|
|
30
|
Yogalingam G and Pendergast AM: Abl
kinases regulate autophagy by promoting the trafficking and
function of lysosomal components. J Biol Chem. 283:35941–35953.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Turcotte S, Chan DA, Sutphin PD, Hay MP,
Denny WA and Giaccia AJ: A molecule targeting VHL-deficient renal
cell carcinoma that induces autophagy. Cancer Cell. 14:90–102.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH,
Fidler IJ and Hung MC: Survival of cancer cells is maintained by
EGFR independent of its kinase activity. Cancer Cell. 13:385–393.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare
S, Kondo S, Kondo Y, Yu Y, Mills GB, et al: The tumor suppressor
gene ARHI regulates autophagy and tumor dormancy in human ovarian
cancer cells. J Clin Invest. 118:3917–3929. 2008.PubMed/NCBI
|