Introduction
The endoplasmic reticulum (ER) is an essential
organelle responsible for protein maturation and quality control
and is the primary site of oxidative protein folding. During
dysfunctional oxidative protein folding in the ER, misfolded
proteins accumulate in the lumen accompanied by increased reactive
oxygen species (ROS) production, a condition known as 'ER/oxidative
stressʼ (1). Several studies have
indicated that induction of ER stress is closely associated with
oxidative stress during the pathogenesis of multiple human diseases
such as diabetes, neurodegenerative diseases and cancer (2–5).
However, the role of ROS and related signals during ER stress is
not well understood.
Though the mammalian response to stress is complex,
often involving multiple signaling pathways acting in concert to
influence cell fate, mitogen activated protein kinase (MAPK) family
members are crucial for cell maintenance. There are three types of
MAP kinases, the extracellular signal-regulated kinases (ERKs),
c-Jun N-terminal kinases (JNKs) and p38. Activating different
combinations of MAPKs can produce distinct biological responses.
JNK and p38 MAPK are activated by a diverse array of ER
stress-inducing agents such as thapsigargin, tunicamycin or
dithiothreitol (DTT), which may cause depletion of ER
Ca2+, inhibition of N-linked protein glycosylation or
impairment of disulfide bond formation (6). Sustained activation of JNK and p38 is
dependent on apoptosis signaling regulating kinase 1 (ASK1), a
molecule implicated in the apoptosis mediated by ER stress
(7). Inhibition of MEK blocks
glucose-regulated protein-78 (GRP78) upregulation and enhances
apoptosis induced by ER stress in gastric cancer cells (8). In another study, the ER stress
response modulated the balance between ERK and JNK signaling
pathways to prevent cell death after oxidative injury (9). In addition, ROS may influence cell
fate by regulating MAPK signaling pathways (10). These findings imply that ROS
produced during ER stress may play a vital role, via regulation of
MAPKs, in determining whether cells survive.
Autophagy is a conserved cellular protein
degradation process (11) and it
has been reported that ER stress is one of its major inducers
(12). Most evidence indicates that
autophagy activation during ER stress primarily promotes survival,
clearing unfolded proteins that have accumulated in the ER lumen
(13). However, various studies
have suggested that autophagy switches from an anti-apoptotic to a
pro-apoptotic process with increased ER stress (14). The molecular mechanisms involved in
autophagy induction by ER stress are diverse, including the
inositol requiring enzyme 1-α (IRE1-α)-JNK and the [RNA-dependent
protein kinase (PKR)-like ER kinase (PERK)]-activating
transcription factor 4 (ATF4) pathways (15). In other studies, ROS regulated
autophagy through MAPK signaling pathways (16). JNK activation regulated expression
of autophagy-related genes (17,18)
and ERK activation influenced the fusion of autophagosomes and
lysosomes (19). The protein p38
itself was implicated as a substrate of the autophagy pathway
(6). Overall, these findings
suggest that MAPK signaling pathways are involved in the process of
autophagy occurring during ER stress.
The aim of the present study was to explore effects
of ROS and MAPK signaling pathways on autophagy and apoptosis
during DTT-induced ER/oxidative stress. Our findings suggested
that, in HeLa cells, ROS was switched from a pro-survival to a
pro-apoptotic signal as ER stress increased. The activation profile
of MAPKs signaling pathways changed as ROS production increased,
resulting in inhibition of autophagy flux. Impaired autophagic
flux, in turn, aggravated ER stress, ultimately leading to cell
death. Regulation of autophagy through ROS and MAPKs during
ER/oxidative stress suggests novel targets for cancer therapy.
Materials and methods
Reagents and antibodies
1,4-DTT, N-acetyl-L-cysteine (NAC), Hoechst 33258,
acridine orange (AO) and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma (St. Louis, MO, USA). LysoTracker Red
DND-99, fetal bovine serum (FBS), Roswell Park Memorial Institute
(RPMI)-1640 culture medium, penicillin, streptomycin, propidium
iodide (PI) and Annexin V-FITC were purchased from Invitrogen
(Carlsbad, CA, USA). 5-(6)-carboxy-2′,7′-dichlorofluorescein
diacetate (DCFH-DA) was purchased from Sigma Chemical Co. (St.
Louis, MO, USA). Antibodies against β-actin, Grp78, Chop,
caspase-3, cleaved-caspase-3, p62, LC3-II, JNK, phospho-JNK, p38,
phospho-p38, ERK and phosphor-ERK were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Enhanced chemiluminescence
(ECL) reagents were from Thermo Scientific (Rockford, IL, USA).
Cell culture
Human cervical carcinoma cells (HeLa cells) were
purchased from the American Type Culture Collection (ATCC;
Rockville, MD, USA) and cultured in RPMI-1640 medium supplemented
with 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin
(complete medium) and were grown in a humidified cell culture
incubator equilibrated with 95% air and 5% CO2 at
37°C.
Cell viability assays
Cells were plated into 96-well plates at a density
of 1×104 cells/well in 200 µl complete medium.
Each treatment was repeated in six separate wells. MTT reagent [10
µl of 10 mg/ml MTT in phosphate-buffered saline (PBS)] was
added to each well and cells were returned to the cell culture
incubator for 4 h. After incubation, the formazan crystals were
dissolved in 150 µl dimethylsulfoxide (DMSO). Absorbance was
recorded at a wavelength of 490 nm.
Apoptosis analysis by Hoechst 33258
staining
Apoptotic morphological alterations in nuclear
chromatin were detected by Hoechst 33258 staining. Briefly, HeLa
cells were cultured in 24-well plates and treated as indicated for
24 h. Cells were washed with ice-cold PBS and fixed with 4% (w/v)
paraformaldehyde overnight. The plates were then incubated with 10
µM Hoechst 33258 staining solution for 10 min. The cells
were visualized under a fluorescence microscope (IX-71; Olympus,
Tokyo, Japan).
Morphological assessment
Cells were seeded at 2.0×105 cells/well
in 6-well cell culture dishes and treated with experimental
conditions, as indicated in the individual images, during their
logarithmic growth phase. After various treatments and time points,
morphological alterations were analyzed under an inverted phase
contrast microscope (Olympus) at a magnification of ×20.
Western blotting
After exposure to experimental conditions,
expression of selected proteins in each sample was determined by
western blotting. Briefly, after quantitating protein in each
sample with the Bio-Rad protein reagent (40 µg protein/well)
they were separated by SDS-PAGE and transferred onto an Immun-Blot
PVDF membrane (both from Bio-Rad Laboratories, Hercules, CA, USA).
After blocking in Tris-buffered saline containing 5% (w/v) non-fat
dry milk at room temperature for 1 h, the membranes were incubated
with specific primary antibodies overnight at 4°C. Following
washing with PBS-Tween-20, membranes were incubated with
horseradish-peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology) at room temperature for 1 h. Membranes were then
incubated in ECL reagents and images were captured by Syngene Bio
Imaging (Synoptics, Cambridge, UK). Densitometric quantitation of
bands was performed using Syngene Bio Imaging tools.
Apoptosis assay
After exposure to experimental conditions, cells
were trypsinised and incubated with PI (1 µg/ml) and Annexin
V-FITC (1 µg/ml) for 15 min at 37°C. Samples were then
analyzed for apoptosis with a FACScan flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA).
Measurement of intracellular ROS
production
Cells were plated in black cell culture fluorometric
plates at a density of 1×104 cells/well in 200 µl
complete medium. Each treatment was repeated in six separate wells.
After cells were treated under the various experimental conditions,
the cells were incubated with 100 µM DCFH-DA in PBS for 30
min at 37°C in the dark followed by a PBS wash step. The
fluorescence of the cells from each well was measured with a
microplate reader (BioTek Synergy™ HT, USA) at 525 nm. The
percentage increase in fluorescence per well was calculated as
previously described (20).
Acidic compartment detection with AO
Autophagy is characterized by formation of acidic
vesicular organelles (autophagosomes and autolysosomes). AO, a
fluorescent weak base, causes such acidic compartments to fluoresce
bright red, and the cytoplasm and nucleolus to fluoresce bright
green and dim green, respectively. Cells were plated on coverslips,
washed in PBS with 5% FBS and then stained with AO (100
µg/ml) in the dark for 15 min at room temperature. Staining
was performed in the presence of various drugs or their vehicles,
as indicated. Cells were then washed twice with PBS and analyzed
under an inverted fluorescence microscope (Olympus) with a ×40
objective.
Acidic compartment detection with
LysoTracker Red
Cells were incubated for 15 min in PBS containing
LysoTracker Red DND-99 (100 nM), a fluorescent acidotropic probe
with a high selectivity for acidic organelles and showing good
retention after aldehyde fixation. After washing with PBS, cells
were fixed with 4% paraformaldehyde in PBS for 30 min at room
temperature, washed twice with PBS, and then analyzed under the
inverted fluorescence microscope (Olympus) with a ×40
objective.
Statistical analysis
Results are expressed as means ± standard deviation
(SD) or means ± standard error of mean (SEM), as indicated in the
figure legends. Data are representative of three independent
experiments performed in triplicate. Statistical analysis of the
data was performed using one-way ANOVA. The Tukey's post hoc test
was used to determine the significance for all pairwise comparisons
of interest. Differences were considered statistically significant
for values of p<0.05.
Results
Severe ER stress induced by DTT
contributes to HeLa cell death
HeLa cells were treated with increasing doses of DTT
(1.6, 3.2, 6.4 and 9.6 mM) for 12 or 24 h and then the survival
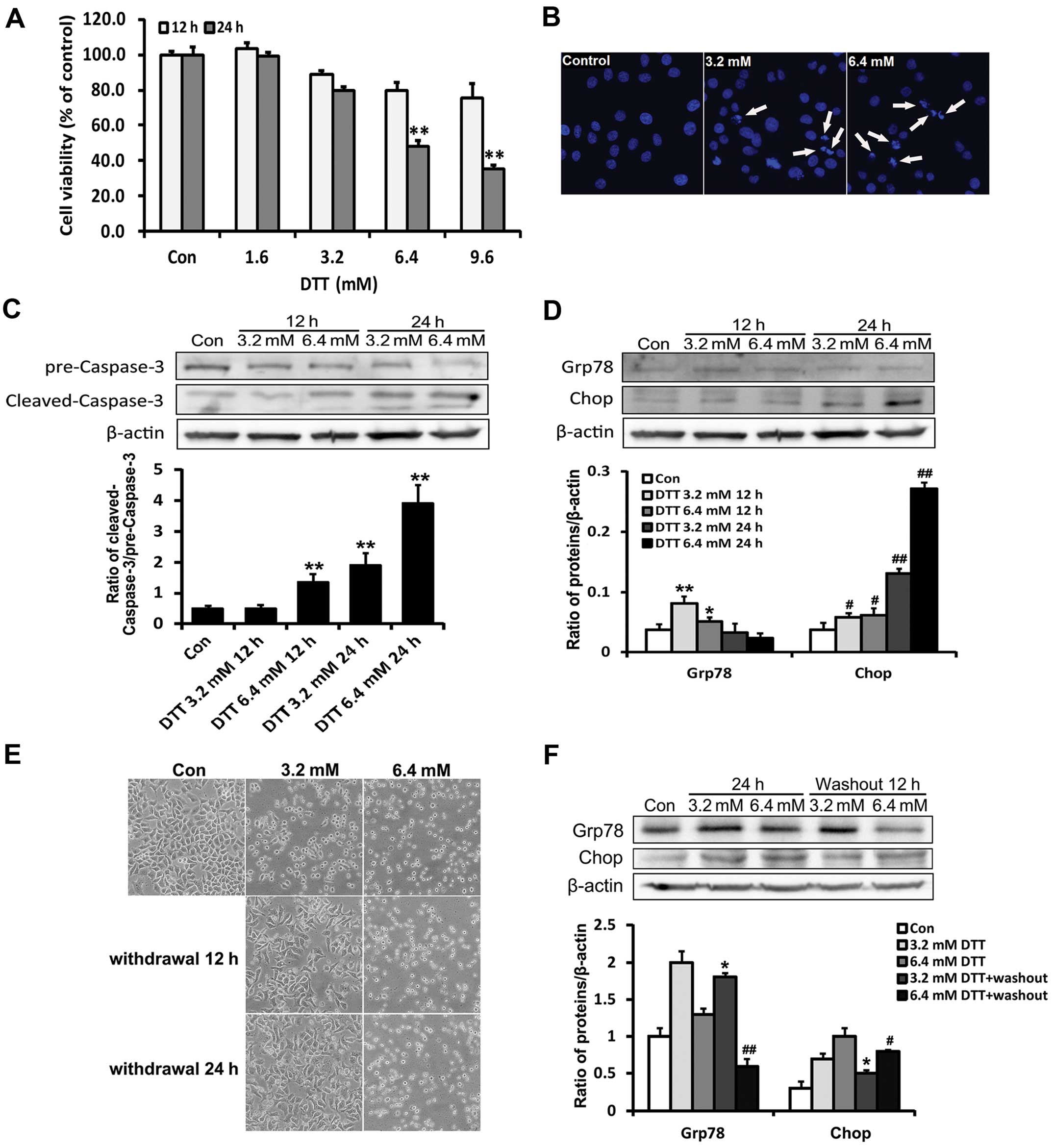
rate was detected using MTT assay. The result showed that the
viability of HeLa cells was decreased by DTT in a time- and
dose-dependent manner (Fig. 1A).
Based on the MTT results, we treated HeLa cells with DTT (3.2 and
6.4 mM) for 12 or 24 h and then assessed apoptosis based on Hochest
33258 staining and caspase-3 activation. Compared with the control
group, apoptotic chromatin condensation was obvious in DTT-treated
HeLa cells (Fig. 1B). DTT enhanced
the expression of cleaved caspase-3 (Fig. 1C).
As DTT is a strong inducer of ER stress, we
investigated whether ER stress was partially responsible for the
cell death induced by DTT, through assessing expression of ER
stress marker proteins Grp78 and Chop. Grp78 is an ER chaperone
protein believed to increase the ER protein folding capacity and
suppress pro-apoptotic pathways (21). Chop is the product of a gene induced
by growth arrest and DNA damage, also known as the 153/C/EBP
(GADD153) homology protein, involved in cell death induced by ER
stress (22). We found that DTT
decreased the expression of Grp78 and enhanced that of Chop
(Fig. 1D). These results indicated
that DTT efficiently induced intracellular apoptosis in HeLa cells,
partially through an ER stress-mediated apoptotic pathway.
Previous studies showed that cell death was
unavoidable only when the ER stress was too prolonged or severe
(23). We observed the morphology
of HeLa cells after treatment with different doses of DTT for 12 h,
followed by replacement with fresh medium and incubation for an
additional 12 or 24 h (Fig. 1E).
Compared with controls, DTT-treated cells were rounded, displaying
a higher refractive index after treatment. After DTT removal, the
morphology of cells previously exposed to 3.2 mM DTT became normal,
while those previously exposed to 6.4 mM DTT remained rounded. Chop
expression in cells previously exposed to 3.2 mM DTT decreased
whereas Grp78 expression remained elevated after DTT removal. In
contrast, in cells previously exposed to 6.4 mM DTT, Chop
expression remained elevated whereas that of Grp78 was decreased
after DTT removal (Fig. 1F). These
expression patterns likely reflected cellular adaptation, including
attempted restoration of ER homeostasis, occurring under milder ER
stress conditions.
These results, overall, indicated that severe ER
stress induced by DTT contributed to HeLa cell death.
The effect of ROS changes from
pro-survival to pro-death as ER stress increased
Since DTT induces ER stress primarily by disrupting
the redox state of the ER, we next investigated whether ROS were
involved in DTT-induced cell death. We first assessed intracellular
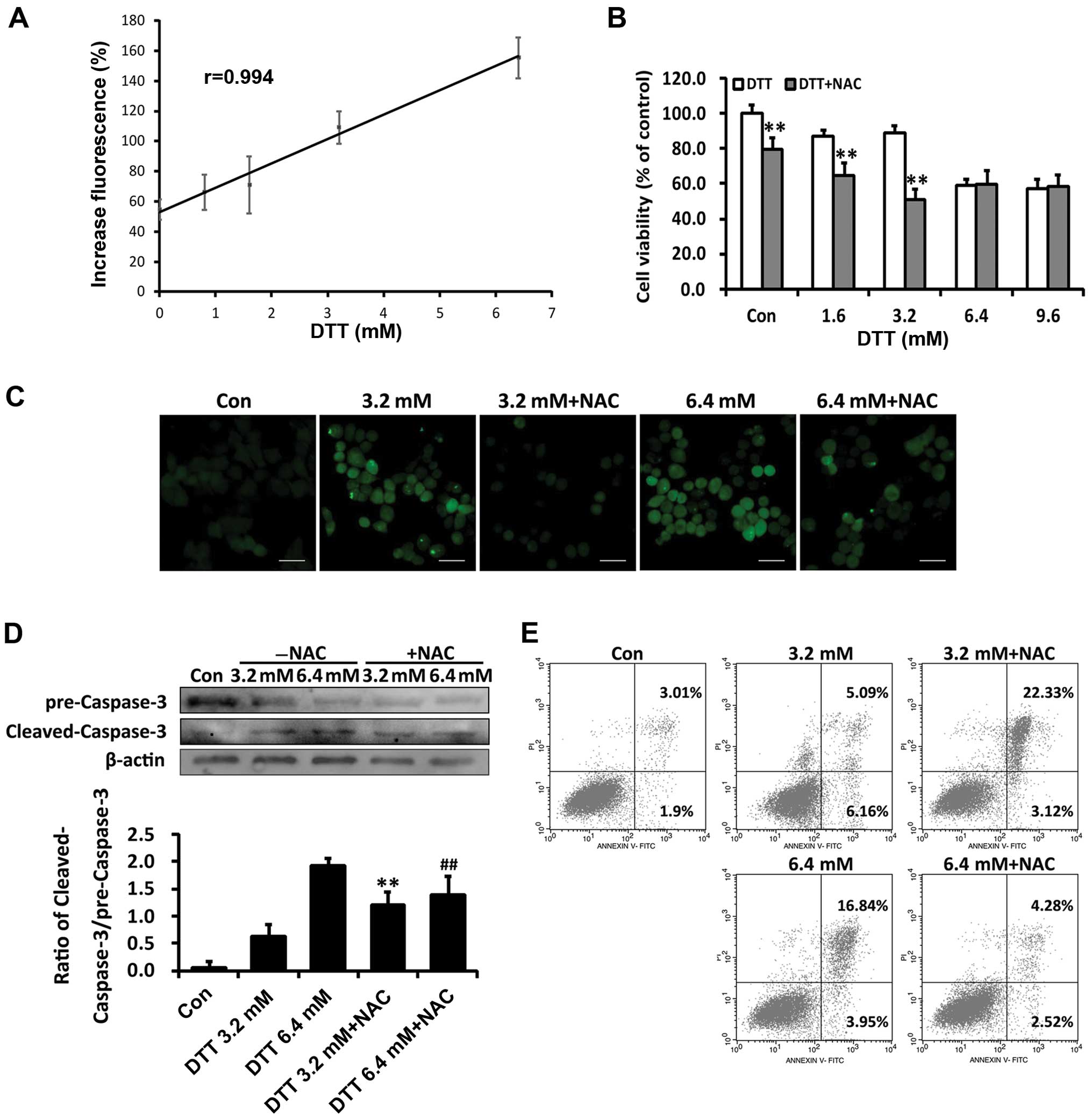
ROS levels in HeLa cells loaded with DCFH-DA. As shown in Fig. 2A, DTT treatment increased ROS
production in HeLa cells and ROS levels were linearly correlated
with DTT concentrations. Data from the MTT assay showed that NAC
exacerbated cell death induced by the lower dose (3.2 mM) DTT, but
attenuated that induced by 6.4 mM DTT (Fig. 2B). The antioxidant NAC reduced the
ROS levels induced by DTT (Fig.
2C).
We assessed involvement of ROS in apoptosis induced
by DTT. As shown in Fig. 2D,
compared with cells treated with DTT (3.2 mM) alone, cells
pretreated with NAC showed enhanced expression of cleaved
caspase-3. In contrast, NAC decreased cleaved caspase-3 levels in
cells exposed to 6.4 mM DTT. DTT-treated HeLa cells with or without
NAC pretreatment were stained with PI and Annexin V-FITC and
apoptotic cell populations quantified by flow cytometry. In cells
pretreated with NAC, the percentage of apoptosis induced by 3.2 mM
DTT was increased by 17.24% (p<0.05; Fig. 2E). In contrast, in cells treated
with 6.4 mM DTT, NAC pretreatment decreased the apoptotic cell
population by 12.56% (p<0.05; Fig.
2F).
The extent of ER stress induced by DTT was
dose-dependent. Therefore, these findings indicated that ROS
changed from a pro-survival to pro-death signal as the ER stress
increased.
Differential activation of the MAP-kinase
p38, JNK and ERK is involved in ROS-mediated cell death
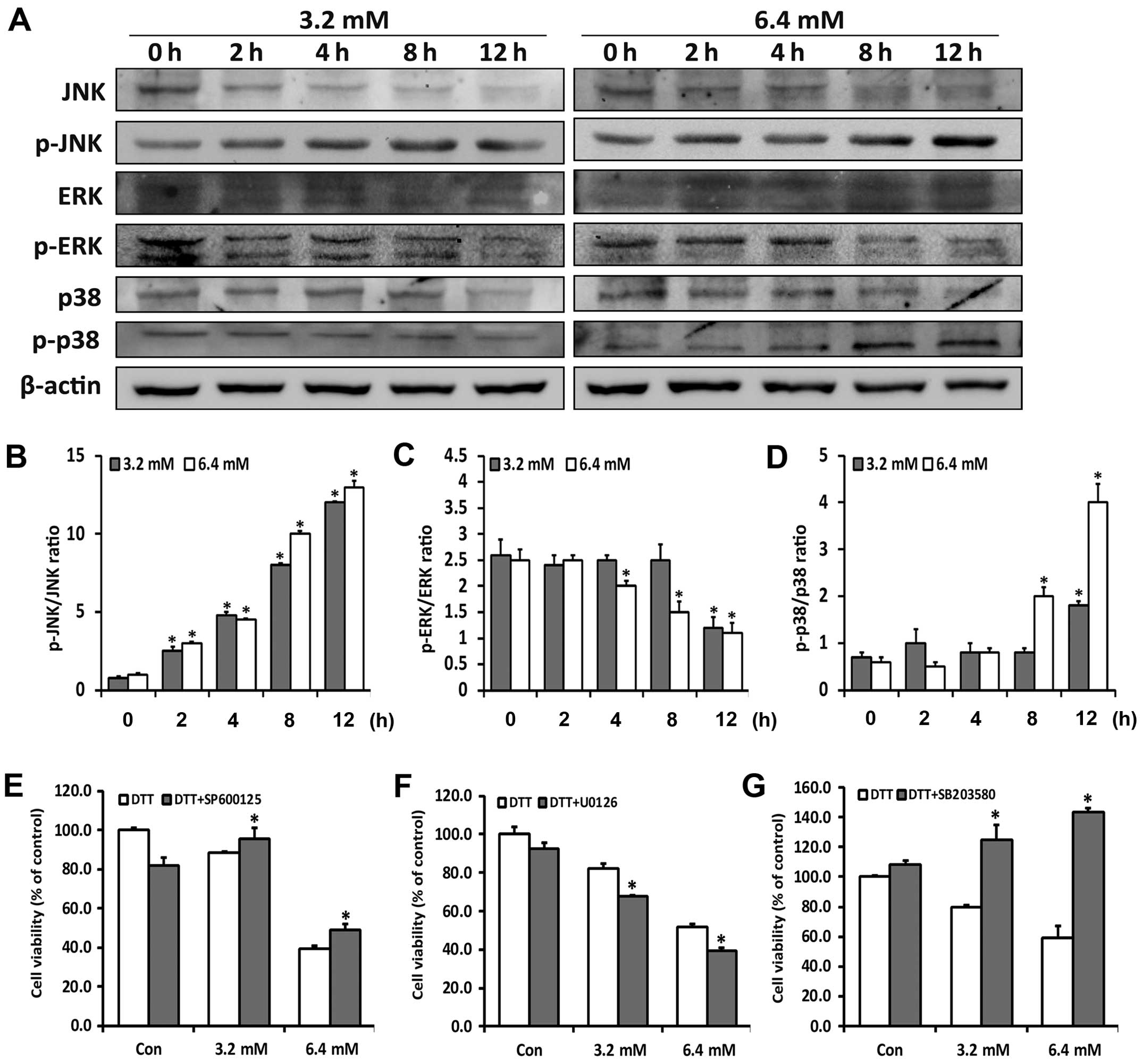
The MAPKs are downstream signaling molecules
activated by ROS and ER stress, and have been shown to be crucial
for maintenance of cells (24). To
investigate whether the MAPKs were related to the diverse roles of
ROS in DTT-induced cell death, we examined activation of p38, JNK
and ERK by western blotting. Low cytotoxic doses of DTT transiently
activated JNK and p38, whereas high dose of DTT persistently
activated JNK and p38 and simultaneously reduced ERK activity
(Fig. 3A–D). Cells were next
treated with DTT with or without corresponding inhibitors of JNK
(SP600125), p38 (SB203580) and ERK (U0126) (Fig. 3E–G) for 24 h. Based on the MTT
assays, SP600125 or SB203580 decreased and U0126 increased,
DTT-induced cytotoxity in HeLa cells. The inhibitors alone
exhibited no significant cytotoxity.
These results suggested that DTT differentially
activated the MAPK signaling pathways in a dose-dependent manner,
with this balance determining the final fate of the cells.
Severe ER stress inhibits autophagic
flux
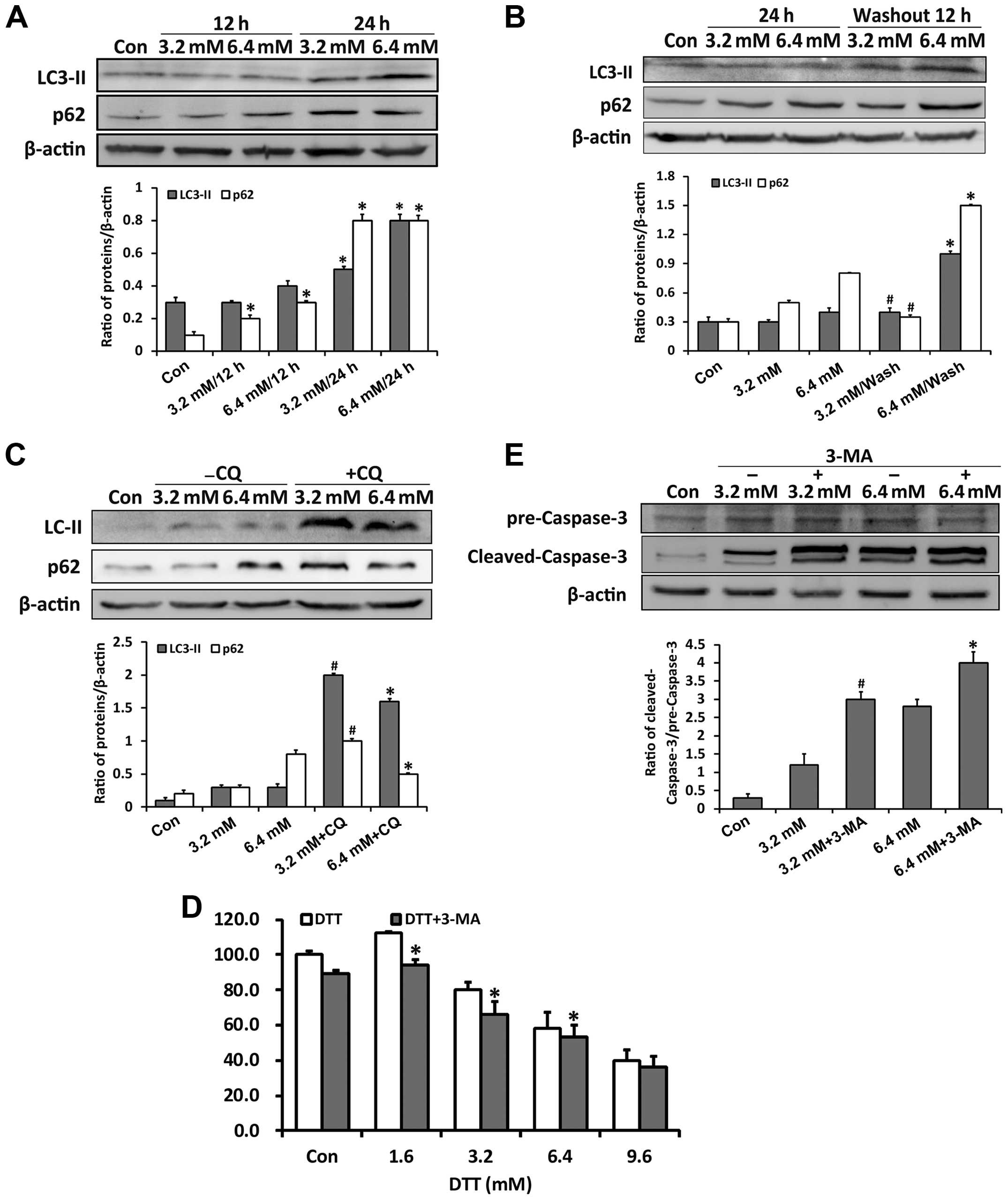
Previous studies showed that ROS and ER stress may
regulate autophagy through the MAPKs (25,26).
We examined whether the role of autophagy changed when the ER
stress increased. Through detecting expressions of the autophagy
markers p62 and LC3-II, we found that DTT activated autophagy
(Fig. 4A). After DTT removal, p62
expression decreased in cells previously exposed to 3.2 mM DTT,
while it persistently increased in those previously exposed to 6.4
mM DTT (Fig. 4B). Since p62 is one
of the substrate proteins degraded through autophagy, its
expression is an indicator of autophagy flux (27,28).
These data implied that autophagy flux changed with increased ER
stress. Therefore, we examined autophagy flux further by assessing
p62 expression in cells treated with DTT in combination with the
autophagy inhibitor chloroquine (CQ) (Fig. 4C). CQ, which inhibits fusion of
autophagosomes and lysosomes, enhanced the p62 expression induced
by DTT. However, the increase in p62 in cells exposed to DTT with
CQ was negatively correlated with DTT dose. Our explanation is that
the autophagy flux in cells exposed to the lower dose of DTT was
relatively normal and efficient, whereas that in cells exposed to
the higher dose was partially blocked. To check the role of
autophagy in DTT-induced cell death, we used the compound 3-MA,
which inhibits initiation of autophagy. Based on the MTT assay,
3-MA enhanced the cell death induced by DTT (Fig. 4D). We next detected cleaved
caspase-3, as an indicator of apoptosis activation. 3-MA enhanced
the expression of cleaved caspase-3 induced by DTT in HeLa cells
(Fig. 4E).
These results indicated that, in HeLa cells,
autophagy flux was partially blocked during severe ER stress and
that inhibition of autophagy increased cytotoxicity induced by
DTT.
Downregulated ERK phosphorylation
mediates inhibition of autophagic flux
To elucidate whether blocked autophagy flux was the
result of downregulated ERK phosphorylation, controlled by ROS, we
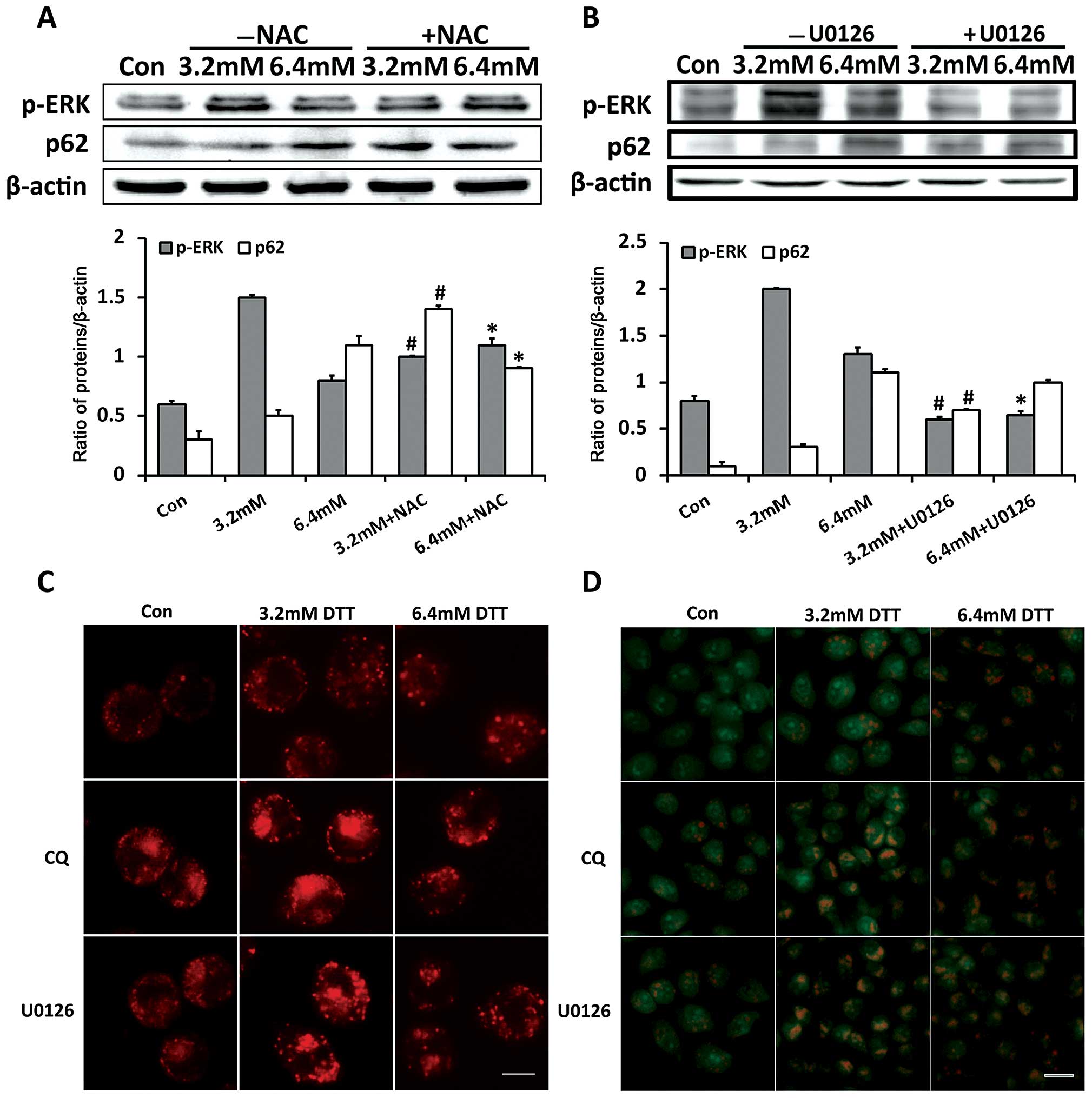
first examined effects of ROS on autophagy flux and ERK activation.
As shown in Fig. 5A, compared with
cells exposed to 3.2 mM DTT alone, cells exposed to 3.2 mM DTT with
NAC had decreased p-ERK and increased p62 expression. In contrast,
p-ERK expression in cells exposed to 6.4 mM DTT was further
increased, while that of p62 was decreased, after pretreatment with
NAC. This indicated that ROS inactivated the ERK signaling pathway
and blocked the autophagy flux when the stress was severe.
To determine whether ERK is involved in regulating
autophagy flux, we monitored p62 expression in cells treated with
DTT and the ERK inhibitor U0126. As shown in Fig. 5B, compared with cells exposed to DTT
alone, cells exposed to DTT with U0126 had higher p62 expression.
However, the increase in p62 expression in the presence of U0126
was inversely correlated with DTT dose. Similar to that observed in
cells treated with DTT and CQ, U0126 changed the expression profile
of p62 induced by DTT. This indicated that ERK inactivation led to
blockade of autophagy flux. We next monitored accumulation of
cellular acidic compartments in cells treated by DTT with or
without CQ or U0126, using LysoTracker Red and AO staining. The
lysosome specific stain LysoTracker Red and the pH-sensitive probe
AO have been frequently used to detect autophagic flux (29). In cells stained with LysoTracker
Red, the red-dotted structures are lysosomes or structures formed
by fusion of autophagosomes and lysosomes. In cells stained with
AO, red fluorescence indicates acid vesicular structures such as
lysosomes and fused autophagosomes. As shown in Fig. 5C and D, compared with cells exposed
to DTT alone, cells exposed to DTT with CQ or U0126 showed clearly
increased numbers of red-dotted structures and enhanced red
fluorescence. These observations indicated that U0126 and CQ
similarly increased accumulation of cellular acidic components
induced by DTT.
These results indicated that ERK inhibition during
severe ER stress contributed to blockade of autophagy flux by
affecting the fusion between autophagosomes and lysosomes.
Discussion
Numerous studies have explored the relationship
between ER stress and autophagy. However, the signaling pathways
responsible, during ER stress, for the outcome of life-or-death in
autophagy appear to vary with cell types and stimuli. Our study
explored the roles of ROS and downstream events in the autophagy
and apoptosis that follow DTT-triggered ER stress in HeLa cells.
Our results indicated that: i) severe ER stress induced by DTT
contributes to cell death, ii) the role of ROS changes from
pro-survival to pro-death with increased ER stress, iii)
differential activation of the MAP kinases p38 and ERK are involved
in ROS-mediated cell death and iv) ERK phosphorylation inhibited by
ROS is involved in blockade of autophagy flux.
Consistent with the results we report in this study,
the extent and duration of ER stress is often regarded as closely
related to the final fate of the cells (30). Our results confirmed that human
cervical cancer or HeLa cells could, partially or fully, restore ER
homeostasis after exposure to low concentrations of DTT while, with
exposure to higher concentrations, they continued to be stressed.
The apoptosis induced by DTT in HL-60 cells was associated with
early caspase-3 activation and was independent of mitochondria
(31). To understand whether the
DTT-induced cell damage in HeLa cells was related to over-extended
ER stress, we investigated expression of Grp78 and Chop, whose
levels are hallmarks of ER stress. Downregulation of Grp78 with
elevated Chop expression exacerbated retinal deterioration and
apoptosis during retinal ischemia/reperfusion (I/R) injury
(32). Our findings were consistent
with this report. During mild ER stress, cells that displayed
higher Grp78 and lower Chop expression might adapt to the stress.
After withdrawal of the stressor, cells with relatively low Grp78
and high Chop expression would ultimately undergo apoptosis. Our
results suggested that the severe ER stress induced by DTT
contributed to cell death in HeLa cells.
ROS were involved in DTT-induced cell death under
severe ER stress. Hydrogen peroxide (H2O2) is
generated as a by-product in the ER during disulfide bond formation
driven by the Ero1/PDI system (33). Schuiki et al reported that
DTT quickly and potently reduced the normally oxidizing ER
environment (1). This implies that
oxidative stress was upstream in DTT-exposed cells and ROS may have
determined the final impact of ER stress on cell fate. In HT1080
cells treated with DTT, H2O2 in the ER lumen
was markedly increased, leading to peroxiredoxin IV (PrxIV)
hyperoxidation (33). In our study,
we found that H2O2 production was increased
in DTT-treated HeLa cells in a DTT dose-dependent manner. Removal
of ROS with the antioxidant NAC promoted DTT-induced cytotoxity
under milder ER stress, but attenuated its effects when ER stress
was severe. Our results are consistent with the concept that
intracellular ROS levels at any moment can determine the fate of
the cells. Low levels of ROS function as regulators of cell
signaling while accumulation of higher ROS levels induces apoptosis
(34,35). The implication is that a prolonged
H2O2 signal is responsible for shifting the
outcome of ER stress from cell survival to cell death.
Activation of the ASK1-JNK/p38 pathway, among the
signaling pathways responsible for apoptosis following ER stress
was dependent on ROS (36,37). In Eca109 cells, p38 MAPK kinase was
essential for DTT- and cisplatin-induced apoptosis (38). Our results showed that JNK and p38
activation persisted with increased ER stress, while ERK activation
was attenuated early in HeLa cells undergoing prolonged exposure to
DTT. In gastric cancer cells, MEK inhibition blocked Grp78
upregulation and enhanced apoptosis induced by ER stress (8). Our results showed that JNK/p38
inhibition attenuated DTT-induced cytotoxity while ERK inhibition
promoted DTT-induced cell death. These results indicated that
differential activation of MAPK pathways occurred during various
extent of ER stress and this balance may determine the final
outcome of the cells.
Autophagy is important for alleviating ER stress
(39). Numerous studies have
indicated that ER stress was induced simultaneously with activation
of autophagy (40,41). Our findings suggested that the
autophagy flux decreased with increased ER stress. ROS removal with
NAC simultaneously affected ERK activation and autophagy flux.
Previous studies showed that ERK activation was involved in the
process of autophagosome and lysosome fusion (19). Our data demonstrated that inhibition
of ERK affected autophagy flux in a similar manner as did the
classic autophagy inhibitor CQ, which inhibits the fusion process
of autophagosome and lysosome. When the autophagy pathway was
inhibited by the specific inhibitor 3-MA, DTT-induced cytotoxity
was increased. These results indicated that autophagy flux was
negatively correlated with the extent of ER stress and that ROS
blocked autophagy flux through inhibiting ERK, potentially
contributing to cell death during severe ER stress.
As ER stress and autophagy are targets for cancer
therapy (42), there is a need for
more research to explore the mechanisms linking these two
processes. Our findings indicate that the MAPKs are vital to
regulating autophagy during ER-associated oxidative stress. ROS may
control autophagy flux through regulating ERK phosphorylation.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (nos. 81272876 and
81202552).
References
|
1
|
Schuiki I, Zhang L and Volchuk A:
Endoplasmic reticulum redox state is not perturbed by
pharmacological or pathological endoplasmic reticulum stress in
live pancreatic β-cells. PLoS One. 7:e486262012. View Article : Google Scholar
|
|
2
|
Bhandary B, Marahatta A, Kim HR and Chae
HJ: An involvement of oxidative stress in endoplasmic reticulum
stress and its associated diseases. Int J Mol Sci. 14:434–456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cameron NE: Role of endoplasmic reticulum
stress in diabetic neuropathy. Diabetes. 62:696–697. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mota SI, Costa RO, Ferreira IL, Santana I,
Caldeira GL, Padovano C, Fonseca AC, Baldeiras I, Cunha C, Letra L,
et al: Oxidative stress involving changes in Nrf2 and ER stress in
early stages of Alzheimer's disease. Biochim Biophys Acta.
1852:1428–1441. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yadav RK, Chae SW, Kim HR and Chae HJ:
Endoplasmic reticulum stress and cancer. J Cancer Prev. 19:75–88.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar
|
|
7
|
Matsuzawa A and Ichijo H: Redox control of
cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase
pathway in stress signaling. Biochim Biophys Acta. 1780:1325–1336.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang LJ, Chen S, Wu P, Hu CS, Thorne RF,
Luo CM, Hersey P and Zhang XD: Inhibition of MEK blocks GRP78
up-regulation and enhances apoptosis induced by ER stress in
gastric cancer cells. Cancer Lett. 274:40–46. 2009. View Article : Google Scholar
|
|
9
|
Han J, Back SH, Hur J, Lin YH,
Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M,
et al: ER-stress-induced transcriptional regulation increases
protein synthesis leading to cell death. Nat Cell Biol. 15:481–490.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: How can ROS activate MAPK pathways? J Signal
Transduct. 2011:7926392011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yorimitsu T, Nair U, Yang Z and Klionsky
DJ: Endoplasmic reticulum stress triggers autophagy. J Biol Chem.
281:30299–30304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar
|
|
15
|
Høyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sridharan S, Jain K and Basu A: Regulation
of autophagy by kinases. Cancers. 3:2630–2654. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li C and Johnson DE: Bortezomib induces
autophagy in head and neck squamous cell carcinoma cells via JNK
activation. Cancer Lett. 314:102–107. 2012. View Article : Google Scholar
|
|
18
|
Haberzettl P and Hill BG: Oxidized lipids
activate autophagy in a JNK-dependent manner by stimulating the
endoplasmic reticulum stress response. Redox Biol. 1:56–64. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kao C, Chao A, Tsai CL, Chuang WC, Huang
WP, Chen GC, Lin CY, Wang TH, Wang HS and Lai CH: Bortezomib
enhances cancer cell death by blocking the autophagic flux through
stimulating ERK phosphorylation. Cell Death Dis. 5:e15102014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang H and Joseph JA: Quantifying cellular
oxidative stress by dichlorofluorescein assay using microplate
reader. Free Radic Biol Med. 27:612–616. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo B and Lee AS: The critical roles of
endoplasmic reticulum chaperones and unfolded protein response in
tumorigenesis and anticancer therapies. Oncogene. 32:805–818. 2013.
View Article : Google Scholar
|
|
22
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rubio C, Pincus D, Korennykh A, Schuck S,
El-Samad H and Walter P: Homeostatic adaptation to endoplasmic
reticulum stress depends on Ire1 kinase activity. J Cell Biol.
193:171–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rutkowski DT and Kaufman RJ: That which
does not kill me makes me stronger: Adapting to chronic ER stress.
Trends Biochem Sci. 32:469–476. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar
|
|
26
|
Matsumoto H, Miyazaki S, Matsuyama S,
Takeda M, Kawano M, Nakagawa H, Nishimura K and Matsuo S: Selection
of autophagy or apoptosis in cells exposed to ER-stress depends on
ATF4 expression pattern with or without CHOP expression. Biol Open.
2:1084–1090. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Komatsu M and Ichimura Y: Physiological
significance of selective degradation of p62 by autophagy. FEBS
Lett. 584:1374–1378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang X, Xiang X, Xia M, Su J, Wu Y, Shen
L, Xu Y and Sun L: Inhibition of JNK3 promotes apoptosis induced by
BH3 mimetic S1 in chemoresistant human ovarian cancer cells. Anat
Rec. 298:386–395. 2015. View
Article : Google Scholar
|
|
29
|
Degtyarev M, Reichelt M and Lin K: Novel
quantitative autophagy analysis by organelle flow cytometry after
cell sonication. PLoS One. 9:e877072014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.PubMed/NCBI
|
|
31
|
Tartier L, McCarey YL, Biaglow JE,
Kochevar IE and Held KD: Apoptosis induced by dithiothreitol in
HL-60 cells shows early activation of caspase 3 and is independent
of mitochondria. Cell Death Differ. 7:1002–1010. 2000. View Article : Google Scholar
|
|
32
|
Li H, Zhu X, Fang F, Jiang D and Tang L:
Down-regulation of GRP78 enhances apoptosis via CHOP pathway in
retinal ischemia-reperfusion injury. Neurosci Lett. 575:68–73.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tavender TJ and Bulleid NJ: Peroxiredoxin
IV protects cells from oxidative stress by removing
H2O2 produced during disulphide formation. J
Cell Sci. 123:2672–2679. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scherz-Shouval R and Elazar Z: ROS,
mitochondria and the regulation of autophagy. Trends Cell Biol.
17:422–427. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Noguchi T, Ishii K, Fukutomi H, Naguro I,
Matsuzawa A, Takeda K and Ichijo H: Requirement of reactive oxygen
species-dependent activation of ASK1-p38 MAPK pathway for
extracellular ATP-induced apoptosis in macrophage. J Biol Chem.
283:7657–7665. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shore GC, Papa FR and Oakes SA: Signaling
cell death from the endoplasmic reticulum stress response. Curr
Opin Cell Biol. 23:143–149. 2011. View Article : Google Scholar :
|
|
38
|
Zhang QX, Feng R, Zhang W, Ding Y, Yang JY
and Liu GH: Role of stress-activated MAP kinase P38 in cisplatin-
and DTT-induced apoptosis of the esophageal carcinoma cell line
Eca109. World J Gastroenterol. 11:4451–4456. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kapuy O, Vinod PK and Bánhegyi G: mTOR
inhibition increases cell viability via autophagy induction during
endoplasmic reticulum stress - An experimental and modeling study.
FEBS Open Bio. 4:704–713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xi H, Kurtoglu M, Liu H, Wangpaichitr M,
You M, Liu X, Savaraj N and Lampidis TJ: 2-Deoxy-D-glucose
activates autophagy via endoplasmic reticulum stress rather than
ATP depletion. Cancer Chemother Pharmacol. 67:899–910. 2011.
View Article : Google Scholar :
|
|
42
|
Verfaillie T, Salazar M, Velasco G and
Agostinis P: Linking ER stress to autophagy: Potential implications
for cancer therapy. Int J Cell Biol. 2010:9305092010. View Article : Google Scholar : PubMed/NCBI
|