Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of
the most lethal solid malignancies and the fourth leading cause of
cancer-related death (1). Surgical
resection is the only potentially curative treatment. However, most
patients present with advanced disease stages and are not
candidates for surgery at the time of diagnosis due to the presence
of distant metastases, peritoneal seeding, or invasion into
adjacent vital structures (2,3). For a
decade, gemcitabine is the standard chemotherapy for advanced PDAC
(4). Most significant problem of
gemcitabine treatment for PDAC is the chemoresistance. Although
several studies reported that upregulation or downregulation of the
multiple membrane transporters, enzymes involved in the metabolism
of gemcitabine and alterations in the apoptotic pathways may confer
sensitivity and resistance (5), the
overall survival remains dismal with chemotherapy using
gemcitabine. Therefore, there is an urgent need for new therapeutic
strategies which could overcome the chemoresistance to gemcitabine
in PDAC.
The Janus kinase/signal transducer and activator of
transcription (JAK/STAT) pathway is known as one of the key
pathways that affect cell growth, proliferation and survival in
many human cancers (6,7). The STAT family members (seven members
comprising STAT1, STAT2, STAT3, STAT4, STAT5a/5b and STAT6) play
dual roles as cytoplasmic signaling proteins and as nuclear
transcription factors (6,8). Critical interactions leading to
modulation of STAT activity may occur in the cytoplasm where: JAK
kinases, coupled to cytokine receptors, phosphorylate STAT, thereby
promoting nuclear translocation. Furthermore, JAK-mediated STAT
phosphorylation leads to the formation of stable homodimers and
heterodimers, resulting in their nuclear translocation (9). Once in the nucleus, STAT molecules
bind specific promoter DNA sequences causing gene transcription
that regulates cell proliferation, differentiation and apoptosis
(10).
Of these STAT members, STAT3 has been frequently
identified by its constitutive activation in many human cancers,
including pancreatic cancer (7,11,12).
In addition to STAT3, STAT5 has also been implicated in several
cancers for tumor proliferation, apoptosis and invasion (7,9). STAT5
has two highly homologous isoforms, STAT5a and STAT5b. STAT5a was
originally identified as a mammary gland factor that augments milk
protein expression on prolactin induction (13) and has been reported to be associated
with breast (14,15) and prostate cancer (16,17).
STAT5b, which is highly homologous with STAT5a at the amino acid
level, has been associated with advanced tumor stages, venous
infiltration, and poor prognosis in hepatocellular carcinoma
(18); with tumor size in lung
cancer (19); with TNM stage in
colorectal cancer (20) and with
tumorigenesis and progression in glioblastoma multiforme (21). As for pancreatic diseases, STAT5 has
been reported to be associated with diabetes mellitus (22) and activated STAT5b (but not STAT5a)
was found in intraductal papillary mucinous neoplasm (IPMN) but not
in benign adenomas (23). In
contrast, the expression and biological role of STAT5b in PDAC are
less clearly defined. We, therefore, hypothesized that elucidating
the biological role of STAT5b in PDAC may pave the way for novel
therapeutic options in treating pancreatic cancer.
In the present study, we examined the expression and
activation of STAT5b in human pancreatic cancer cell lines. We
downregulated STAT5b with shRNA in human pancreatic cancer cells
and investigated the effect of STAT5b suppression on reducing cell
growth, gemcitabine chemosensitivity, apoptosis, invasion and
adhesion. We also investigated the clinicopathological
characteristics of STAT5b expression in PDAC.
Materials and methods
Antibodies
The following items were purchased: anti-STAT5a
(sc1081) rabbit polyclonal antibodies, anti-STAT5b (sc1656) mouse
monoclonal antibodies, and anti-phosphotyrosine (sc7020) mouse
monoclonal antibodies from Santa Cruz Biotechnology (Santa Cruz,
CA, USA); Alexa Fluor 488 goat anti-mouse IgG (A-11001), Hoechst
33258, pentahydrate (bisbenzimide), and anti-V5 Tag (#46-0705)
mouse monoclonal antibodies from Invitrogen (Life Technologies;
Carlsbad, CA, USA); anti-lamin B1 (ab16048) rabbit polyclonal
antibodies from Abcam plc (Cambridge, UK); anti-PARP (#9542) rabbit
polyclonal antibodies, anti-cleaved caspase-3 (#9664) rabbit
monoclonal antibodies, and anti-Bcl-xL (#2746) rabbit monoclonal
antibodies from Cell Signaling Technology (Danvers, MA, USA); and
anti-β-actin (A1978) mouse monoclonal antibodies from Sigma-Aldrich
(St. Louis, MO, USA).
Reagents
The following items were purchased: fetal bovine
serum (FBS), DMEM, RPMI, MEM and trypsin solution from Gibco (Life
Technologies); RNeasy Mini kit from Qiagen (Hilden, Germany); High
Capacity RNA-to-cDNA kit from Applied Biosystems (Darmstadt,
Germany); NE-PER Nuclear and Cytoplasmic Extraction reagents and
SuperSignal West Pico and Femto chemiluminescent substrates from
Thermo Fisher Scientific (Rockford, IL, USA); FuGENE HD
Transfection reagent from Promega (Madison, WI, USA); G418
(Geneticin) from Roche Diagnostics Deutschland GmbH (Mannheim,
Germany); laminin, collagen from human placenta type IV [(collagen
IV) from Sigma-Aldrich]; Cell Counting kit-8 from Dojindo
Laboratories (Kumamoto, Japan); BioCoat Matrigel Invasion Chamber
(8-µm pore size) and fibronectin from BD Biosciences
(Franklin Lakes, NJ, USA); gemcitabine hydrochloride from Wako Pure
Chemical Industries (Osaka, Japan); Fluorescent Mounting Medium
from Dako Japan (Tokyo, Japan); Histofine Simple Stain Max PO (M)
or (R) kit from Nichirei Biosciences, Inc., (Tokyo, Japan); and
recombinant human epidermal growth factor (EGF, 236-EG) and
recombinant human platelet-derived growth factor (PDGF, 220-BB)
from R&D Systems (Minneapolis, MN, USA).
Pancreatic cancer cell lines
AsPC-1, BxPC3, Capan-1, HPAF-2, MIA PaCa-2, PANC-1
and SW1990 PDAC cell lines were purchased from the American Type
Culture Collection and the PK-45H cell line was obtained from the
Cell Resource Center for Biomedical Research, Institute of
Development, Aging and Cancer, Tohoku University.
Cell culture
PANC-1, MIA PaCa-2 and SW1990 cells were grown in
DMEM, AsPC-1, Capan-1, BxPC3 and PK-45H cells were grown in RPMI,
and HPAF-2 cells were grown in MEM. All of the cultures were
supplemented with 10% FBS, 100 units/ml penicillin, 100
µg/ml streptomycin and 250 ng/ml amphotericin B. Cells were
maintained at 37°C in a humid atmosphere with 5%
CO2.
Quantitative RT-PCR analysis
To evaluate the STAT5b expression levels in the
pancreatic cancer cell lines, TaqMan quantitative RT-PCR was
performed. Total RNA was isolated with RNeasy Mini kit according to
the manufacturer's protocol. For cDNA synthesis, the High Capacity
RNA-to-cDNA kit was used according to the manufacturer's protocol.
Quantitative RT-PCR was performed using the 7500 Fast Real-Time PCR
System (Applied Biosystems, Darmstadt, Germany) and for evaluation,
commercial TaqMan® Gene Expression- assays (Applied
Biosystems, AoD, Assay-ID: Hs00273500_m1 for the studied genes
STAT5b and Hs02758991_g1 for GAPDH as the reference gene) were used
with optimized primer and probe concentrations.
Protein extraction and western blot
analysis
To extract protein from cell lines, cells were
washed twice with cold PBS and reacted with lysis buffer Triton
X-100 at 4°C for 30 min. After the reaction, cells were harvested
by cell scraping and centrifuged at 4°C and 15,000 rpm for 30 min.
For western blot analysis, 20 µl of each protein was
separated by gel electrophoresis on a polyacrylamide gel and
transferred to nitrocellulose membranes. The membranes were blocked
with Tris-buffered saline containing 0.1% Tween-20 (TBST) and 5%
skim milk, then incubated overnight at 4°C with the indicated
primary antibodies and then for 60 min with the corresponding
horseradish-conjugated secondary antibodies. The membranes were
washed twice with TBST and incubated with goat anti-mouse or
anti-rabbit IgG-horseradish peroxidase conjugated secondary
antibodies for 1 h at 4°C. The blots were washed three times with
TBST and an enhanced chemiluminescence system (ImageQuant LAS 4000
mini; GE Healthcare Japan, Tokyo, Japan) was used to detect the
bands. The band densities of PARP and Bcl-xL were measured with
ImageJ (National Institutes of Health, Bethesda, MD, USA), and
densitometry analysis was performed as previously described
(24).
Immunofluorescence and confocal
analysis
Cells were plated onto chamber slides and allowed to
adhere overnight. Slides were fixed for 10 min in 2%
paraformaldehyde and free aldehydes were quenched with 50 mM/l
NH4Cl in PBS for 10 min. Then, the cells were
permeabilized in 0.1% Triton X-100 in PBS-2% BSA for 15 min. Slides
were then incubated at 23°C for 1 h with anti-STAT5b antibody (in
1:50 dilution). After washing with PBS, slides were incubated for
30 min with Alexa Fluor 488-conjugated goat anti-mouse secondary
antibody (in 1:500 dilution), and the nuclei were counterstained
with 0.2 µg/ml Hoechst 33258 dye for 1 min at 23°C, as per a
previously reported method (25).
Slides were next washed 3 times in PBS and then mounted in a
fluorescent mounting medium. Immunofluorescence scans were taken
with a Nikon TE2000-E inverted fluorescence microscope system
equipped with a Nikon Digital Eclipse C1 laser scanning confocal
microscope (Nikon, Tokyo, Japan).
Cell fractionation and
immunoprecipitation
Cell fractionation was carried out utilizing NE-PER
Nuclear and Cytoplasmic Extraction reagents according to the
manufacturer's protocol. Preparation of cell lysates and western
blotting were performed as described above with primary antibodies
against STAT5b (in 1:500 dilution) and against lamin B1 (1:10,000
dilution).
For immunoprecipitation, a total of 1 mg of cell
lysate was incubated overnight at 4°C against STAT5b antibody
followed by 2-h incubation with protein A/G plus agarose, as per a
previously reported method (23).
The beads were then washed three times with TBST, then boiled and
subjected to western blotting as described above with primary
antibodies against phosphotyrosine (1:500 dilution) and STAT5b
(1:500 dilution).
Constructions and transfection of
expression plasmids
To construct expression vectors for human STAT5b
short hairpin (sh) RNA containing the sense target sequence for
STAT5b (5′-GGA CAC AGA GAA TGA GTT A-3′), the antisense target
sequence was synthesized and inserted into a pBAsi-hU6 Neo DNA
vector. Likewise, the scrambled sequence (5′-TCT TAA TCG CGT ATA
AGG C-3′) was used to construct the sham vectors that served as
negative controls. PANC-1 cells (2×106 cells/well) were
plated on a 10-cm plate and grown in DMEN medium at 37°C.
Transfection of STAT5b shRNA expression vectors and sham vectors
was performed using FuGENE HD Transfection reagent according to the
manufacturer's instructions, whereby 2×106 cells/well
were transfected with 19 µg of DNA using FuGENE HD and cells
were passaged and cultured with 500 µg/ml of G418. Cell
lysates were collected and protein levels were measured by western
blotting in the same manner as described above. In order to
construct the STAT5b overexpression clones, the STAT5b full-length
cDNA was subcloned into pcDNA3.1/V5-His TOPO vector, which
expresses a V5 tag fusion protein. Authenticity was confirmed by
sequencing. AsPC-1 cells (2×106 cells/well) were
transfected with FuGENE HD Transfection reagent and stably
transfected clones were selected with 800 µg/ml of G418 in
RPMI. Individual clones were isolated, and expression of the
V5-tagged STAT5b protein was determined by western blotting with
anti-STAT5b and anti-V5 Tag antibodies as described above.
Cell proliferation assay
To assess cell proliferation, cells were seeded in a
96-well plate at a density of 8.0×103 cells/well in DMEN
and incubated at 37°C for 24, 48 or 72 h. After the indicated time,
the medium was replaced with fresh DMEN and 10 µl of Cell
Counting kit-8 was added to the wells. The plate was incubated for
2 h at 37°C followed by an absorbance measurement of the wells at
450 nm using a microtiter plate reader. Analysis was performed in
triplicate.
Sensitivity of the antineoplastic
agents
To assess cell proliferation after treatment with
the antineoplastic agent, 8.0×103 cells were seeded in a
96-well plate in DMEN and incubated at 37°C for 24 h similarly to
the cell proliferation assay. After incubation, the medium was
replaced with 100 µl of 0.1% BSA containing 0, 10, 100 or
1,000 µM of gemcitabine reagent and incubated at 37°C for 48
or 72 h. After treatment with gemcitabine for the indicated time,
the medium was changed to 10 µl of Cell Counting kit-8.
Absorbance measurements at 450 nm were taken after 2 h of
incubation with a microtiter plate reader. Analysis was performed
in triplicate.
Apoptosis assay
PARP and cleavage of caspase-3 were monitored to
assess apoptosis. After treatment with absence or presence of 1,000
µM gemcitabine for 72 h at 37°C, protein extraction and
western blotting (as described above) were performed with
antibodies against PARP (in 1:1,000 dilution), cleaved caspase-3
(in 1:1,000 dilution) and β-actin (in 1:10,000 dilution).
Adhesion assay
Adhesion assay was performed as per a previously
reported method with some modification (26). Cells were suspended in serum-free
medium containing 0.1% BSA and were seeded at a density of
3×104 cells/plate on non-adhesive NUNC 96-well culture
plates (Thermo Fisher Scientific) coated with fibronectin, laminin
or collagen IV. The cells were then incubated for 3 h at 37°C and
washed with PBS. Adherent cells were fixed in 50 µl of 96%
ethanol for 10 min, stained with 50 µl of 0.1% crystal
violet, rinsed with water and dried at room temperature for 30 min.
Stained cells were solubilized with 50 µl of 0.2% Triton
X-100 and absorbance was measured at 595 nm with a microtiter plate
reader. Adhesion assays were performed in triplicate.
Invasion assay
The invasiveness of stable transfected PANC-1 cells
was measured as per a previously reported method with some
modification (27). Cells
(2.5×104 in quantity) were suspended in 500 µl of
serum-free medium [0.1% bovine serum albumin (BSA)] and placed onto
the upper compartment of Matrigel-coated Transwell chambers
(8-µm pore size, BioCoat Matrigel Invasion Chambers;
Becton-Dickinson Labware). The lower compartment was filled with
750 µl of medium containing 1% FBS, 1 nM EGF or 1 nM PDGF.
After 20 h, cells on the upper surface of the filter were carefully
removed with a cotton swab and membranes were fixed in methanol and
stained with crystal violet. The cells that had migrated through
the membrane to the lower surface of the filter were counted using
a microscope. Each assay was performed in duplicate and experiments
were repeated three times.
Patients and tissue samples
Tissues from 44 patients with invasive PDAC were
obtained for the present study. The patients received treatment at
Nippon Medical School Hospital (Tokyo, Japan) from 2009 to 2013.
All patients underwent either a pancreatoduodenectomy or a distal
pancreatectomy, and received adjuvant or neoadjuvant chemotherapy
with gemcitabine alone or gemcitabine combination regimen. Patients
were 27 males and 17 females, and median age was 69 years (range,
37–88 years). Clinicopathological stage was determined according to
the TNM classification system of the International Union Against
Cancer (UICC) and additionally characterized according to the Japan
Pancreas Society classification (Table
I). The median follow-up period was 21 months.
Paraffin-embedded samples were prepared for immunohistochemical
analysis as previously described (28). This study was carried out in
accordance with the principles embodied in the Declaration of
Helsinki, 2008, and informed consent for the use of pancreatic
tissues was obtained from each patient.
 | Table INo correlation of clinicopathological
characteristics and STAT5b expression in pancreatic cancer. |
Table I
No correlation of clinicopathological
characteristics and STAT5b expression in pancreatic cancer.
| Variables | No. | STAT5b-strong | STAT5b-weak | P-value |
|---|
| Gender |
| Male | 27 | 9 | 18 | |
| Female | 17 | 5 | 12 | NS |
| Age (years) |
| <65 | 14 | 4 | 10 | |
| ≥65 | 30 | 10 | 20 | NS |
| UICC |
| N0 | 18 | 6 | 12 | |
| N1 | 26 | 8 | 18 | NS |
| UICC stage |
| IA/IB/II A | 18 | 6 | 12 | |
| IIB/III/IV | 26 | 8 | 18 | NS |
| Main pancreatic
duct invasion |
| Negative | 33 | 7 | 26 | |
| Positive | 11 | 7 | 4 | 0.014 |
| Pancreatic cut end
margin |
| Negative | 40 | 11 | 29 | |
| Positive | 4 | 3 | 1 | 0.088 |
Immunohistochemistry
Paraffin-embedded sections (3 µm) were
immunostained using a Histofine Simple Stain MAX PO (R) or (M) kit.
After deparaffinization, the tissue sections were preheated in 10
mM citrate buffer solution (pH 6.0) for 5 min at 121°C. Then,
endogenous peroxidase activity was blocked by incubation for 30 min
with 0.3% hydrogen peroxide in methanol. The tissue sections were
then incubated with the anti-STAT5b antibody (1:50 in dilution) in
phosphate-buffered saline (PBS) containing 1% bovine serum albumin
(BSA) overnight at 4°C. Bound antibodies were detected with the
Simple Stain MAX PO (R) or (M) reagent, using diaminobenzidine
tetrahydrochloride as the substrate. The sections were then
counterstained with Mayer's hematoxylin. Negative control tissue
sections were prepared by omitting the primary antibody. As for the
evaluation of immunostaining, both intensity and proportion of
positively stained cancer cells were analyzed at magnification
×200.
Statistical analysis
Student's t-test was used for statistical analysis
of the cell proliferation assay, sensitivity of antineoplastic
agents assay, adhesion assay, invasion assay and densitometry
analysis. χ2 test and Fisher's exact test were used to
analyze the correlation between STAT5b expression and
clinicopathological characteristics. Cumulative survival rates were
calculated by the Kaplan-Meier method. Significant difference was
set at P<0.05. All statistical tests were determined using the
Software Package SPSS for Windows (version 12.0; SPSS, Inc.,
Chicago, IL, USA).
Results
Expression of STAT5b mRNA and protein in
pancreatic cancer cells
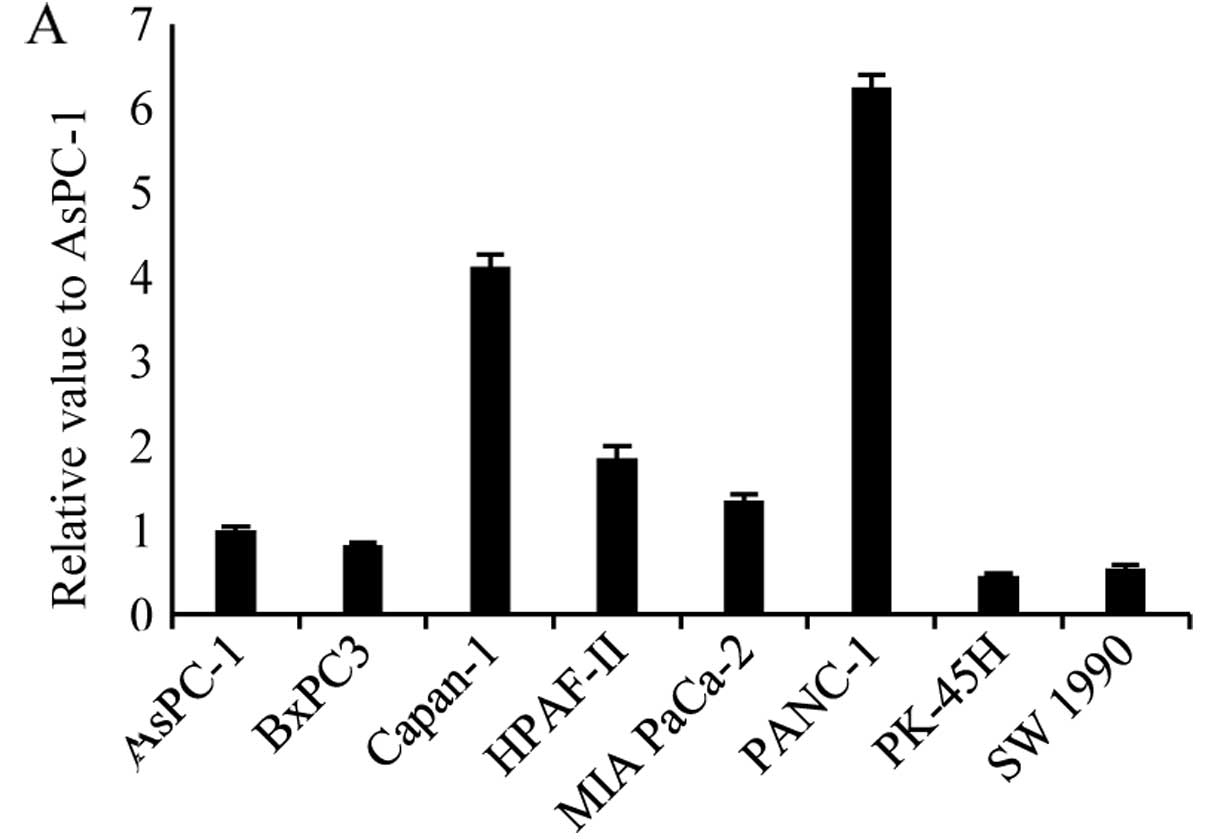
We performed qRT-PCR analysis to investigate the
relative expression levels of STAT5b mRNA in pancreatic cancer cell
lines, including AsPC-1, BxPC3, Capan-1, HPAF-2 MIA PaCa-2, PANC-1,
PK-45H and SW1990. STAT5b mRNA was detected in all pancreatic
cancer cell lines, and the relative levels were highest in PANC-1
cells, second highest in Capan-1 cells and lowest in PK-45H cells.
In PANC-1 cells, STAT5b mRNA levels were 13.5-fold higher than in
PK-45H cells (Fig. 1A). Western
blot analysis was also performed to examine the STAT5b protein
levels with the same eight pancreatic cancer cell lines. Consistent
with our qRT-PCR results, STAT5b protein was detected in all cell
lines, with the highest protein levels in PANC-1 cells and second
highest levels in Capan-1 cells (Fig.
1B).
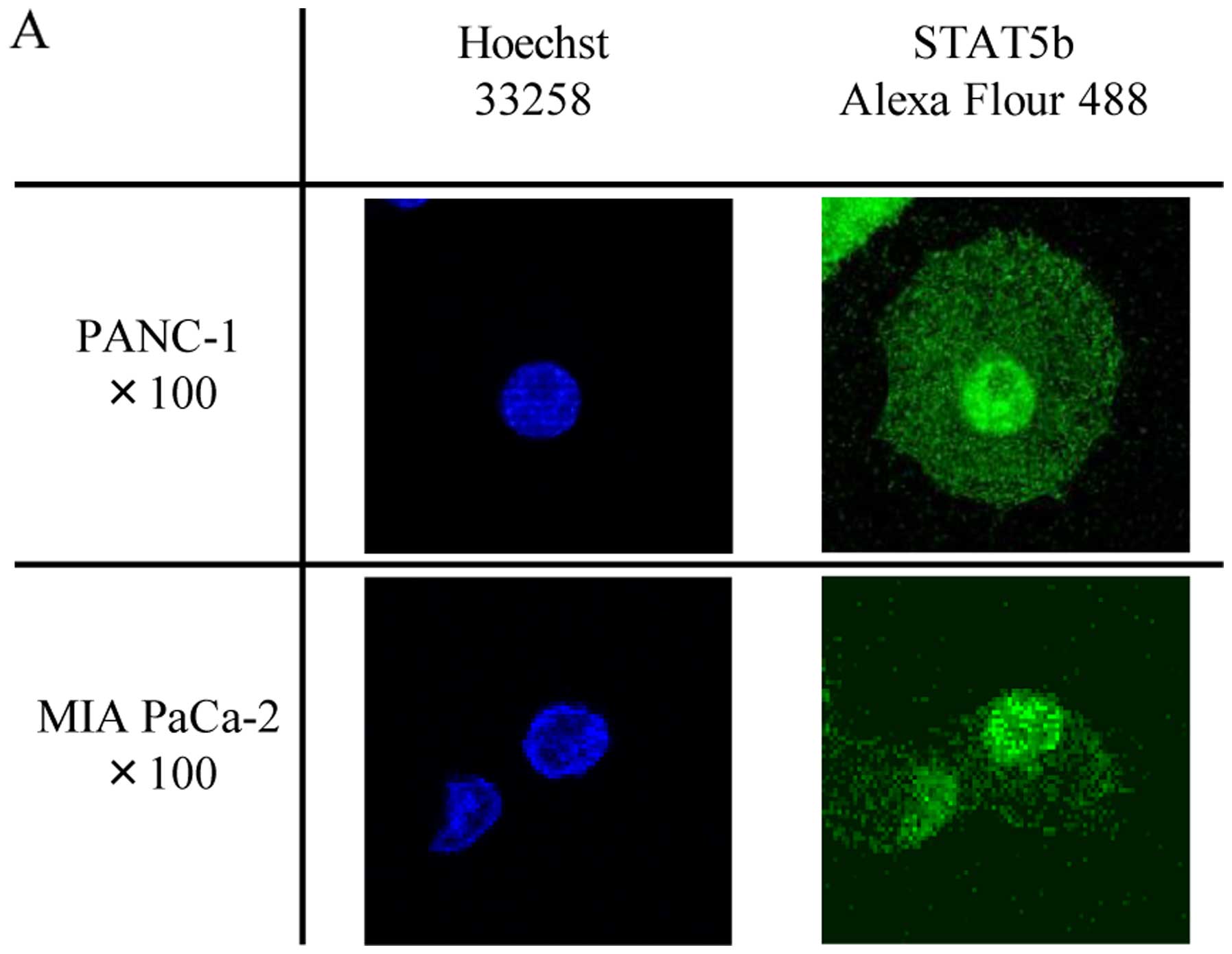
Localization and activation of STAT5b in
pancreatic cancer cells
Confocal immunofluorescence microscopy analysis was
next carried out to determine the subcellular localization of
STAT5b in PANC-1 and MIA PaCa-2 cells. This analysis revealed that
STAT5b was distributed in the nuclei and cytoplasm in both cell
types (Fig. 2A). Similarly, western
blotting using nuclear and cytoplasmic fractions detected STAT5b
protein in both the nuclei and the cytoplasm (Fig. 2B). To investigate the activation of
STAT5b in the two cell types, the cell lysates were
immunoprecipitated with anti-STAT5b antibody followed by western
blotting with anti-phosphotyrosine or anti-STAT5b antibodies. This
analysis revealed the phosphorylation of STAT5b in the same two
pancreatic cancer cell types (Fig.
2C).
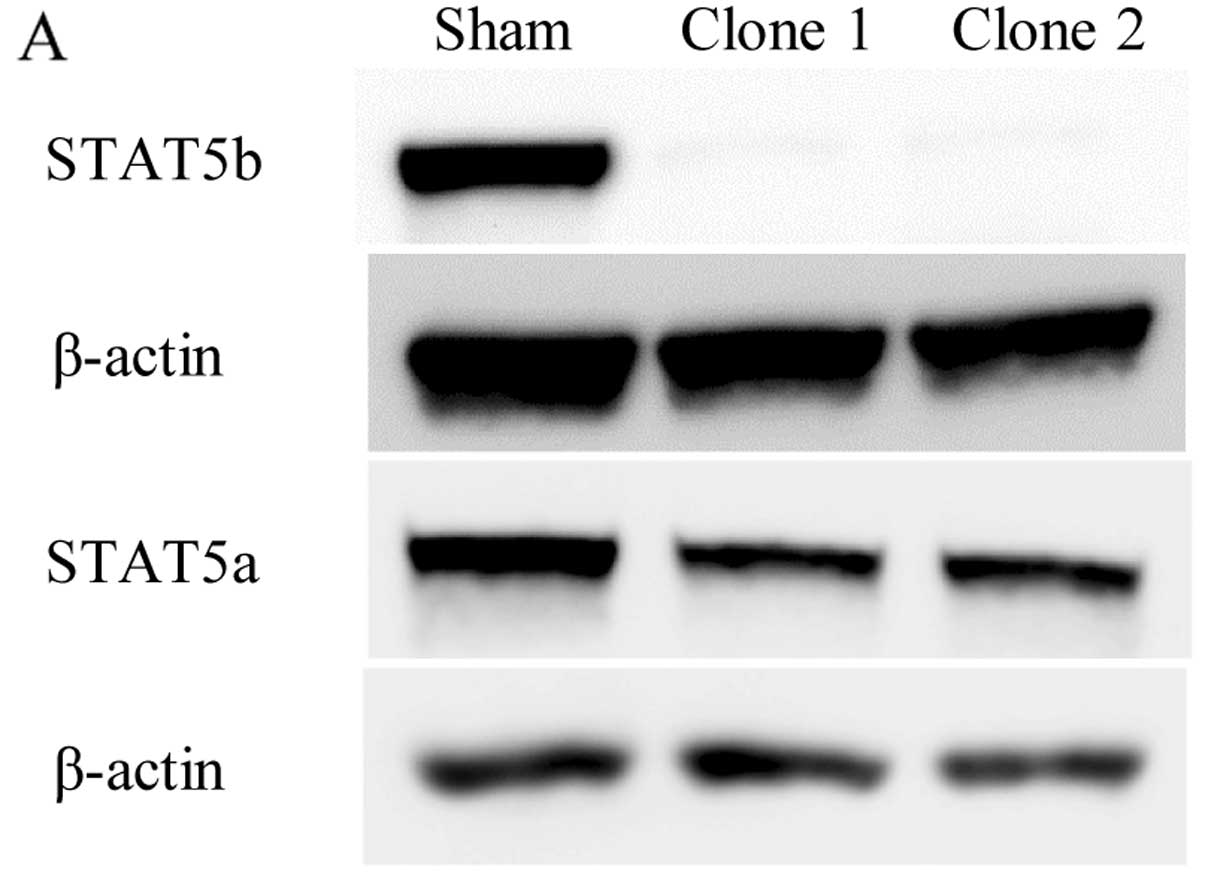
Effects of STAT5b shRNA on STAT5b levels
on the growth of PANC-1 cells
PANC-1 cells are ideal for assessing the role of
endogenous STAT5b in pancreatic cancer cells because they express
relatively high levels of STAT5b protein. Therefore, PANC-1 cells
were stably transfected with a plasmid vector encoding shRNAs
targeting the STAT5b transcripts. Clones transfected with the
STAT5b shRNAs showed inhibition of STAT5b, and uninhibited STAT5a
protein expression by western blotting (Fig. 3A). To assess the consequence of
reduced STAT5b expression on proliferation ability, cell growth was
investigated by cell proliferation assay. There was no significant
difference between the sham-transfected cells and the STAT5b shRNA
clones (Fig. 3B).
Effects of STAT5b suppression on
gemcitabine-treated actions
The chemotherapeutic agent gemcitabine has been the
standard treatment in patients with PDAC for more than a decade
since Burris et al (4). We
therefore, compared the effects of gemcitabine on growth and
apoptosis in sham-transfected and STAT5b shRNA clones by cell
proliferation assay as described above to investigate the effect of
gemcitabine treatment on proliferation ability with STAT5b
suppression. Treatment with gemcitabine for 48 or 72 h resulted in
a dose-dependent reduction in cell growth. In the sham-transfected
cells, a maximum decrease of 19.4% occurred at a concentration of
1,000 µM gemcitabine and 72-h incubation. By contrast, both
STAT5b shRNA clones exhibited a significantly greater growth
inhibitory effect of 32.6 and 52%, respectively with the same
concentration of gemcitabine and incubation time (Fig. 4).
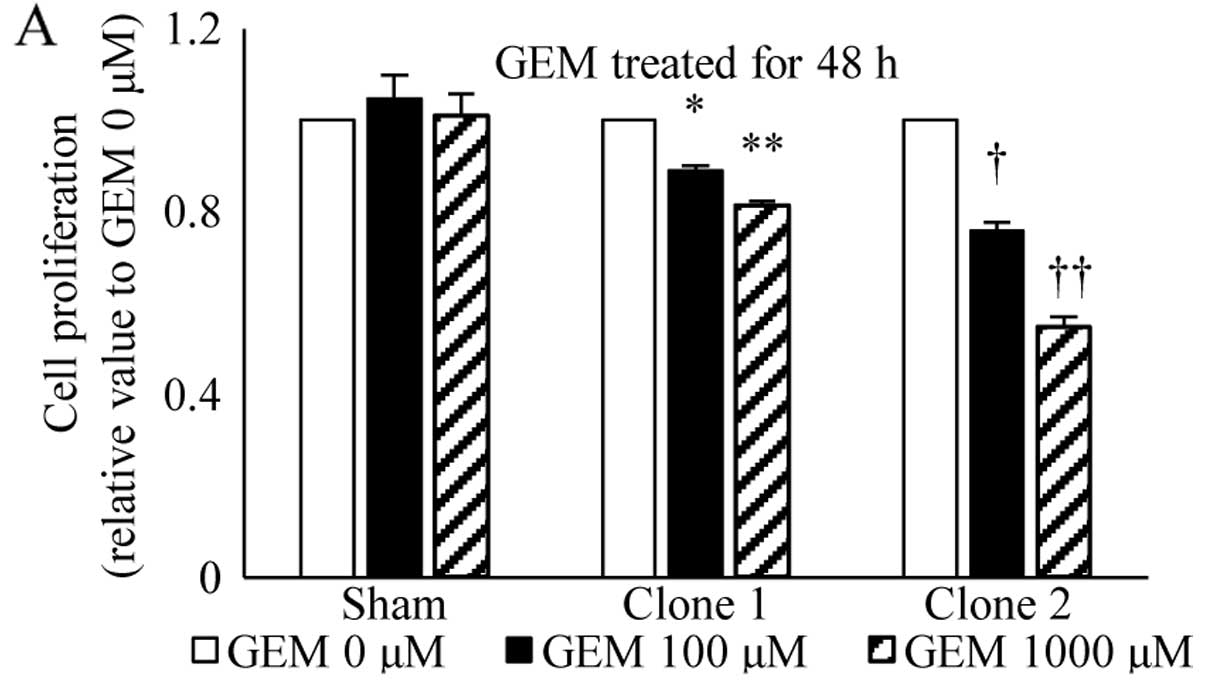 | Figure 4Effects of STAT5b suppression on
gemcitabine-treated actions. Sham and STAT5b shRNA clones, seeded
in the same way as the cell proliferation assay, were treated with
absence or presence of 100 or 1,000 µM gemcitabine for 48 or
72 h (A and B). Treatment with gemcitabine resulted in a
dose-dependent reduction in cell growth. In the sham-transfected
cells, a maximum decrease of 19.4% occurred at a concentration of
1,000 µM gemcitabine and 72-h incubation. By contrast, both
STAT5b shRNA clones exhibited a significantly greater growth
inhibitory effect of 32.6% (P<0.01) and 52% (P<0.001),
respectively with the same concentration of gemcitabine and
incubation time. (A) *P<0.05, **P<0.02,
†P<0.01 and ††P<0.001. (B)
*P<0.02, **P<0.01,
†P<0.01 and ††P<0.001. |
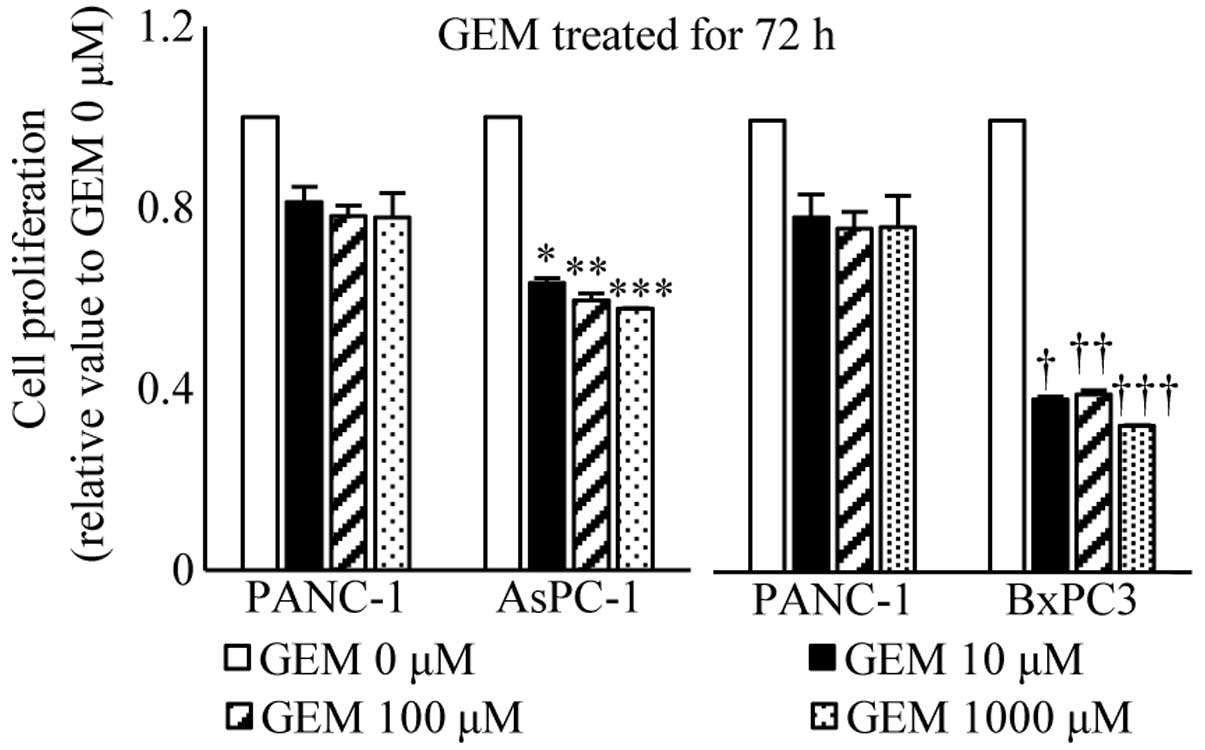
Effects of endogenous STAT5b levels on
gemcitabine-treated actions in pancreatic cancer cells
We next investigated the effects of gemcitabine on
growth in PANC-1, AsPC-1 and BxPC cells by cell proliferation assay
as described above, in order to compare the effect of gemcitabine
treatment on proliferation of pancreatic cancer cells which express
high and low levels of STAT5b. Treatment with gemcitabine for 72 h
resulted in a dose-dependent reduction in cell growth. In PANC-1
cells which expressed relatively high levels of STAT5b, a maximum
decrease of 22.1% occurred at a concentration of 1,000 µM
gemcitabine and 72-h incubation. Both AsPC-1 and BxPC3 cells, which
express relatively low levels of STAT5b, exhibited a significant
decrease in growth by treatment with gemcitabine compared to PANC-1
cells for 72 h (Fig. 5).
Effects of overexpressed STAT5b levels on
the growth of AsPC-1 cells and on gemcitabine-treated actions
AsPC-1 cells, which express relatively low levels of
STAT5b protein, were stably transfected with a plasmid vector
encoding STAT5b full-length cDNA, in order to investigate and
compare the role of STAT5b in pancreatic cancer cells. Clones
transfected with the STAT5b cDNA exhibited overexpression of
STAT5b, verified by western blotting (Fig. 6A). To assess the effect of
overexpressed STAT5b expression on proliferation ability, cell
growth was measured by cell proliferation assay. No significant
difference between the sham-transfected cells and the STAT5b
overexpressing clones was observed (Fig. 6B). We performed cell proliferation
assay with gemcitabine treatment, as described above, to determine
the effect of gemcitabine treatment on proliferation ability with
STAT5b overexpression clones. Treatment with gemcitabine for 72 h
resulted in a significant increase in growth of overexpressed
clones compared to sham-transfected cells (Fig. 6C).
Effects of gemcitabine on pro-apoptotic
actions and apoptosis-regulating protein Bcl-xL
The deregulation of apoptosis (29) is an indicator of carcinogenesis and
the induction of apoptosis is a standard strategy for anticancer
therapies (30). Therefore, we
examined the expression of known apoptosis-related proteins,
cleaved caspase-3 and PARP (31),
by western blotting to investigate whether gemcitabine treatment
for STAT5b shRNA clones induced greater apoptotic actions than the
sham-transfected cells. After treatment with gemcitabine for 72 h,
both STAT5b shRNA clones exhibited a stronger expression of cleaved
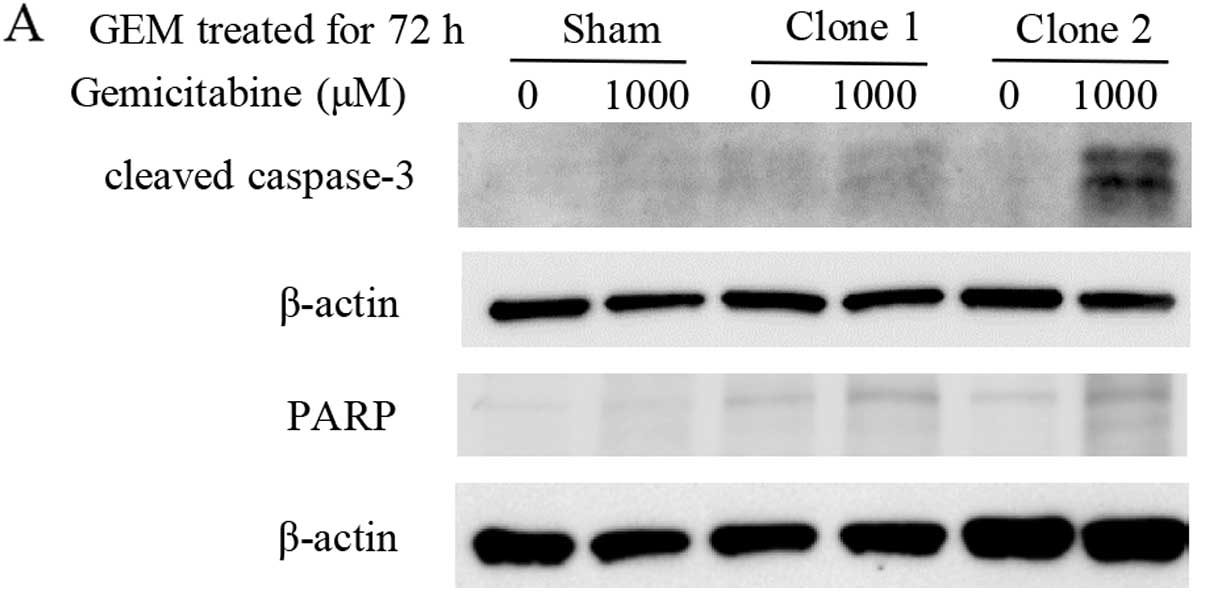
caspase-3 and PARP than the sham-transfected cells (Fig. 7A). STAT5 is known to upregulate
Bcl-xL expression and STAT5 has been implicated in the regulation
of apoptosis (32). Therefore, we
next sought to determine whether the levels of these transcription
factors were altered in our cells. Gemcitabine markedly reduced
Bcl-xL levels in the STAT5b shRNA cells compared with the
sham-transfected cells (Fig. 7B).
Relative values of PARP and Bcl-xL proteins were calculated by
quantitative densitometry. Band densities of PARP protein
significantly increased in STAT5b shRNA clones compared to
sham-transfected cells (Fig. 7C).
Band densities of Bcl-xL protein significantly decreased in STAT5b
shRNA clones compared to sham-transfected cells (Fig. 7D).
Effects of reduced STAT5b levels in
PANC-1 cells on adhesion and invasion
Cancer cell adhesion is a process that involves
cancer cells interacting with adjacent cancer cells as well as with
extracellular matrix components (33). For tumors to metastasize and grow,
neoplastic and endothelial cells must invade into surrounding
tissues. The ability to block the invasive capacity of tumor cells
therefore offers a new approach to treating patients with malignant
disease (34). Hence, we next
compared the effects of STAT5b suppression on cell adhesion and
invasion. In comparison to sham-transfected cells, the adhesion
assay for both STAT5b shRNA clones exhibited a significantly
reduced adhesion to fibronectin (P<0.001), laminin (P<0.001),
and collagen IV (P<0.002), which are known as major types of
extracellular matrices (ECM) (Fig.
8A). Similarly, compared to sham-transfected cells, both clones
exhibited significantly reduced ability to invade across a Matrigel
membrane treated with FBS (P<0.02), EGF (P<0.01) and PDGF
(P<0.02) in the invasion assay (Fig.
8B).
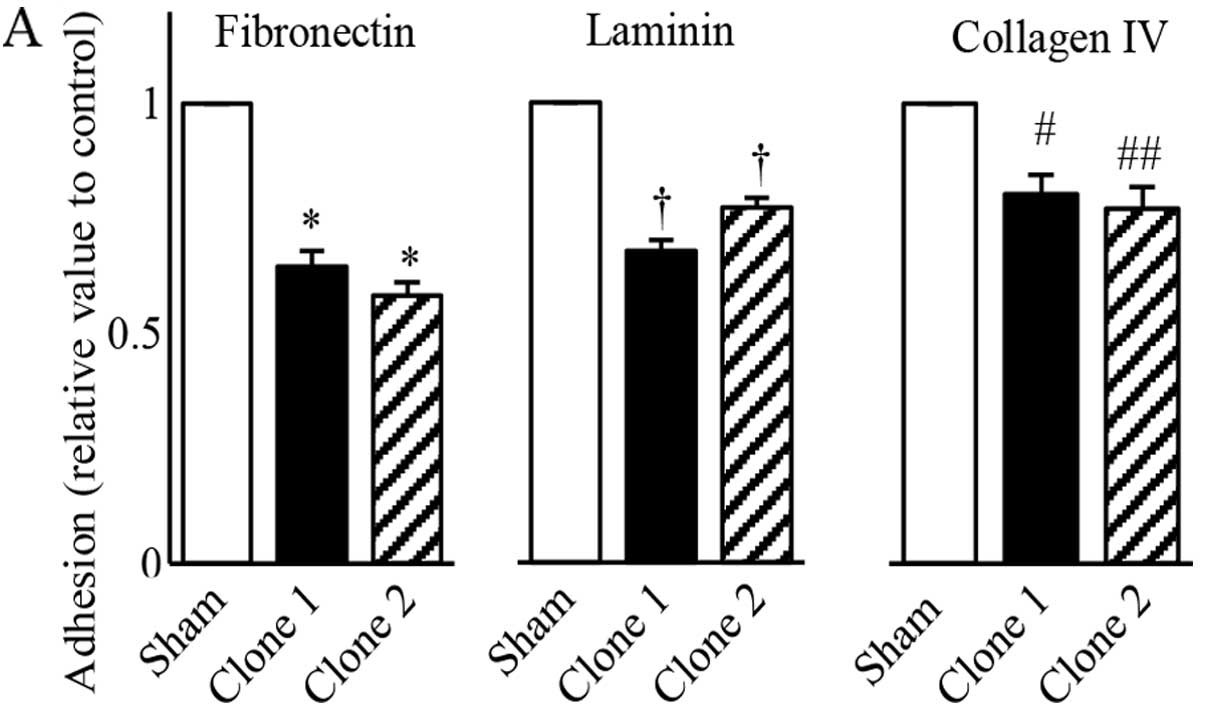 | Figure 8Effects of reduced STAT5b levels in
PANC-1 cells on adhesion and invasion. (A) Adhesion assay with
sham-transfected and STAT5b shRNA expressing clones. In comparison
with sham-transfected cells, both STAT5b shRNA expressing clones
exhibited a significantly reduced adhesion to fibronectin, laminin
and collagen IV, which are known as major types of extracellular
matrices. *P<0.001, †P<0.001,
#P<0.02 and ##P<0.01. (B) Invasion assay with
sham-transfected and STAT5b shRNA expressing clones. In comparison
with sham-transfected cells, both STAT5b shRNA clones exhibited
significantly reduced ability to invade across a Matrigel membrane
treated with FBS, EGF and PDGF. *P<0.02,
**P<0.01, †P<0.01, #P<0.02 and
##P<0.01. |
Cumulative Kaplan-Meier survival curve
and correlation of clinicopathological characteristics with STAT5b
expression
Immunohistochemical results for STAT5b were
evaluated as the expression index; 'staining percentage of the
tumor cell' multiplied by 'staining intensity (0–2)'. We set 0–20
index as weak (29 cases) (Fig. 9A),
and 30–120 index as strong (15 cases) (Fig. 9B). However, there was a trend toward
reduced overall survival of patients in the STAT5b-strong group,
but a significant difference in the overall survival rate between
the STAT5b-weak group and STAT5b-strong group was not observed
(P=0.35) (Fig. 9C).
Clinicopathologically, a significant correlation between STAT5b
expression in the cancer cells and main pancreatic duct invasion
(P=0.014). (Table I) was
observed.
Discussion
In the present study, we demonstrated expression of
STAT5b in human pancreatic cancer cells by RT-PCR (Fig. 1A) and western blot analysis
(Fig. 2A). STAT5b is activated by
cytokines and some growth factors. Phosphorylation of STAT5b by
tyrosine kinases is so far the best-documented mechanism of STAT5b
activation. We also showed that STAT5b is localized in both the
cytoplasm and nuclei by confocal microscopy (Fig. 2A) and by western blot analysis using
cell fractionation (Fig. 2B).
Immunoprecipitation analysis further revealed tyrosine
phosphorylation of STAT5b (Fig.
2C). These results indicate that STAT5b in pancreatic cancer
cells is constitutively activated.
STAT5b are frequently expressed in the nuclei of
tumor cells in borderline intraductal papillary mucinous neoplasms
(IPMNs) and intraductal papillary mucinous carcinomas but not in
intraductal papillary mucinous adenomas (23). Since the nuclear expression of
STAT5b protein correlated to the Ki-67 labeling index of the IPMNs,
STAT5b protein can contribute to the progression and proliferation
of IPMNs (23). On the contrary, we
could not detect significant proliferation differences between sham
and STAT5b-shRNA clones in pancreatic cancer cells in the present
study. In PDAC, STAT5b protein may not contribute to
proliferation.
Substantial evidence suggests that STAT5 is
associated with chemoresistance and apoptosis in PDAC. First,
curcubitacin B, an experimental drug for pancreatic cancer, causes
dose- and time-dependent G(2)-M-phase arrest and apoptosis of
pancreatic cancer cells in association with inhibition of activated
STAT5, STAT3 and JAK2 (35).
Second, downregulation in pancreatic cancer cells of neuropilin-1,
a coreceptor for vascular endothelial growth factor-A (VEGF-A), is
associated with increased sensitivity to an antineoplastic agent,
with pro-apoptotic actions, and with reduced STAT5 and Bcl-xL
protein levels (26). Third, Bcl-xL
plays a vital role in pancreatic cancer chemoresistance; the
increased expression of this antiapoptotic protein is associated
with poor survival (36,37). Fourth, STAT5 has a crucial role in
antiapoptotic signaling of the erythropoietin receptor and mediates
the immediate-early induction of Bcl-xL (32). In the present study, although there
was no significant difference of proliferation between the sham and
STAT5b shRNA clones without gemcitabine treatment (Fig. 3B), as for gemcitabine resistance,
PANC-1 STAT5b shRNA clones with gemcitabine treatment exhibited a
significantly greater growth inhibitory effect compared to
sham-transfected cells (Fig. 4).
Compared with PANC-1 cells which expressed relatively high levels
of STAT5b, both AsPC-1 and BxPC3 cells which express relatively low
levels of STAT5b exhibited significantly decreased growth by
treatment with gemcitabine (Fig.
5). Moreover, treatment with gemcitabine resulted in
significant increase in the growth of AsPC-1 STAT5b overexpression
clones compared to sham-transfected cells (Fig. 6B). These results suggest that STAT5b
in pancreatic cancer cells play an important role in gemcitabine
chemoresistance.
In this study, a significant clinicopathological
correlation between STAT5b expression and overall survival rates of
PDAC was observed (Fig. 9C).
However, even with the limited number of patients, a tendency
toward reduced overall survival for patients receiving neoadjuvant
or adjuvant gemcitabine treatments in the STAT5b-strong group.
These results suggest a correlation between STAT5b
expression/activation and clinical gemcitabine resistance. In our
immunohistochemical study, a correlation between expression of
STAT5b in PDAC and main pancreatic duct invasion was observed
(Table I). This suggests that
STAT5b may also play an important role in the invasiveness of
PDAC.
Regarding apoptosis, the caspase-3 activity was
upregulated in gemcitabine-treated STAT5b-shRNA clones with
subsequent cleavage of PARP protein (Fig. 7A). This activation was associated
with increased levels of apoptosis in the pancreatic cancer cells.
Gemcitabine also decreased Bcl-xL levels in the STAT5b-shRNA
expressing cells, but not in the sham-transfected cells (Fig. 7B). These observations suggest that
STAT5 may contribute to the antiapoptotic effects of pancreatic
cancer cells by inducing Bcl-xL.
EGF is capable of activating STAT5b, signaling of
which is mediated by the EGF receptor (38). PDGF is also capable of activating
STAT5, mediated by the PDGFβ-receptor (39). Both EGF and PDGF-induced activation
of STAT5 are independent of JAK family kinases and both EGF
receptor and PDGFβ-receptor have been shown to activate STAT5
(38,39). Thus, several tyrosine kinase
receptors have the ability to directly activate STAT5. In the
present study, downregulation of STAT5b resulted in reduced
adhesion and invasion of pancreatic cancer cells in vitro
(Fig. 8). Notably, EGF- and
PDGF-induced invasion were inhibited by downregulation of STAT5b.
These results may indicate that STAT5b has a pivotal role in both
EGF and PDGF signaling pathways in PDAC.
Taken together, our results suggest that targeting
STAT5b in PDAC may enhance the effectiveness of other therapeutic
modalities by enhancing gemcitabine chemosensitivity, increasing
apoptosis and suppressing cellular adhesion and invasion.
Acknowledgments
The authors would like to thank Mrs. Sumie Etoh of
Nippon Medical School for her excellent technical assistance. The
present study was supported by a Grant-in-Aid for Scientific
Research (C) from the Japan Society for the Promotion of Science
(No. 26462075 to A.M.).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stathis A and Moore MJ: Advanced
pancreatic carcinoma: Current treatment and future challenges. Nat
Rev Clin Oncol. 7:163–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997.PubMed/NCBI
|
|
5
|
Mini E, Nobili S, Caciagli B, Landini I
and Mazzei T: Cellular pharmacology of gemcitabine. Ann Oncol.
17(Suppl 5): v7–v12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bowman T, Garcia R, Turkson J and Jove R:
STATs in oncogenesis. Oncogene. 19:2474–2488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darnell JE Jr: STATs and gene regulation.
Science. 277:1630–1635. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ferbeyre G and Moriggl R: The role of
Stat5 transcription factors as tumor suppressors or oncogenes.
Biochim Biophys Acta. 1815:104–114. 2011.
|
|
10
|
Levine RL, Pardanani A, Tefferi A and
Gilliland DG: Role of JAK2 in the pathogenesis and therapy of
myeloproliferative disorders. Nat Rev Cancer. 7:673–683. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Scholz A, Heinze S, Detjen KM, Peters M,
Welzel M, Hauff P, Schirner M, Wiedenmann B and Rosewicz S:
Activated signal transducer and activator of transcription 3
(STAT3) supports the malignant phenotype of human pancreatic
cancer. Gastroenterology. 125:891–905. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sahu RP and Srivastava SK: The role of
STAT-3 in the induction of apoptosis in pancreatic cancer cells by
benzyl isothiocyanate. J Natl Cancer Inst. 101:176–193. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wakao H, Gouilleux F and Groner B: Mammary
gland factor (MGF) is a novel member of the cytokine regulated
transcription factor gene family and confers the prolactin
response. EMBO J. 13:2182–2191. 1994.PubMed/NCBI
|
|
14
|
Ren S, Cai HR, Li M and Furth PA: Loss of
Stat5a delays mammary cancer progression in a mouse model.
Oncogene. 21:4335–4339. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vafaizadeh V, Klemmt P, Brendel C, Weber
K, Doebele C, Britt K, Grez M, Fehse B, Desriviéres S and Groner B:
Mammary epithelial reconstitution with gene-modified stem cells
assigns roles to Stat5 in luminal alveolar cell fate decisions,
differentiation, involution, and mammary tumor formation. Stem
Cells. 28:928–938. 2010.PubMed/NCBI
|
|
16
|
Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi
M, Alanen K, Rui H and Nevalainen MT: Inhibition of transcription
factor Stat5 induces cell death of human prostate cancer cells. J
Biol Chem. 278:27287–27292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kazansky AV, Spencer DM and Greenberg NM:
Activation of signal transducer and activator of transcription 5 is
required for progression of autochthonous prostate cancer: Evidence
from the transgenic adenocarcinoma of the mouse prostate system.
Cancer Res. 63:8757–8762. 2003.PubMed/NCBI
|
|
18
|
Lee TK, Man K, Poon RT, Lo CM, Yuen AP, Ng
IO, Ng KT, Leonard W and Fan ST: Signal transducers and activators
of transcription 5b activation enhances hepatocellular carcinoma
aggressiveness through induction of epithelial-mesenchymal
transition. Cancer Res. 66:9948–9956. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pastuszak-Lewandoska D, Domańska D,
Czarnecka KH, Kordiak J, Migdalska-Sęk M, Nawrot E, Kiszałkiewicz
J, Antczak A, Górski P and Brzeziańska E: Expression of STAT5,
COX-2 and PIAS3 in correlation with NSCLC histhopathological
features. PLoS One. 9:e1042652014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Du W, Wang YC, Hong J, Su WY, Lin YW, Lu
R, Xiong H and Fang JY: STAT5 isoforms regulate colorectal cancer
cell apoptosis via reduction of mitochondrial membrane potential
and generation of reactive oxygen species. J Cell Physiol.
227:2421–2429. 2012. View Article : Google Scholar
|
|
21
|
Liang QC, Xiong H, Zhao ZW, Jia D, Li WX,
Qin HZ, Deng JP, Gao L, Zhang H and Gao GD: Inhibition of
transcription factor STAT5b suppresses proliferation, induces G1
cell cycle arrest and reduces tumor cell invasion in human
glioblastoma multiforme cells. Cancer Lett. 273:164–171. 2009.
View Article : Google Scholar
|
|
22
|
Jackerott M, Møldrup A, Thams P, Galsgaard
ED, Knudsen J, Lee YC and Nielsen JH: STAT5 activity in pancreatic
beta-cells influences the severity of diabetes in animal models of
type 1 and 2 diabetes. Diabetes. 55:2705–2712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kataoka TR, Ioka T, Tsukamoto Y, Matsumura
M, Ishiguro S and Nishizawa Y: Nuclear expression of STAT5 in
intraductal papillary mucinous neoplasms of the pancreas. Int J
Surg Pathol. 15:277–281. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Canales NA, Marina VM, Castro JS, Jiménez
AA, Mendoza-Hernández G, McCARRON EL, Roman MB and Castro-Romero
JI: A1BG and C3 are overexpressed in patients with cervical
intraepithelial neoplasia III. Oncol Lett. 8:939–947.
2014.PubMed/NCBI
|
|
25
|
Matsushita A, Götze T and Korc M:
Hepatocyte growth factor-mediated cell invasion in pancreatic
cancer cells is dependent on neuropilin-1. Cancer Res.
67:10309–10316. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukasawa M, Matsushita A and Korc M:
Neuropilin-1 interacts with integrin beta1 and modulates pancreatic
cancer cell growth, survival and invasion. Cancer Biol Ther.
6:1173–1180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rowland-Goldsmith MA, Maruyama H, Kusama
T, Ralli S and Korc M: Soluble type II transforming growth
factor-beta (TGF-beta) receptor inhibits TGF-beta signaling in
COLO-357 pancreatic cancer cells in vitro and attenuates tumor
formation. Clin Cancer Res. 7:2931–2940. 2001.PubMed/NCBI
|
|
28
|
Kawamoto M, Ishiwata T, Cho K, Uchida E,
Korc M, Naito Z and Tajiri T: Nestin expression correlates with
nerve and retroperitoneal tissue invasion in pancreatic cancer. Hum
Pathol. 40:189–198. 2009. View Article : Google Scholar :
|
|
29
|
Kang N, Zhang JH, Qiu F, Tashiro S,
Onodera S and Ikejima T: Inhibition of EGFR signaling augments
oridonin-induced apoptosis in human laryngeal cancer cells via
enhancing oxidative stress coincident with activation of both the
intrinsic and extrinsic apoptotic pathways. Cancer Lett.
294:147–158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Strasser A, Cory S and Adams JM:
Deciphering the rules of programmed cell death to improve therapy
of cancer and other diseases. EMBO J. 30:3667–3683. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schimmer AD, Hedley DW, Penn LZ and Minden
MD: Receptor-and mitochondrial-mediated apoptosis in acute
leukemia: A translational view. Blood. 98:3541–3553. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Socolovsky M, Fallon AE, Wang S, Brugnara
C and Lodish HF: Fetal anemia and apoptosis of red cell progenitors
in Stat5a−/−5b−/− mice: A direct role for Stat5 in Bcl-X(L)
induction. Cell. 98:181–191. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liotta LA, Steeg PS and Stetler-Stevenson
WG: Cancer metastasis and angiogenesis: An imbalance of positive
and negative regulation. Cell. 64:327–336. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hood JD and Cheresh DA: Role of integrins
in cell invasion and migration. Nat Rev Cancer. 2:91–100. 2002.
View Article : Google Scholar
|
|
35
|
Thoennissen NH, Iwanski GB, Doan NB,
Okamoto R, Lin P, Abbassi S, Song JH, Yin D, Toh M, Xie WD, et al:
Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT
pathway and potentiates antiproliferative effects of gemcitabine on
pancreatic cancer cells. Cancer Res. 69:5876–5884. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ghaneh P, Kawesha A, Evans JD and
Neoptolemos JP: Molecular prognostic markers in pancreatic cancer.
J Hepatobiliary Pancreat Surg. 9:1–11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takahashi H, Chen MC, Pham H, Matsuo Y,
Ishiguro H, Reber HA, Takeyama H, Hines OJ and Eibl G: Simultaneous
knock-down of Bcl-xL and Mcl-1 induces apoptosis through Bax
activation in pancreatic cancer cells. Biochim Biophys Acta.
1833:2980–2987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kloth MT, Catling AD and Silva CM: Novel
activation of STAT5b in response to epidermal growth factor. J Biol
Chem. 277:8693–8701. 2002. View Article : Google Scholar
|
|
39
|
Paukku K, Valgeirsdóttir S, Saharinen P,
Bergman M, Heldin CH and Silvennoinen O: Platelet-derived growth
factor (PDGF)-induced activation of signal transducer and activator
of transcription (Stat) 5 is mediated by PDGF beta-receptor and is
not dependent on c-src, fyn, jak1 or jak2 kinases. Biochem J.
345:759–766. 2000. View Article : Google Scholar : PubMed/NCBI
|