Introduction
Adrenocortical carcinoma (ACC) is a rare, highly
aggressive endocrine malignancy derived from cells of the adrenal
cortex and is often accompanied by unfavorable prognosis (1,2).
Despite significant advances in the last decade, its pathogenesis
is only incompletely understood and overall therapeutic means are
unsatisfactory (3). Therefore, it
is urgent to find new treatment targets for ACC.
ω-3 (n-3) and ω-6 (n-6) polyunsaturated fatty acids
(PUFAs) are essential fatty acids necessary for human health.
Epidemiological studies suggest that diet with a high n-6/n-3 ratio
contributes to cardiovascular disease, inflammation and cancer
(4–6), while consumption of more fish or fish
oil with a high n-3/n-6 ratio could reduce the risk of colon,
prostate and breast cancers (7,8).
Laboratory and animal studies have shown that n-3 PUFAs inhibit the
growth of various types of cancers both in vitro and in
vivo and affect emotions through the
hypothalamic-pituitary-adrenal (HPA) axis (9,10), as
well as catecholamine handling through adrenal chromaffin cells
(11), suggesting that n-3 PUFAs
may affect adrenal structure and function. However, the effect of
n-3 PUFAs on ACC is not known.
Mammalian target of rapamycin (mTOR) is a highly
conserved Ser/Thr kinase composed of two functionally distinct
signaling complexes, mTOR complex 1 (mTORC1) and complex 2 (mTORC2)
(12). mTOR is a key molecule for
controlling cell growth, proliferation, survival and metabolism
(12,13). Upregulation of mTOR signaling has
been found in many cancers and is thought to play important roles
in carcinogenesis and tumor progression (14–16).
The mTOR pathway is also activated in a subset of adrenal tumors,
but its role remains unclear.
We previously developed a transgenic mouse model
that expresses fat-1, a desaturase that converts n-6 PUFAs
to n-3 PUFAs and have applied it to study the effects of n-3 PUFAs
on breast cancer (17,18). In the present study, we investigated
the potential effects of exogenous and endogenous n-3 PUFAs on the
growth of ACC cell lines and tumor xenografts and explored the
possible signaling mechanisms.
Materials and methods
Materials
All cell culture reagents were obtained from
Gibco-BRL Life Technologies (Grand Island, NY, USA). Arachidonic
acid (AA) and docosahexenoic acid (DHA) were obtained from Cayman
Chemical (Ann Arbor, MI, USA) and prepared as dimethyl sulfoxide
(DMSO) stock solutions following the manufacturer's instructions.
Insulin, primary antibody against β-actin and HRP-conjugated
anti-mouse and anti-rabbit IgG were from Sigma (St. Louis, MO,
USA). Primary antibodies against phospho-S6 (S235/236) were
purchased from Cell Signaling Technology (Danvers, MA, USA).
Anti-S6, phospho-Akt (S473) and Akt antibodies were obtained from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). pST180
control vector and pST180-fat-1 vector were chemically
synthesized by GenePharma Co., Ltd. (Shanghai, China).
Cell lines and culture conditions
The human ACC cell lines SW13 and H295R were used in
the present study. SW13 was preserved in our laboratory in L-15
supplemented with 10% fetal bovine serum (FBS) (Invitrogen Corp.,
Grand Island, NY, USA) and cultured in a 37°C humidified atmosphere
containing 100% air and no CO2. The H295R cell line was
obtained from the Cell Center of the Chinese Academy of Sciences,
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 15% FBS and cultured in a 37°C humidified
atmosphere containing 95% air and 5% CO2. Cells were
trypsinized upon confluency and propagated to passage 2 before
being subcultured in 6-, 12- or 96-well plates for the
experiments.
Cell proliferation assay
Cells were seeded at 2×103 cells/well in
96-well plates. After being cultured in medium containing 10% FBS
for 24 h, the cells were treated with DHA at different
concentrations (10–80 μM) for 72 h with refreshing every 24
h. Cell viability was then assessed using Cell Counting Kit-8
(CCK-8) (cat no. KGA317; KeyGen Biotech, Nanjing, China) following
the manufacturer's instructions. The viability of the SW13 cells
was similarly assessed at 48 h after transfection with the
pST180-fat-1 or control vector.
Colony formation assay
Colony formation assays were performed as previously
described (19). Briefly, the cells
were seeded in 6-well plates in triplicate at a density of 200
cells/well containing 2 ml medium with 10% FBS. Twenty-four hours
later, the cells were cultured in fresh medium containing 5% FBS
alone as control or with DHA at different concentrations (5 and 20
μM) for 14 days at 37°C. The formed colonies were stained
for 15 min with a solution containing 0.5% crystal violet and 25%
methanol, followed by washing with water to remove excessive dye.
The colony numbers were counted by Quantity One® 1-D
analysis software (Bio-Rad Laboratories Inc., Hercules, CA,
USA).
Flow cytometric analysis
Flow cytometric analysis was performed as previously
described to determine the effects of DHA on the cell cycle
(19). Briefly, SW13 cells grown in
6-well plates (2×105 cells/well) were starved for 24 h
in basal medium to synchronize at the G1/S boundary, and were then
incubated in medium containing 10% FBS alone or with 20 μM
DHA. Twenty-four hours later, the cells were harvested by
trypsinization and fixed with 70% ethanol. Cell numbers were
assessed following the manufacturer's protocol (KeyGen Biotech),
and cell cycle distribution was analyzed by flow cytometry
(FACSCalibur; BD Biosciences, Bedford, MA, USA). DNA histograms
were plotted using FCS Express software, and the percentage of
cells at the G0/G1, S and G2/M stages were calculated.
Apoptosis assessment of cells and
tissues
The Annexin V-FITC apoptosis detection kit was used
for the apoptosis assay (KeyGen Biotech). In brief, SW13 cells
(1×106 cells/ml) were treated with 25 μM DHA for
12 h, harvested by trypsinization, washed twice with
phosphate-buffered saline (PBS), and resuspended in 500 μl
of binding buffer. The cells were then incubated with 5 μl
of Annexin V-FITC and 5 μl of propidium iodide (PI) for 10
min at room temperature in the dark and evaluated immediately by
flow cytometry (FACSCalibur).
Apoptotic cell death in paraffin-embedded tumor
tissue sections was examined using the DeadEnd™ Colorimetric TUNEL
System (Promega, Madison, WI, USA) according to the manufacturer's
protocol. Apoptotic cells were identified as dark brown nuclei
under a light microscope. The number of apoptotic cells was counted
in five random fields (magnification, ×400) in a blinded
manner.
Western blot analysis
Protein expression levels were determined by western
blot analysis as previously described (20). Briefly, after treatment for the
indicated time, cells were lysed immediately in Laemmli buffer
(62.5 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 50 mM
dithiothreitol, 0.01% bromophenol blue) for 5 min at 95°C. The cell
lysates were resolved by SDS/PAGE and transferred
electrophoretically to nitrocellulose membranes (Bio-Rad). The
membranes were incubated with specific antibodies and
immunoreactive proteins were revealed using an enhanced
chemiluminescence (ECL) kit (Santa Cruz Biotechnology, Inc.).
Xenografts in nude mice
Four-week-old female nude mice were purchased from
the Experimental Animal Center of Southern Medical University
(Guangzhou, China) and housed in a specific pathogen-free facility
in accordance with guidelines established by the Committee on
Animal Research of the Southern Medical University. SW13 cells at
early passages were harvested, prepared as 3×106
cells/100 μl PBS suspension and implanted subcutaneously
into the right flanks of each mouse. The animals were then randomly
assigned into two groups with 6 mice each and fed with a normal
(low) or high n-3 PUFA diet (21).
Bidimensional tumor measurements were taken every 2 days. Tumor
volume (V) was calculated as: V = 0.52 (length ×
width2). On day 29 after SW13 cells were implanted, all
animals were sacrificed and the tumors were collected and weighed.
The volume of the excised tumors was calculated as: V = 0.52
(length × width × depth). Various tumor specimens were minced and
lysed for western blot analysis, while others were fixed in 4%
formaldehyde, embedded in paraffin and cut in 4-μm sections
for immunohistochemical analysis.
Xenografts in severe combined immune
deficiency (SCID) and fat-1-SCID mice
Homozygous SCID mice (Jax no. 001131) (BALB/c
background) were bred with fat-1 transgenic mice (originally
on the C57BL/6 background) produced previously to generate
fat-1-SCID double-hybrid mice. These mice were backcrossed
with BALB/c mice for 10 generations. Female littermates lacking the
fat-1 transgenic gene were used as controls. DNA extractions
from the tail tips of offspring were subjected to PCR for
genotyping in accordance with the protocol on the Jackson
Laboratory webpage, using primers listed in Table I. Four-week-old female homozygous
SCID mice (n=6) with or without the fat-1 gene were injected
with 100 μl (1×105) of the SW13 cell suspension
and were subjected to bidimensional tumor measurement and tumor
sample analysis as described above.
 | Table IPrimer sequences for PCR
amplification. |
Table I
Primer sequences for PCR
amplification.
| Gene | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| fat-1 |
GGACCTGGTGAAGAGCATCCG |
GCCGTCGCAGAAGCCAAAC |
| SCID |
GGAAAAGAATTGGTATCCAC |
AGTTATAACAGCTGGGTTGGC |
Immunohistochemistry
Tumor allografts were removed. After being weighed,
they were immediately fixed in 2.5% glutaraldehyde-polyoxymethylene
solution and processed as paraffin sections using a standard
method. Three sequential sections (5 μm) of each sample were
used for hematoxylin and eosin (H&E) staining,
immunohistochemical (IHC) detection of phospho-S6 (S235/S236)
(1:300; Cell Signaling Technology) and IHC detection of phospho-Akt
(S473) (1:100; Santa Cruz Biotechnology, Inc.), respectively. Cells
with yellow brown cytoplasm or nuclei were considered positive. The
percentage of positive cells was calculated after counting 1,000
cells at higher magnification (×400) according to the following
formula: Percentage of positive cells = (number of positive
cells/number of total cells) × 100%.
Statistical analysis
Statistical analyses were performed using SPSS
(version 13.0). Data are presented as mean ± SD of at least three
independent experiments. Differences between two groups were
analyzed using the Student's t-test, and differences among more
than two groups were analyzed using one-way ANOVA and post hoc LSD
tests. A P<0.05 was considered as statistically significant. In
the case of western blot analysis, one representative set of data
is shown.
Results
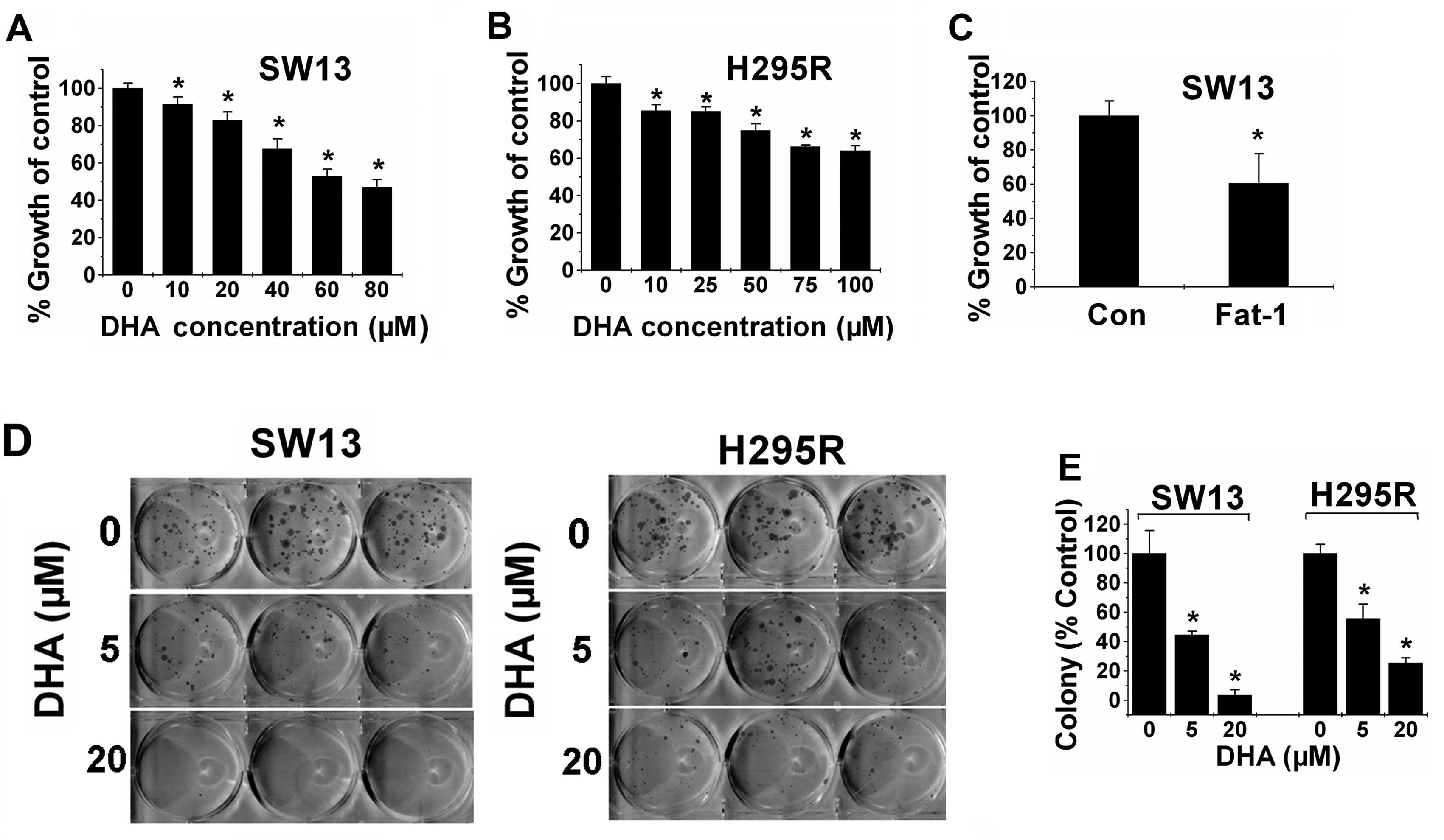
n-3 PUFAs inhibit ACC cell proliferation
and colony formation and induce G0/G1 cell cycle arrest
To investigate the potential protective role of n-3
PUFAs against ACC, we examined the effect of DHA on the
proliferation of ACC cell lines. We treated SW13 and H295R cells
with various concentrations (0–100 μM) of DHA and assessed
the effects after 48 h of treatment. As shown in Fig. 1A and B, DHA significantly inhibited
the proliferation of both ACC cell lines in a dose-dependent
manner. Transgenic expression of fat-1 is capable of
converting n-6 PUFAs to n-3 PUFAs, leading to an increase in the
amount of n-3 PUFAs and a decrease in the n-6/n-3 ratio. In
addition, SW13 cells were transfected with the fat-1 or
control vector, and cell viability was assessed 48 h later. As
expected, the proliferation rate of the fat-1-expressing
cells was significantly decreased by almost 40% compared with that
noted in the control cells (Fig.
1C). Together, these findings demonstrate that both exogenous
and endogenous n-3 PUFAs are effective to inhibit ACC cell
proliferation.
We next examined the effects of DHA on cell colony
formation of ACC cells. Our results showed that DHA prevented
colony formation of the ACC cells in a dose-dependent manner. When
used at 5 μM DHA, DHA inhibited the cell colony formation of
the SW13 and H295R cells by 60 and 50%, respectively. Additionally,
20 μM DHA decreased the colony formation of the SW13 and
H295R cells by 97 and 75%, respectively, compared with the control
(Fig. 1D and E).
We also examined the effects of DHA on cell cycle
progression by flow cytometric analysis. The SW13 cells were
serum-starved for 24 h to arrest cells at the G0/G1 phase. After
refreshing the medium, FBS was added to the arrested cells for 48 h
in the presence or absence of DHA. DHA-treated cells were arrested
at G0/G1, whereas FBS-treated cells progressed to the S phase (data
not shown). However, this effect was not significant.
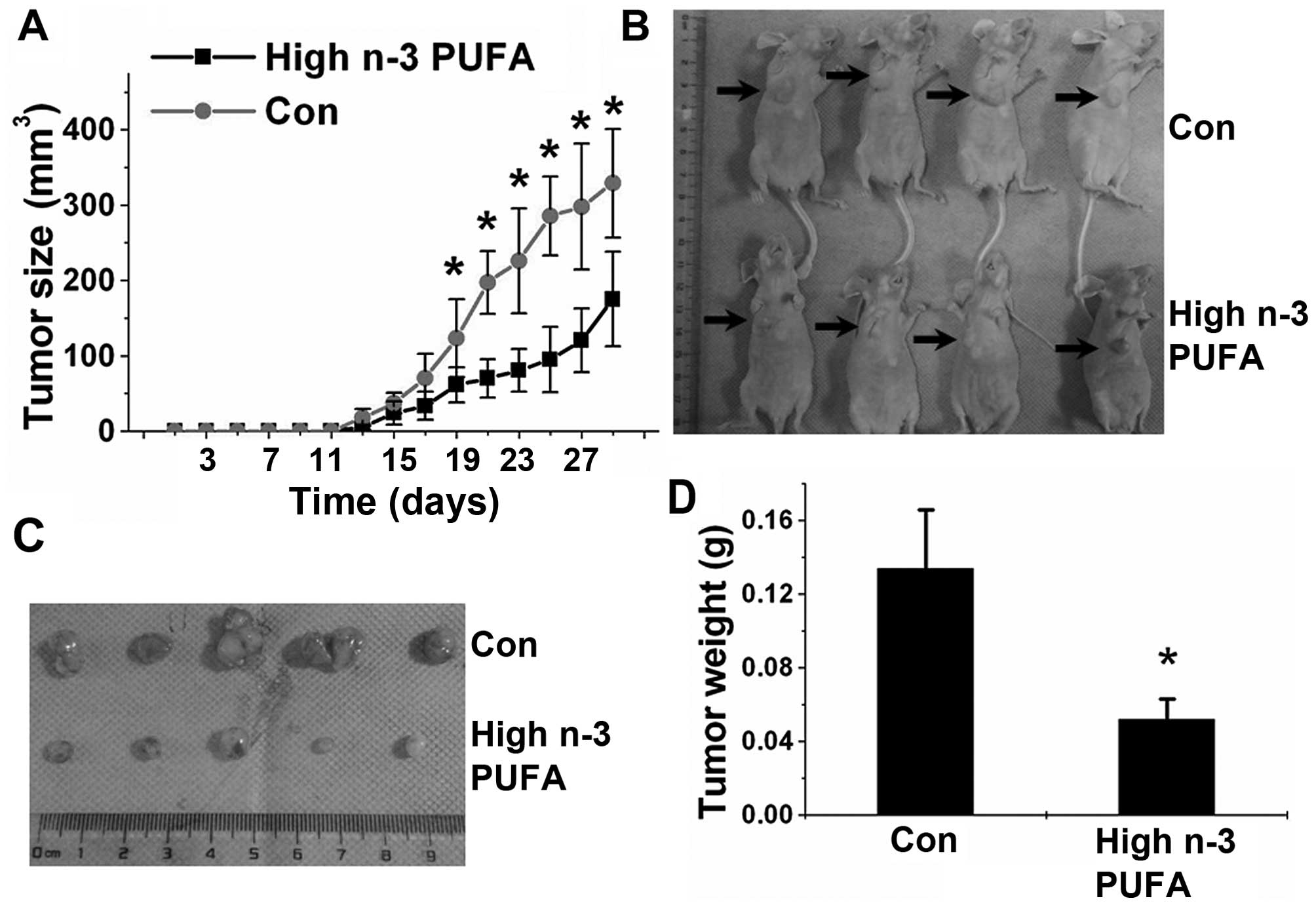
Dietary n-3 PUFAs inhibit the growth of
ACC xenografts in nude mice
After showing the inhibitory effects of n-3 PUFAs on
the proliferation of ACC cells, we examined the ability of n-3
PUFAs to inhibit tumor growth in vivo. We established an ACC
xenograft model by subcutaneously implanting SW13 cells into nude
mice. The animals were administered a normal (low) or high n-3 PUFA
diet. As shown in Fig. 2, a high
n-3 PUFA diet efficiently prevented tumor growth and reduced the
average tumor weight and volume. Fatty acid composition analysis
identified a significantly increased ratio of n-3/n-6 PUFAs in the
tumors of mice fed a high n-3 PUFA diet when compared with that of
mice fed a normal diet (Table
II).
| Table IIFatty acid composition in the nude
mouse tumors. |
Table II
Fatty acid composition in the nude
mouse tumors.
| Type of fatty acids
(mol % of total fatty acid) | n-3 PUFA diet
|
|---|
| Normal | High |
|---|
| C18:3 n-3,
α-linoleic acid | 0.25±0.12 | 0.44±0.08 |
| C20:5, n-3,
eicosapentaenoic acid (EPA) | 0.13±0.02 | 0.96±0.05 |
| C22:5, n-3,
docosapentaenoic acid (DPA) | 0.56±0.11 | 1.23±0.14 |
| C22:6, n-3,
docosahexaenoic acid (DHA) | 2.15±0.38 | 4.37±0.33 |
| n-3, total | 3.08±0.33 | 7.00±0.40a |
| C18:2, n-6,
linoleic acid | 19.24±3.12 | 17.32±2.17 |
| C20:4, n-6,
arachidonic acid (AA) | 1.75±0.37 | 1.22±0.18 |
| n-6, total | 20.99±2.92 | 18.55±2.08 |
| n-3/n-6 | 0.15±0.02 | 0.38±0.04a |
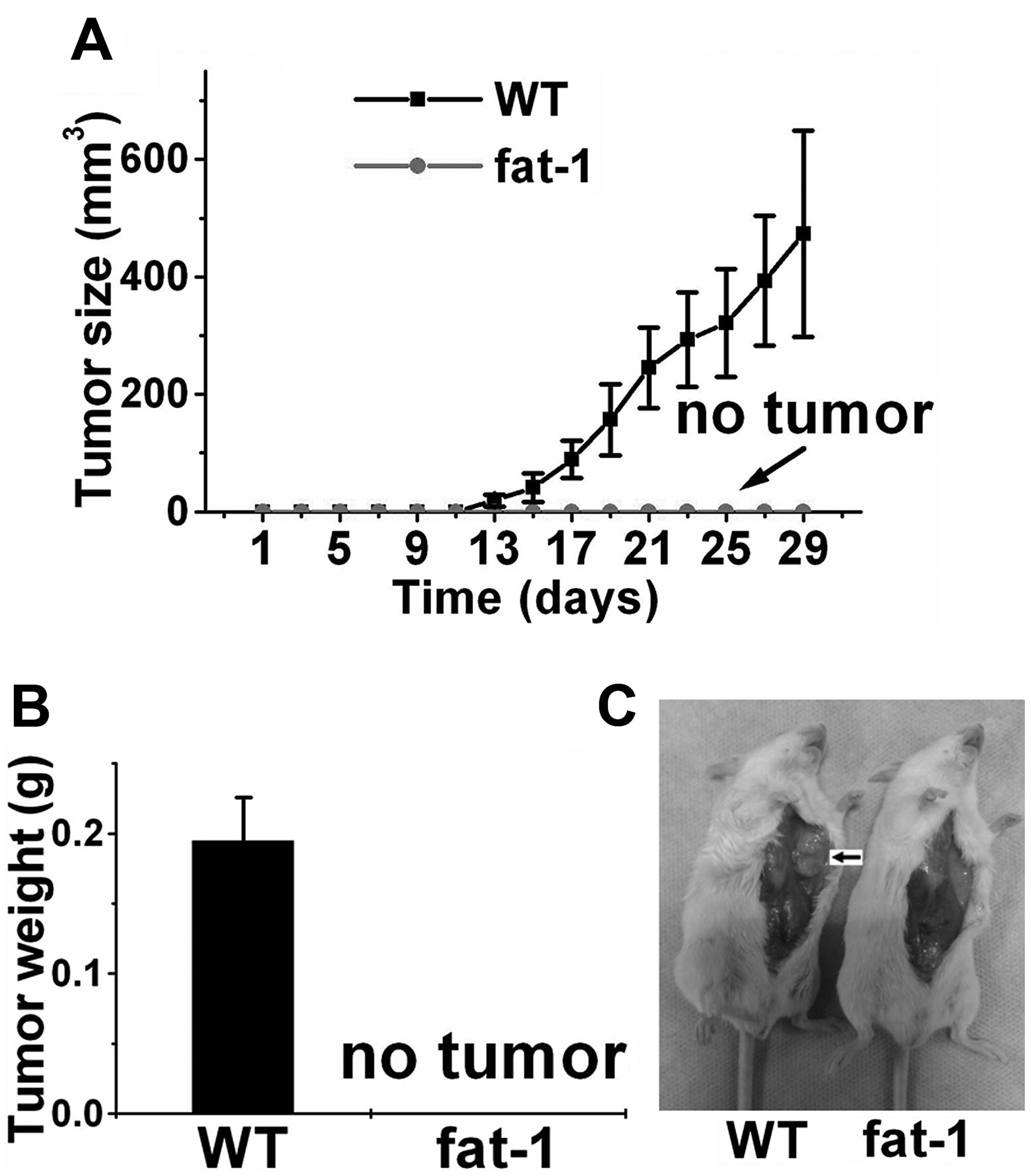
Endogenously produced n-3 PUFAs inhibit
the growth of ACC xenografts in SCID mice
Transgenic expression of fat-1 is capable of
converting n-6 PUFAs to n-3 PUFAs, leading to an increase in n-3
PUFAs and a decrease in the n-6/n-3 ratio, and allows us to perform
well-controlled studies in the absence of restricted diets. For
this purpose, we previously established fat-1 transgenic
SCID mice and confirmed that the ratio of n-3/n-6 PUFAs in the
fat-1 transgenic SCID mice is significantly increased
compared with this ratio in the wild-type (WT) SCID mice lacking
fat-1 expression (22).
Notably, although xenograft tumors with an average volume of 473
mm3 were observed within 4 weeks in the control SCID
mice, we failed to observe tumor growth in any of the fat-1
SCID mice (Fig. 3), suggesting that
ACC cells are unable to proliferative or survive in the presence of
high levels of endogenously produced n-3 PUFAs in vivo.
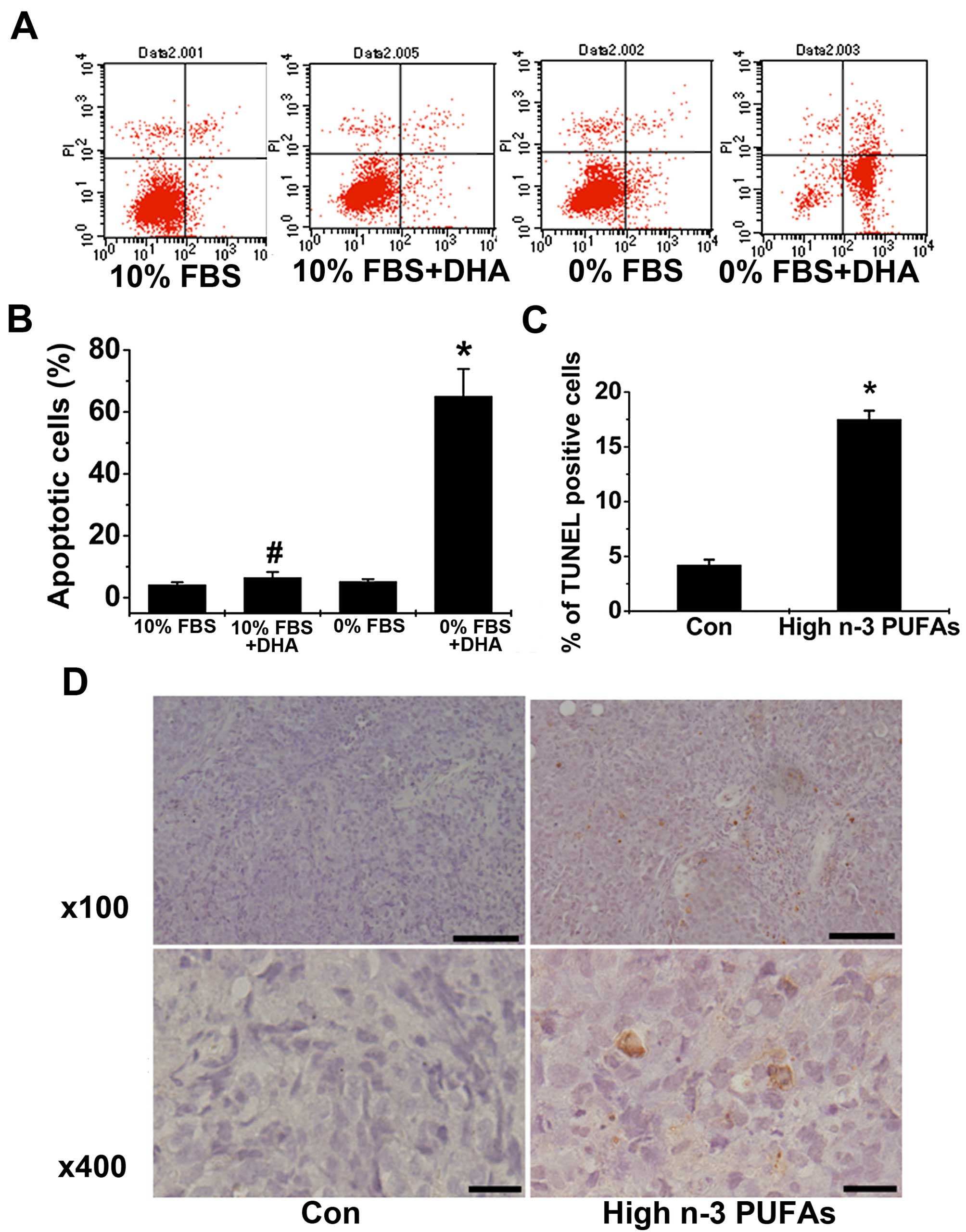
n-3 PUFAs promote ACC cell apoptosis both
in vitro and in vivo
We further examined whether n-3 PUFAs could induce
ACC cell apoptosis in vitro and in xenografts. The results
showed that DHA significantly increased the number of apoptotic
cells in serum-starved SW13 cells but not in SW13 cells cultured
with normal FBS (Fig. 4A and B),
suggesting that DHA promotes ACC cell apoptosis in vitro.
The effect of n-3 PUFAs on apoptosis was further examined in
xenograft models. The results revealed that administration of n-3
PUFAs promoted xenografted ACC tumor apoptosis as manifested by the
increased numbers of TUNEL-positive tumor cells in mice fed a high
n-3 PUFA diet (Fig. 4C and D). We
conclude that n-3 PUFAs promote ACC cell apoptosis both in
vitro and in vivo.
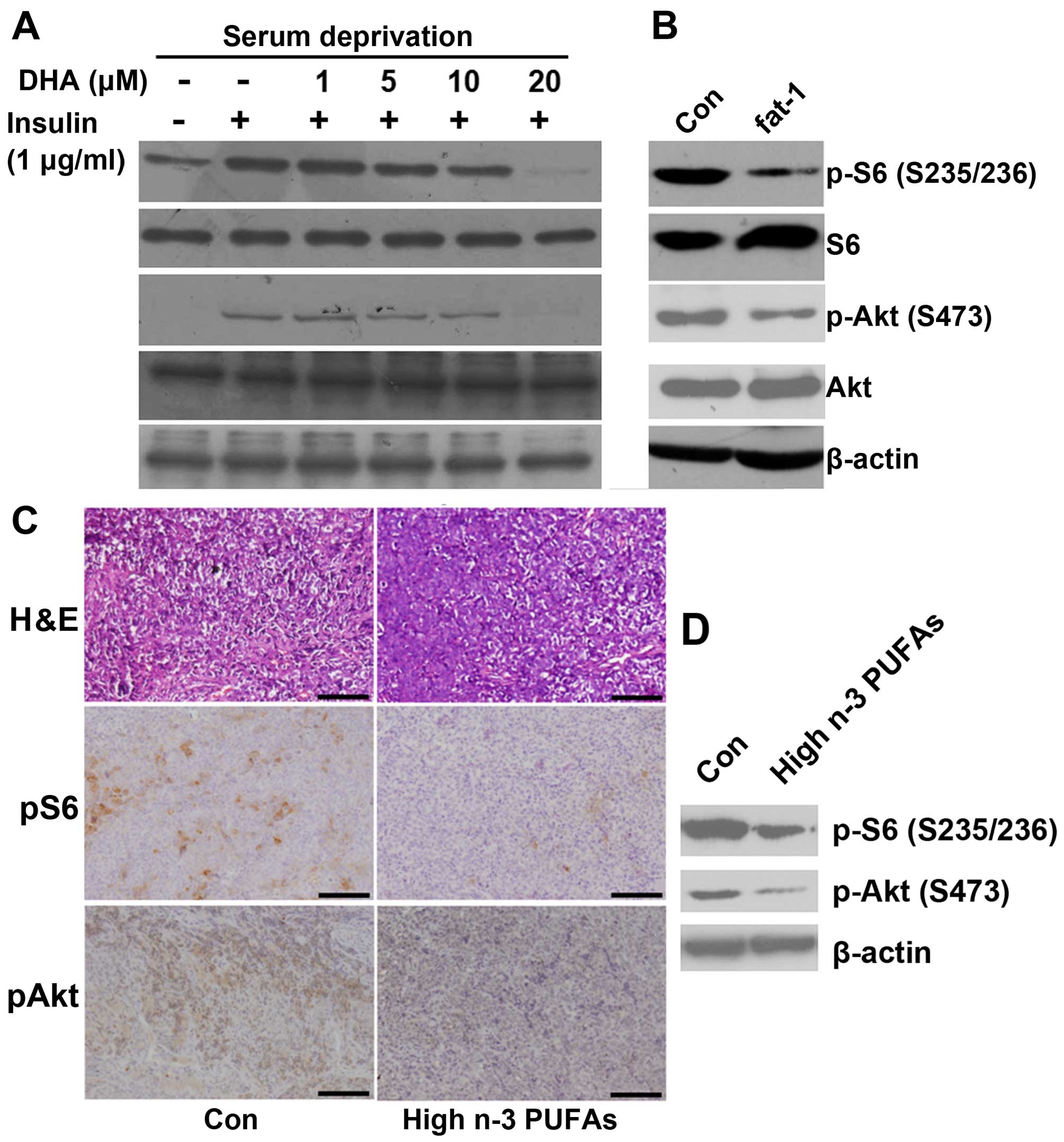
n-3 PUFAs inhibit mTORC1/2 signaling in
ACC cell lines and a xenograft mouse model
To investigate the underlying mechanisms of the
in vitro effect of n-3 PUFAs on mTORC1/2 signaling, we first
examined whether DHA (exogenous n-3 PUFAs) inhibited mTORC1/2 in
the ACC cells. In the SW13 cells, DHA rapidly and dose-dependently
suppressed insulin-stimulated mTORC1-directed phosphorylation of S6
(S235/235) and mTORC2-directed phosphorylation of Akt (S473)
(Fig. 5A). We next examined the
role of endogenously produced n-3 PUFAs in mTORC1/2 signaling. In
the SW13 cells transfected with fat-1 cDNA, phosphorylation
of S6 (S235/235) and Akt (S473) was significantly reduced compared
with the cells transfected with the control vector (Fig. 5B). These results suggest that both
the mTORC1 and mTORC2 signaling pathways are targets of exogenous
and endogenous n-3 PUFAs in ACC cells.
We next determined whether n-3 PUFAs could suppress
mTORC1/2 in vivo. The levels of mTORC1/2 signaling in the
excised tumors of the different groups were examined by
immunohistochemical staining and western blotting. As shown in
Fig. 5C, a high n-3 PUFA diet
decreased the percentage of p-S6 (S235/236) and p-Akt
(S473)-positive cells. Western blot analysis also showed that a
high n-3 PUFA diet resulted in a significant reduction in levels of
phosphorylated S6 (S235/236) and Akt (S473) (Fig. 5D). These data suggest that mTORC1
and mTORC2 signaling are the targets of n-3 PUFAs in vivo
and suppression of mTORC1/2 signaling by n-3 PUFAs may contribute
to their inhibitory effects on ACC tumor growth.
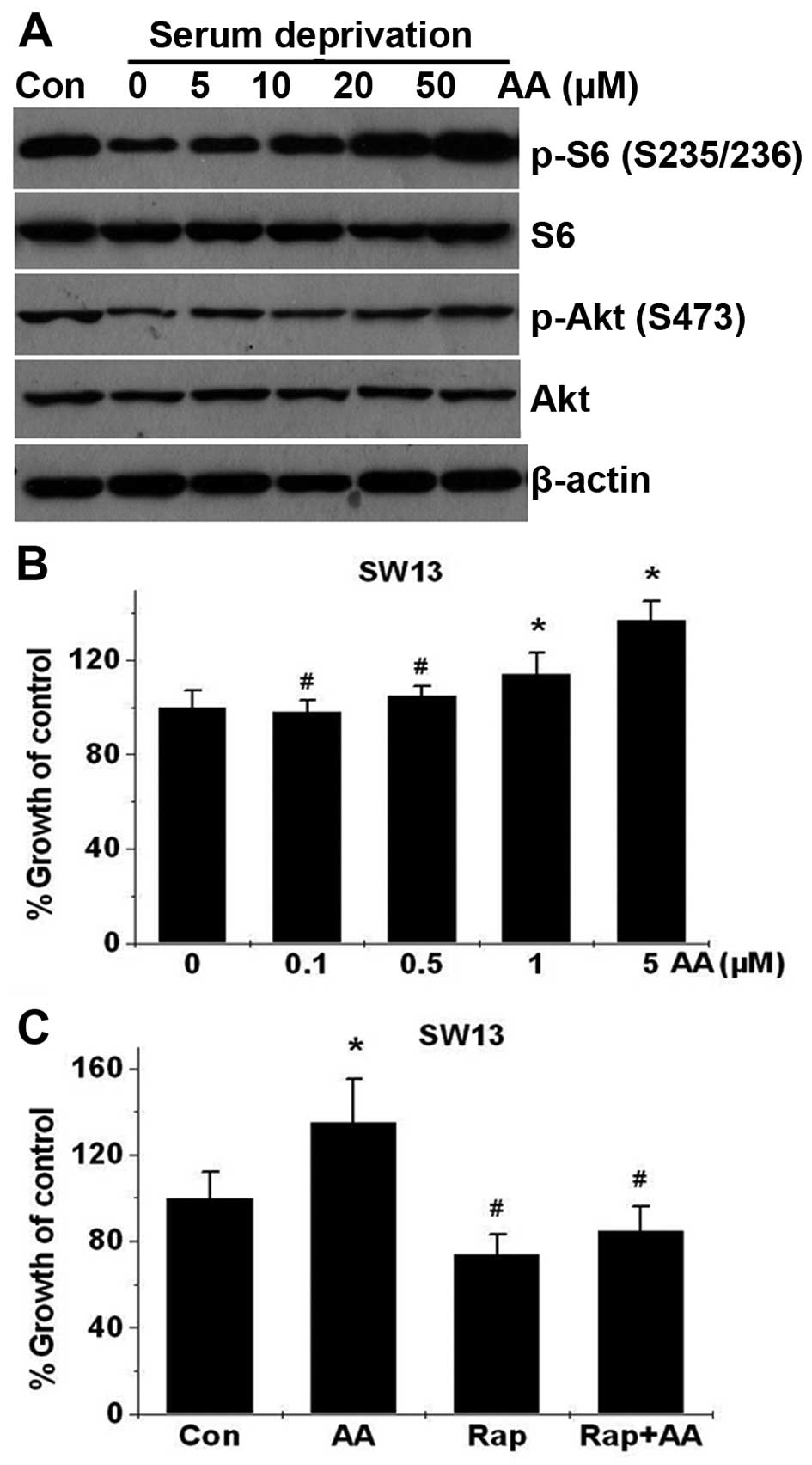
n-6 PUFAs activate mTORC1 signaling in
ACC cells and stimulate ACC cell proliferation
Several previous studies have shown that arachidonic
acid (AA) and its metabolites stimulate the growth and
proliferation of various tumor cells (6,23–25),
suggesting that n-6 PUFAs may have opposite effects on cancer when
compared with n-3 PUFAs. However, whether AA has any effect on ACC
cell proliferation has not been reported. As shown in Fig. 6B, AA dose-dependently stimulated
SW13 cell proliferation from 0.1 to 5 μM, and increased
mTORC1-directed phosphorylation of S6 (S235/235) (Fig. 6A) in the SW13 cells. To further
explore whether mTOR signaling plays a role in AA-induced cell
growth, we examined the effect of rapamycin, an mTORC1 inhibitor,
on SW13 cell proliferation. As shown in Fig. 6C, rapamycin treatment significantly
suppressed AA-stimulated growth, suggesting that mTORC1 is required
for AA-enhanced SW13 cell proliferation.
Discussion
The present study, for the first time, investigated
the roles of n-3 PUFAs in the growth of ACC using in vitro
cultured cells and an animal model. We found that DHA, a n-3 PUFA,
exhibits strong anticancer activity in human ACC cells through a
combination of multiple actions, including inhibition of cell
proliferation and cell cycle progression and induction of cell
apoptosis. The strong anticancer effect was also observed in
vivo in tumor-bearing mice. All this evidence indicates that
exogenous and endogenous n-3 PUFAs inhibit ACC cell growth both
in vitro and in vivo.
Although increasing evidence from animal and in
vitro studies indicate that n-3 PUFAs present in fatty fish and
fish oils inhibit the carcinogenesis of many types of tumors,
inconsistencies remain (4,21). Several factors may account for these
inconsistent results including: i) a wide variations in the amount
and source of n-3 PUFAs consumed in each study; and ii) the ratio
of n-6 to n-3, which may be more important than the absolute amount
of n-3 PUFAs, as suggested by animal and human studies (6). Transgenic expression of fat-1
enables the host to produce n-3 PUFAs endogenously while
concomitantly reducing the levels of n-6 PUFAs (17,21).
In addition, the fat-1 transgenic mouse is capable of
increasing n-3 content with a balanced n-6/n-3 PUFA ratio in all
tissues and allows carefully controlled studies to be performed in
the absence of restricted diets (17,21).
We found that ACC cells did not grow at all in fat-1
transgenic SCID mice possibly since the transplanted cells the in
fat-1 transgenic SCID mice underwent growth inhibition and
apoptosis after uptaking n-3 PUFAs from their immediate
environment. Due to the lack of a reagent available to induce
adrenal tumorigenesis and a genetically induced animal model with
adrenal tumorigenesis, it was not possible for us to investigate
whether n-3 PUFAs have any effect on adrenal carcinogenesis and
progression. However, we can conclude from our results that
exogenous and endogenous n-3 PUFAs can inhibit ACC growth both
in vitro and in vivo.
Understanding n-3 PUFA-regulated signaling pathways
in cancer may provide valuable information for assessing their
potential value in both cancer prevention and treatment. We found
that exogenous and endogenous n-3 PUFAs inhibited mTORC1 and mTORC2
signaling in the ACC cells and in the tissues of tumor-bearing
mice. mTOR is a key molecule which integrates diverse signals
including nutrients, growth factors, energy and stresses to control
cell growth, proliferation, survival and metabolism (12,13).
mTOR signaling is upregulated in many types of cancers and plays
key roles in the carcinogenesis and progression of these diseases,
thus it is regarded as a key target for cancer therapy (14–16).
mTORC1 inhibitors such as rapamycin and envirolumus have been used
for the treatment of ACC in preclinical trials, but the results
were not promising (26). The
reason is that these drugs only block mTORC1 and have little effect
on mTORC2, which has a feedback effect on mTORC1 signaling
(20). The development of dual
inhibitors of mTORC1 and mTORC2 may obviate this issue since they
appear to exhibit a superior effect in tumor treatment than
inhibitors of mTORC1 only (27).
Our findings that n-3 PUFAs target both mTORC1 and mTORC2 pathways
may explain their strong inhibitory effects on ACC in vitro
and in vivo. The mechanisms by which n-3/n-6 PUFAs regulate
mTORC1/2 remain to be identified.
We previously reported that AA-activated mTOR
signaling plays a critical role in breast carcinogenesis and
angiogenesis (25). Not
surprisingly, in the present study, we also found that AA activated
mTOR signaling in ACC cells and promoted ACC cell proliferation,
which requires activation of the mTORC1 signaling pathway.
Unfortunately, no animal model, which can mimic ACC development,
can be used to explore the effects of n-6 PUFAs on adrenal
carcinogenesis. Our results at least showed that n-6 PUFAs may be
related to ACC growth and upregulation of mTOR signaling in ACC may
be partly due to AA uptake. It has been reported that prostate or
breast-specific knockout of TSC1 (which activates mTORC1) can lead
to prostate or breast carcinogenesis (28,29).
Therefore, we can use the adrenal cortical-specific Cre mouse and
TSC1-loxp mouse to investigate whether adrenal mTORC1 activation
could lead to adrenal hyperplasia, adenoma formation or ACC
development and confirm the role of mTORC1 in adrenal
carcinogenesis (30).
In conclusion, our results indicate that both
endogenously synthesized and exogenously uptaken n-3 PUFAs inhibit
ACC growth, while n-6 PUFAs have an opposite effect, the mechanisms
of which may be mediated by mTORC1/2 signaling. However, further
research is required to investigate the key role of mTORC1/2
signaling in adrenal carcinogenesis. In addition, future research
must elucidate whether n-6 PUFA intake has any direct relationship
with mTOR signaling activation in ACC.
Acknowledgments
The present study was supported by the National
Natural Sciences Foundation of China (nos. 81302230 and 31371186),
the China Postdoctoral Science Foundation (2013M542159), the
Natural Science Foundation of Guangdong Province, China
(2014A030313296), and the Guangdong Province Outstanding Young
Teacher Training funds.
Abbreviations:
|
ACC
|
adrenocortical carcinoma
|
|
n-3 PUFAs
|
ω-3 polyunsaturated fatty acids
|
|
n-6 PUFAs
|
ω-6 polyunsaturated fatty acids
|
|
mTOR
|
mammalian target of rapamycin
|
|
mTORC1
|
mTOR complex 1
|
|
mTORC2
|
mTOR complex 2
|
|
SCID
|
severe combined immune deficiency
|
References
|
1
|
Ronchi CL, Kroiss M, Sbiera S, Deutschbein
T and Fassnacht M: EJE prize 2014: Current and evolving treatment
options in adrenocortical carcinoma: Where do we stand and where do
we want to go? Eur J Endocrinol. 171:R1–R11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang W, Hu W, Zhang X, Wang B, Bin C and
Huang H: Predictors of successful outcome after adrenalectomy for
primary aldosteronism. Int Surg. 97:104–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Erickson LA, Rivera M and Zhang J:
Adrenocortical carcinoma: Review and update. Adv Anat Pathol.
21:151–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gerber M: Ω-3 fatty acids and cancers: A
systematic update review of epidemiological studies. Br J Nutr.
107(Suppl 2): S228–S239. 2012. View Article : Google Scholar
|
|
5
|
Update on marine omega-3 fatty acids:
Management of dyslipidemia and current omega-3 treatment options.
Atherosclerosis. 230:381–389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kang JX and Liu A: The role of the tissue
omega-6/omega-3 fatty acid ratio in regulating tumor angiogenesis.
Cancer Metastasis Rev. 32:201–210. 2013. View Article : Google Scholar
|
|
7
|
Zhang F and Chen Y, Long J, Dong L, Wang Y
and Chen Y: Effect of n-3 and n-6 polyunsaturated fatty acids on
lipid metabolic genes and estrogen receptor expression in MCF-7
breast cancer cells. Clin Lab. 61:397–403. 2015.PubMed/NCBI
|
|
8
|
de Roos B and Romagnolo DF: Proteomic
approaches to predict bioavailability of fatty acids and their
influence on cancer and chronic disease prevention. J Nutr.
142:1370S–1376S. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abel S, Riedel S and Gelderblom WC:
Dietary PUFA and cancer. Proc Nutr Soc. 73:361–367. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Larrieu T, Hilal ML, Fourrier C, De
Smedt-Peyrusse V, Sans N, Capuron L and Layé S: Nutritional omega-3
modulates neuronal morphology in the prefrontal cortex along with
depression-related behaviour through corticosterone secretion.
Transl Psychiatry. 4:e4372014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gomes A, Correia G, Coelho M, Araújo JR,
Pinho MJ, Teixeira AL, Medeiros R and Ribeiro L: Dietary
unsaturated fatty acids differently affect catecholamine handling
by adrenal chromaffin cells. J Nutr Biochem. 26:563–570. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Foster KG and Fingar DC: Mammalian target
of rapamycin (mTOR): Conducting the cellular signaling symphony. J
Biol Chem. 285:14071–14077. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sengupta S, Peterson TR and Sabatini DM:
Regulation of the mTOR complex 1 pathway by nutrients, growth
factors, and stress. Mol Cell. 40:310–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dazert E and Hall MN: mTOR signaling in
disease. Curr Opin Cell Biol. 23:744–755. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu K, Liu P and Wei W: mTOR signaling in
tumorigenesis. Biochim Biophys Acta. 1846:638–654. 2014.PubMed/NCBI
|
|
16
|
Efeyan A and Sabatini DM: mTOR and cancer:
Many loops in one pathway. Curr Opin Cell Biol. 22:169–176. 2010.
View Article : Google Scholar :
|
|
17
|
Kang JX: From fat to fat-1: a tale of
omega-3 fatty acids. J Membr Biol. 206:165–172. 2005. View Article : Google Scholar
|
|
18
|
White PJ, Arita M, Taguchi R, Kang JX and
Marette A: Transgenic restoration of long-chain n-3 fatty acids in
insulin target tissues improves resolution capacity and alleviates
obesity-linked inflammation and insulin resistance in high-fat-fed
mice. Diabetes. 59:3066–3073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu J, Li M, Song B, Jia C, Zhang L, Bai X
and Hu W: Metformin inhibits renal cell carcinoma in vitro and in
vivo xenograft. Urol Oncol. 31:264–270. 2013. View Article : Google Scholar
|
|
20
|
Li M, Zhao L, Liu J, Liu A, Jia C, Ma D,
Jiang Y and Bai X: Multi-mechanisms are involved in reactive oxygen
species regulation of mTORC1 signaling. Cell Signal. 22:1469–1476.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Z, Zhang Y, Jia C, Wang Y, Lai P,
Zhou X, Wang Y, Song Q, Lin J, Ren Z, et al: mTORC1/2 targeted by
n-3 polyunsaturated fatty acids in the prevention of mammary
tumorigenesis and tumor progression. Oncogene. 33:4548–4557. 2014.
View Article : Google Scholar
|
|
22
|
Zheng H, Tang H, Liu M, He M, Lai P, Dong
H, Lin J, Jia C, Zhong M, Dai Y, et al: Inhibition of endometrial
cancer by n-3 polyunsaturated fatty acids in preclinical models.
Cancer Prev Res. 7:824–834. 2014. View Article : Google Scholar
|
|
23
|
Kiyabu GY, Inoue M, Saito E, Abe SK,
Sawada N, Ishihara J, Iwasaki M, Yamaji T, Shimazu T, Sasazuki S,
et al JPHC Study Group: Fish, n-3 polyunsaturated fatty acids and
n-6 polyunsaturated fatty acids intake and breast cancer risk: The
Japan Public Health Center-based prospective study. Int J Cancer.
137:2915–2926. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thomasz L, Oglio R, Rossich L, Villamar S,
Perona M, Salvarredi L, Dagrosa A, Pisarev MA and Juvenal GJ: 6
Iodo-δ-lactone: A derivative of arachidonic acid with antitumor
effects in HT-29 colon cancer cells. Prostaglandins Leukot Essent
Fatty Acids. 88:273–280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wen ZH, Su YC, Lai PL, Zhang Y, Xu YF,
Zhao A, Yao GY, Jia CH, Lin J, Xu S, et al: Critical role of
arachidonic acid-activated mTOR signaling in breast carcinogenesis
and angiogenesis. Oncogene. 32:160–170. 2013. View Article : Google Scholar
|
|
26
|
Fraenkel M, Gueorguiev M, Barak D, Salmon
A, Grossman AB and Gross DJ: Everolimus therapy for progressive
adrenocortical cancer. Endocrine. 44:187–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou HY and Huang SL: Current development
of the second generation of mTOR inhibitors as anticancer agents.
Chin J Cancer. 31:8–18. 2012.
|
|
28
|
Hsu JL, Liu SP, Lee CC, Hsu LC, Ho YF,
Huang HS and Guh JH: A unique amidoanthraquinone derivative
displays antiproliferative activity against human
hormone-refractory metastatic prostate cancers through activation
of LKB1-AMPK-mTOR signaling pathway. Naunyn Schmiedebergs Arch
Pharmacol. 387:979–990. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pande M, Bondy ML, Do KA, Sahin AA, Ying
J, Mills GB, Thompson PA and Brewster AM: Association between
germline single nucleotide polymorphisms in the PI3K-AKT-mTOR
pathway, obesity, and breast cancer disease-free survival. Breast
Cancer Res Treat. 147:381–387. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Z, Dong H, Jia C, Song Q, Chen J,
Zhang Y, Lai P, Fan X, Zhou X, Liu M, et al: Activation of mTORC1
in collecting ducts causes hyperkalemia. J Am Soc Nephrol.
25:534–545. 2014. View Article : Google Scholar :
|