Introduction
Lung cancer is the most lethal cancer worldwide,
with an incidence of 1.6 million new cases annually and 1.38
million deaths reported in 2008 (1). In 2014, there were an estimated
1,665,540 new cancer cases diagnosed and 585,720 lung
cancer-related deaths in the US, and lung cancer continues to be
the most common cause of cancer-related mortality in both men and
women (2). In China, lung cancer is
also the leading cause of cancer-related deaths in both men and
women (3). Although tremendous
effort has been put into lung cancer research, the 5-year survival
rate of lung cancer patients is still only 14%, implying the need
for new treatment strategies such as new single effective
anticancer drug intervention or new rationale drug combination
protocols (4).
EGFR and KRAS are the two most common
driver mutations in lung cancer (5), and are the major drug targets for
NSCLC treatment. Interestingly, the frequency of EGFR
mutations is particularly higher in Eastern Asia countries when
compared with that in Western countries (6). It is known that lung cancer treatment
can be achieved by using cytotoxic agents such as platinum
compounds, tubulin inhibitors and molecular-targeting agents to
interrupt the signaling pathways responsible for cell proliferation
and survival, such as the RAS/RAF/MEK/MAPK and the
phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathways (5). However, the effect of inhibition of
single or both pathways on NSCLC cells with EGFR or
KRAS mutations remain unclear. Multiple studies have
reported the close association of the P13K/Akt/mTOR pathway with
cancer cell survival, differentiation, adhesion, migration and
invasion (7). Preclinical studies
suggest that targeting the PI3K pathway can be an effective
treatment strategy for certain patients with NSCLC (8). Recently, a new P13K/Akt/mTOR
inhibitor, BKM120 was developed, which has shown efficacy in
inhibiting lung cancer as well as other types of cancer, including
squamous and non-squamous carcinoma by suppressing Akt and mTOR and
their downstream effectors (8,9).
BKM120 was launched into a phase II clinical trial
and was demonstrated to be safe and well-tolerant for patients,
with a favorable pharmacokinetic profile, clear evidence of target
inhibition and preliminary clinical antitumor activity (8). However, in a study involving a panel
of 353 cancer cell lines, BKM120 exhibited preferential inhibition
of tumor cells bearing PIK3CA mutations, in contrast to
either KRAS or PTEN mutant alone models (10). Thus, it appears that the treatment
effect of BKM120 on NSCLC cells with KRAS or even
EGFR mutation is not completely known. Moreover, PD0325901
is one of the currently investigated MEK inhibitors, and is a
synthetic organic molecule that selectively binds to and inhibits
mitogen-activated protein kinase (MEK) (11). The RAS/MEK pathway plays an
important role in cancer development and progression (7), and often demonstrates pathway
convergence to the PI3K/Akt/mTOR pathway. Recent studies have
demonstrated that co-targeting both pathways can enhance the
therapeutic response in cancer patients (12). However, the types of gene mutations
have not been verified in these patients showing a positive
response. Thus, the precise biomarkers and mechanism of the
antitumor effect are less clear (13).
In the present study, we aimed to investigate the
in vitro cancer inhibitory effect of a newly developed
PI3K/Akt/mTOR pathway inhibitor (BKM120) on NSCLC cell lines with
either EGFR or KRAS mutations, the two most commonly
found mutations in NSCLC. Both PC-9 and H1650 cells habour
EGFR mutations while H358 and A549 cells habour KRAS
mutations. We first examined and compared the effect of the single
use of BKM120 on these cell lines, and then subsequently compared a
combined treatment effect of BKM120 and the MEK inhibitor
PD0325901. Functional assays including apoptosis and cell cycle
analysis as well as treatment mechanism were performed so as to
evaluate their treatment value.
Materials and methods
Reagents
BKM120 was supplied by Roche Diagnostics Ltd.
(Lewes, East Sussex, UK) and was dissolved in dimethyl sulfoxide
(DMSO) and stored at −80°C. PD0325901 was purchased from Sigma (St.
Louis, MO, USA), dissolved in DMSO and stored at −80°C. Primary
antibodies against p-AKT (S473), p-S6 (S235/S236), S6, p-p70s6k,
p70s6k, GAPDH, PARP, P-ERK and ERK were purchased from Cell
Signaling Technology (Danvers, MA, USA). Primary antibodies against
AKT were purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Fluorescein-conjugated goat anti-rabbit and mouse secondary
antibodies were purchased from Odyssey (Belfast, ME, USA). Annexin
V/PI staining dye was purchased from BD Biosciences (San Jose, CA,
USA).
Cell lines and cell culture
The human NSCLC cell lines A549, H1650, H358, PC-9
and normal lung fibroblast cell line CCD19 were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). Both
A549 and H358 cell lines have KRAS gene mutations (14), while H1650 and PC-9 cells have an
in-frame deletion on exon 19 (Del E746-A750) of EGFR
(15); PC-9 is sensitive to
gefitinib while H1650 is resistant to gefitinib. All NSCLC cell
lines were grown in monolayer culture in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 µg/ml streptomycin from Gibco and were cultured at
37°C in a humidified atmosphere containing 5% CO2. The
normal lung fibroblast CCD19 cells were grown in monolayer culture
in Dulbecco's modified Eagle's medium (DMEM).
Cytotoxicity assay
Cell viability was assessed using a standard
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. All NSCLC cells and the normal lung fibroblast cell line
CCD19 were seeded in 96-well plates at a density of
5×103/well overnight. The cells were then treated with a
range of concentrations of BKM120 for 72 h or with DMSO as vehicle
control. Each dosage was repeated in triplicate. After a 72-h
treatment, 10 µl MTT (5 mg/ml) solution was added to each
well, and the plates were placed back into the incubator for 4 h,
and then 100 µl of the resolved solution (10% SDS and 0.1 mM
HCl) was added and the plates were incubated for another 4 h to
dissolve the formazan crystals. Finally, the colorimetric intensity
of the plates was measured at 570 nm (absorbance) and 650 nm
(reference) using a Tecan microplate reader (Tecan US, Inc.,
Morrisville, NC, USA). The cell viability was calculated as the
percent change in absorbance of the treated cells divided by the
absorbance of the untreated cells.
Cell cycle and apoptosis assay using flow
cytometry
All NSCLC cell lines were plated on a 6-well plate
with a cell density of 2×105 cells, and were
serum-starved overnight. The cells were then treated with different
concentrations of BKM120 or co-treated with the MEK inhibitor
(PD0325901) for 24 h. After treatment, the cells were harvested by
trypsin digestion and collected by centrifugation. For apoptosis
analysis, the cells were then washed twice with ice-cold 1X PBS and
stained with 5 µl of propodium iodide (PI, 1 mg/ml) and 5
µl Annexin V fluorescein dye at room temperature in the dark
for 15 min. The cells were then resuspended in 400 µl of
Annexin-binding buffer (BD Biosciences). The percentage of
apoptotic cells was quantitatively determined using a BD FACSAria
III flow cytometer (BD Biosciences). For cell cycle analysis, the
cells were stained with 5 µl of PI (1 mg/ml) alone in the
presence of methanol for fixation. Then, the percentages of cells
at the S, G1 and G2 phases were quantitatively measured using a BD
FACSAria III flow cytometer (BD Biosciences).
Western blot analysis
Preparation of the whole-cell protein lysates and
western blot analysis were carried out as follows. After drug
treatments, the cells were lysed in RIPA buffer (150 mmol/l NaCl,
50 mmol/l Tris-HCl pH 8.0, 1% Triton X-100, 0.1% SDS and 1%
deoxycholate) with a complete protease inhibitor cocktail from
Roche for 10 min on ice. The concentration of the total protein
extract was determined using a Bio-Rad DC™ protein assay kit
(Bio-Rad Laboratories, Philadelphia, PA, USA). Equal amounts of
total protein (30 µg) were resuspended in loading buffer,
boiled at 100°C for 5 min, and separated by SDS-PAGE, and
transferred to NC membranes (Millipore, Billerica, MA, USA). The
membranes were then blocked with 5% milk without fat in TBST for 1
h at room temperature.
The following primary antibodies were used: rabbit
polyclonal antibodies against GAPDH (1:1,000), phosphatase and
p70s6k (1:1,000), phosphatase and S6, phosphatase and ERK, polyADP
ribose polymerase (PARP) (1:1,000) and phosphatase AKT (1:1,000),
which were purchased from Cell Signaling Technology. The mouse
monoclonal antibody against t-AKT (1:1,000) was purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Primary antibodies were incubated overnight at 4°C.
After washing the membrane with TBST for three times (5 min/time),
the secondary fluorescent antibody (Odyssey) was added to the
membrane at a 1:10,000 dilution at room temperature for 1 h. Actin
or GAPDH was used as the loading control and for normalization. The
signal intensity of the membranes was detected using an LI-COR
Odyssey scanner (Belfast, ME, USA)
Statistical analysis
All data are expressed as the mean ± SD of three
individual experiments. Differences between groups were determined
by one way analysis of variance (ANOVA), followed by Bonferroni's
test to compare all pairs of columns. Results were considered to be
statistically significant at P<0.05.
Results
BKM120 exhibits different degrees of
sensitivity in the different NSCLC cell lines and the normal lung
fibroblast cell line
To examine the cytotoxicity of BKM120 on different
NSCLC cell lines with different types of mutations, four NSCLC cell
lines were treated with BKM120 at 0, 1.25, 2.5, 5 and 10 µM,
respectively for 72 h. In addition, the normal lung fibroblast cell
line was treated with BKM120 at 0, 1.25, 2.5, 5, 10, 20, 40 and 80
µM, respectively for 72 h. Susequently, the percentage of
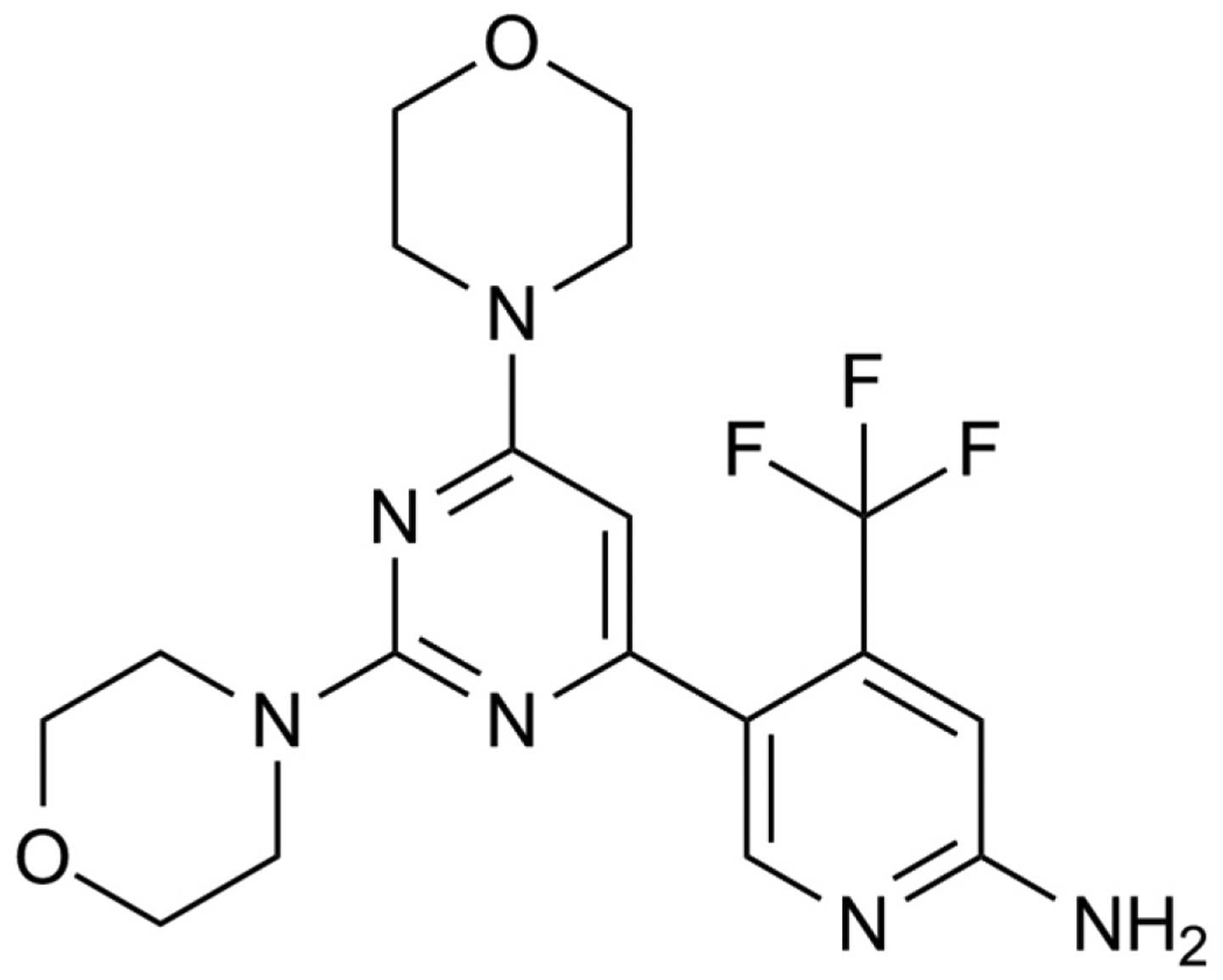
viable cells was determined by MTT assay. Fig. 1 shows the chemical structure of
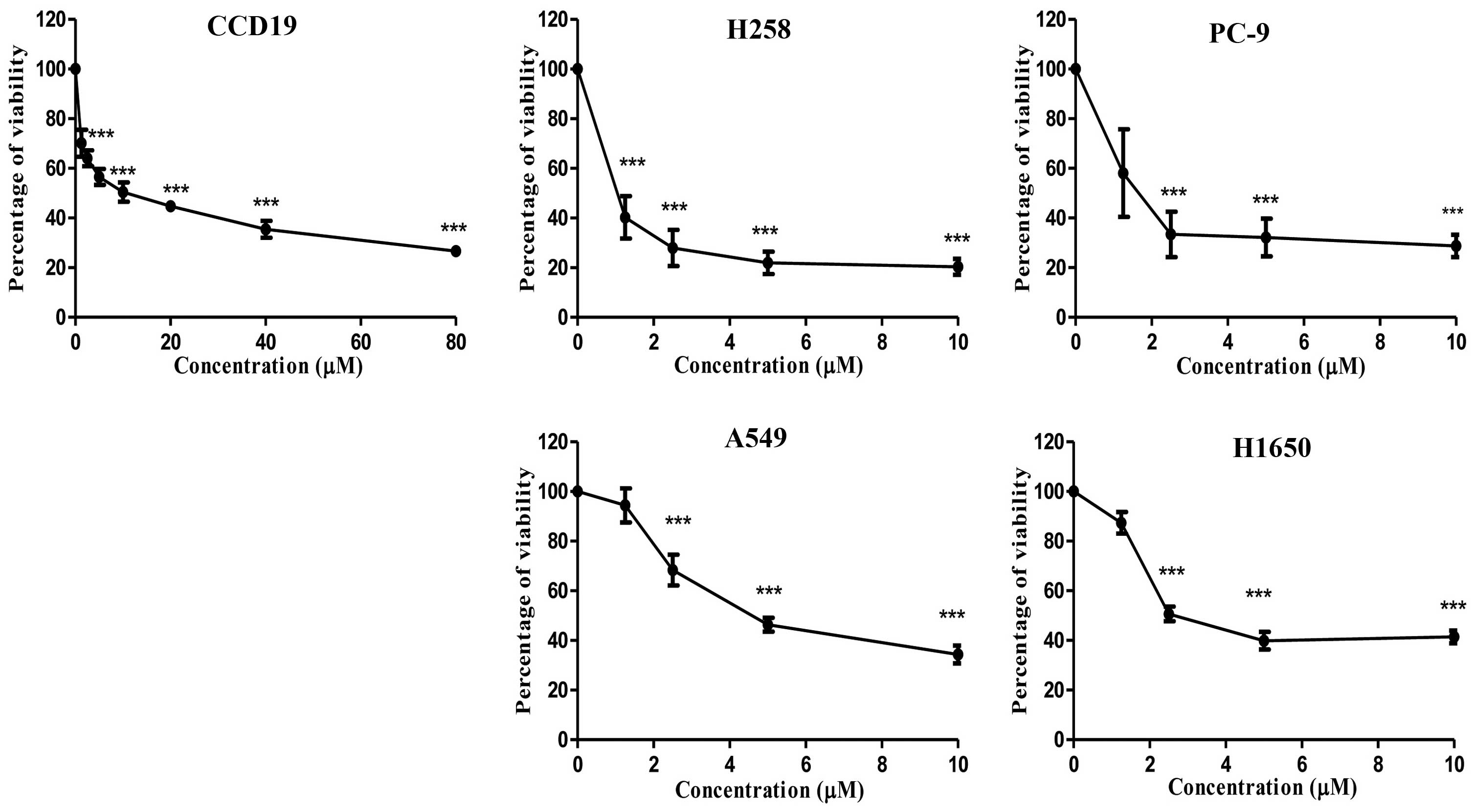
BKM120. Fig. 2 shows the percentage
of viability represented as dose-response curves for H358, A549,
PC-9, H1650 and CCD19 cells after 72 h of BKM120 treatment.
Table I summarizes
the mutation profiles and the IC50 values of BKM120 for
the four NSCLC cell lines and one normal lung fibroblast cell line.
As shown in Table I, among the four
cell lines, H358 and PC-9 were more sensitive to BKM120, with an
IC50 value of 1.15±0.16 and 2.09±0.50 µM,
respectively, while A549 and H1650 were less sensitive, with an
IC50 value of 5.18±0.53 and 6.72±1.42 µM,
respectively. Both H1650 and PC-9 cells harbor the same type of
EGFR-activating mutation, which is exon 19 deletion mutation
(16). However, H1650 is reported
to be resistant to gefitinib and PC-9 is sensitive to gefitinib
(17). Notably, the IC50
value of BKM120 in PC-9 cells was approximately one-third of that
of the H1650 cells, indicating that the EGFR auto-activating
pathway may have a crosstalk pathway in addition to the P13 kinase
pathway in H1650 cells which may contribute to the attenuation of
BKM120 sensitivity. Similarly, both the H358 and A549 cells contain
KRAS mutations (18), yet
H358 was much more sensitive to BKM120 than A549, indicating that
the KRAS/Raf signaling pathway may also have potential crosstalk in
addition to the PI3 kinase pathway in A549 cells. In addition, the
toxicity of BKM120 was further tested in a normal lung fibroblast
cell line (CCD19), and the IC50 value of BKM120 for the
CCD19 cell line was 9.7±2.16 µM. Although the fold different
was not marked, the IC50 of BKM120 for CCD19 was still
the highest among all tested cell lines.
 | Table IThe mutational profile, drug
sensitivity and IC50 value of BKM120 for four NSCLC cell
lines and one normal lung fibroblast cell line (CDD19). |
Table I
The mutational profile, drug
sensitivity and IC50 value of BKM120 for four NSCLC cell
lines and one normal lung fibroblast cell line (CDD19).
| Cell line | KRAS mutation | EGFR mutation | Gefitinib
sensitivity | BKM120
sensitivity | IC50
values to BKM120 (µM) |
|---|
| H358 | G12C | Wild-type | N/A | Sensitive | 1.15±0.16 |
| PC-9 | N/A | delE746-A750 | Sensitive | Sensitive | 2.09±0.50 |
| A549 | G12S | Wild-type | N/A | Resistant | 5.18±0.53 |
| H1650 | N/A | delE746-A750 | Resistant | Resistant | 6.72±1.42 |
| CCD19 | N/A | N/A | N/A | Resistant | 9.74±2.16 |
MTT assay results showed that BKM120 decreased the
cell viability of H358, PC9, A549, H1650 and CCD19 cells after a
72-h treatment. BKM120 inhibited H358, PC-9, A549, H1650 and CCD19
cell growth in a dose-dependent manner.
BKM120 significantly induces apoptosis in
the H358 and PC-9 BKM120-sensitive cell lines
Previous studies have demonstrated that BKM120
induces apoptosis in some NSCLC cell lines at the concentration
range of 2 to 4 µM (18). To
test the biological effect of BKM120, a similar dosage range was
taken into reference to treat the cells, and the level of apoptosis
was determined. As shown in Fig. 3,
flow cytometric analysis showed that when BKM120 was used at or
below 1 µM, no significant increase in apoptosis was
observed in all four cell lines. However, when BKM120 were used at
or above 2 µM, it induced a significant level of late stage
apoptosis in both the H358 and PC-9 cells. However, both A549 and
H1650 cells exhibited no significant apoptotic effect by BKM120 at
all dosages.
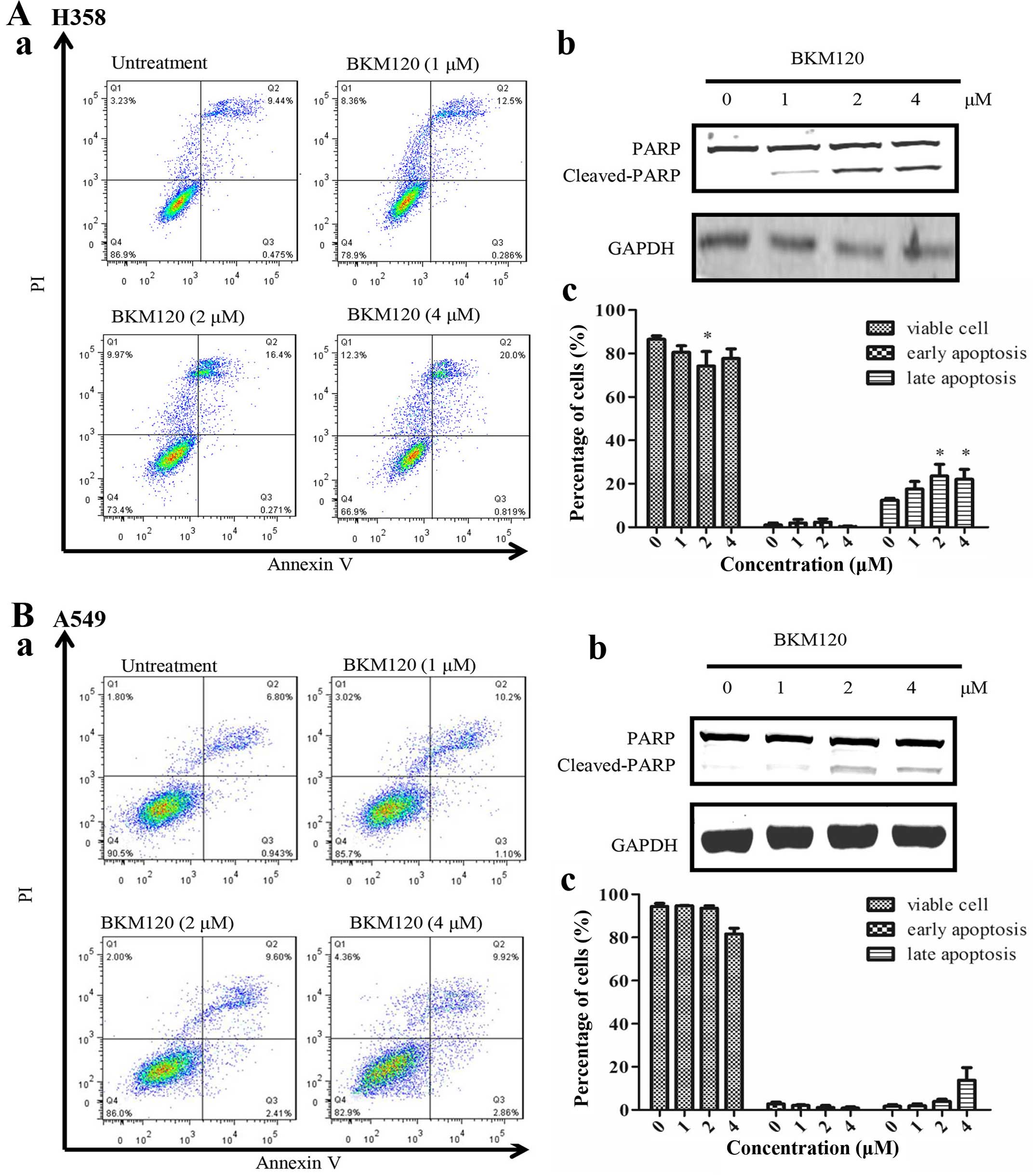 | Figure 3The apoptotic effect induced by
BKM120 in four NSCLC cell lines (H358, A549, PC-9 and H1650). Flow
cytometric analysis of (A) H358 and (B) A549 cell apoptosis induced
by BKM120 (0, 1, 2 and 4 µM). The percentage of cells in
early or late apoptosis are presented in the lower right and upper
right quadrants. Western blot analysis of PARP (full length 110
kDa, and cleaved 89 kDa) activation in (A-b) H358 and (B-b) A549
cells was investigated after BKM120 administration. GAPDH served as
the loading control. The percentage of early and late apoptotic
cells was calculated: (A-c) H358 and (B-c) A549 cells. Results are
expressed as mean ± SD (n=3, *p<0.05,) The apoptosis
effect induced by BKM120 in four NSCLC cell lines (H358, A549, PC-9
and H1650). Flow cytometric analysis of (C) PC-9 and (D) H1650 cell
apoptosis induced by BKM120 (0, 1, 2 and 4 µM). Percentage
of cells in early or late apoptosis is presented in the lower right
and upper right quadrants. Western blot analysis of PARP (full
length 110 kDa, and cleaved 89 kDa) activation in (C-b) PC-9 and
(D-b) H1650 cells was investigated after BKM120 administration.
GAPDH served as the loading control. The percentage of early and
late apoptotic cell was calculated: (C-c) PC-9 and (D-c) H1650
cells. Results are expressed as mean ± SD (n=3,
*P<0.05, **P<0.01,
***P<0.001). |
Although PARP cleavage was shown in all four cell
lines at 2 µM of BKM120 treatment, still, no significant
level of apoptosis could be detected in the A549 and H1650 cells.
This may be due to the fact that PARP cleavage is only an indicator
of active caspase activity, but it may not eventually lead to
apoptotic effects.
BKM120 induces cell cycle arrest at G2
phase
Previous studies demonstrated that BKM120 induced
cell cycle arrest at G1 phase in three KRAS-mutated lung
cancer cell lines, Calu-1, H157 and A549 (18). In order to test whether a similar
effect was shown in our NSCLC cell lines with EGFR and
KRAS mutations, we examined the cell cycle by using flow
cytometry. Fig. 4 shows that BKM120
inhibited the growth of H358, PC-9 and H1650 NSCLC cells at the
concentration starting at 2 µM, while BKM120 enhanced G2
arrest. For A549 cells, cell cycle arrest was only shown at a high
dose (4 µM).
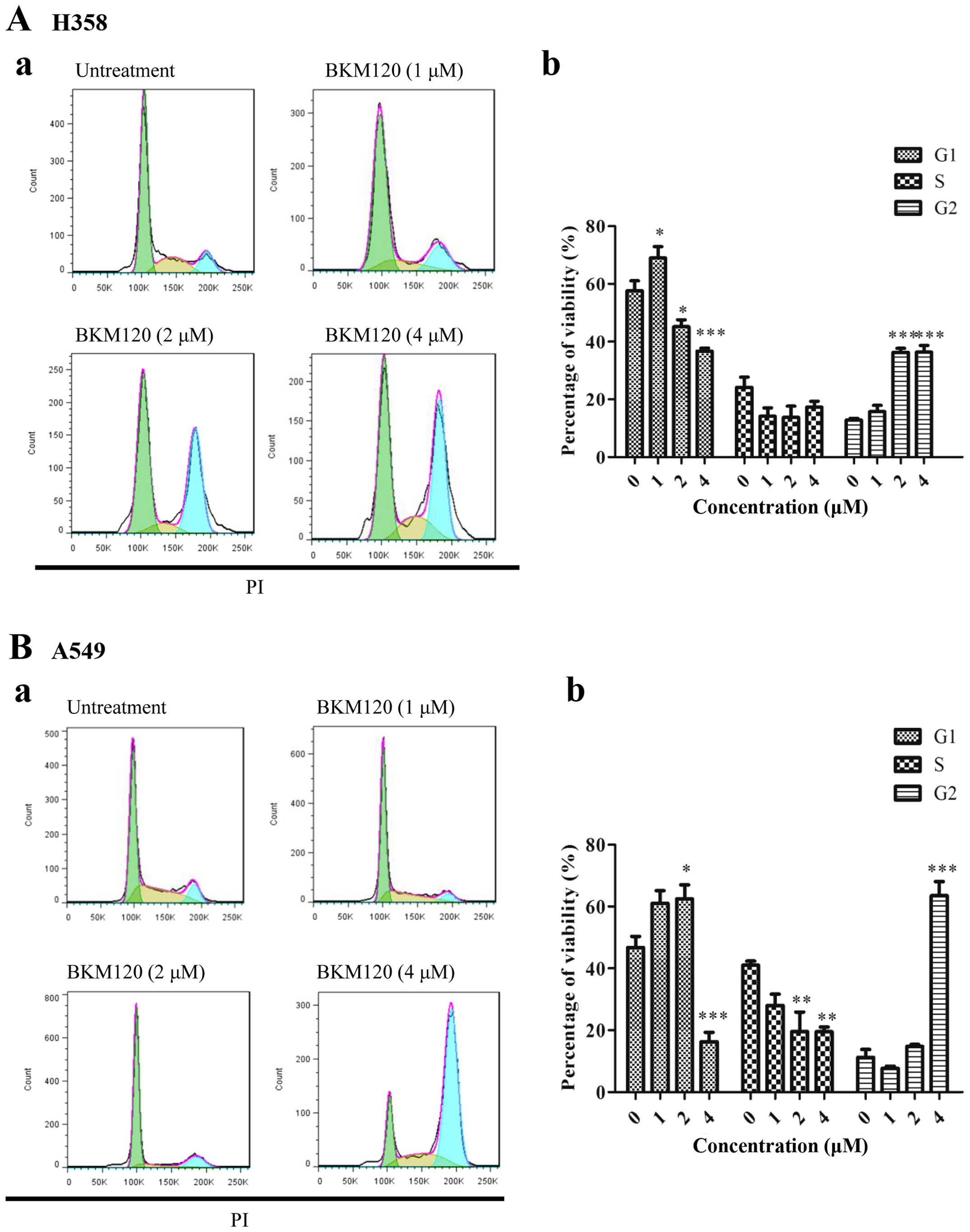 | Figure 4The effect of BMK120 on the cell
cycle arrest in four NSCLC cell lines (H358, A549, PC-9 and H1650).
Flow cytometric analysis of the cell cycle in (A-a) H358 and (B-a)
A549 cells after treatment with different concentrations of BKM120
for 24 h was studied. Percentages of (A-b) H358 and (B-b) A549
cells at the G1, S and G2 phases were statistically analyzed. Each
experiment was repeated at least three times. Results are expressed
as mean ± SD (n=3, *P<0.05, **P<0.01,
***P<0.001). The effect of BMK120 on the cell cycle
arrest in four NSCLC cell lines (H358, A549, PC-9 and H1650). Flow
cytometric analysis of cell cycle in (C-a) PC-9 and (D-a) H1650
cells after treatment with different concentrations of BKM120 for
24 h was studied. Percentages of (C-b) PC-9 and (D-b) H1650 cells
at the G1, S and G2 phases were statistically analyzed. Each
experiment was repeated at least three times. Results are expressed
as mean ± SD (n=3, *P<0.05, **P<0.01,
***P<0.001). |
BKM120 inhibits the mTOR signaling
pathway
EGFR and KRAS were found to be hyperactivated in
certain types of cancer, such as lung and breast (19), while the PI3K signaling pathway is
the downstream of EGFR and KRAS, which may play an important role
in the regulation of cell survival and proliferation. Therefore, we
determined whether BKM120 can inhibit the mTOR signaling pathway in
our cell lines by examining the phosphorylation level of the mTOR
signaling pathway proteins, such as AKT, ribosomal S6 and p70s6k.
After treatment of the cells with BKM120 for 24 h, we determined
the phosphorylation levels of several key proteins involved in this
pathway. As shown in Fig. 5,
western blot results showed that BKM120 reduced the levels of
phosphorylation of AKT, p70S6K and S6K in a dose-dependent manner
in all the NSCLC cell lines, suggesting involvement of the mTOR
pathway. The level of ERK phosphorylation suppression was more
prominent in the H358 than that in the A549 cells although both
cell lines are KRAS mutants. However, there were no significant
differences in the phosphorylation of ERK in the H1650 and PC-9
cells following BKM120 treatment.
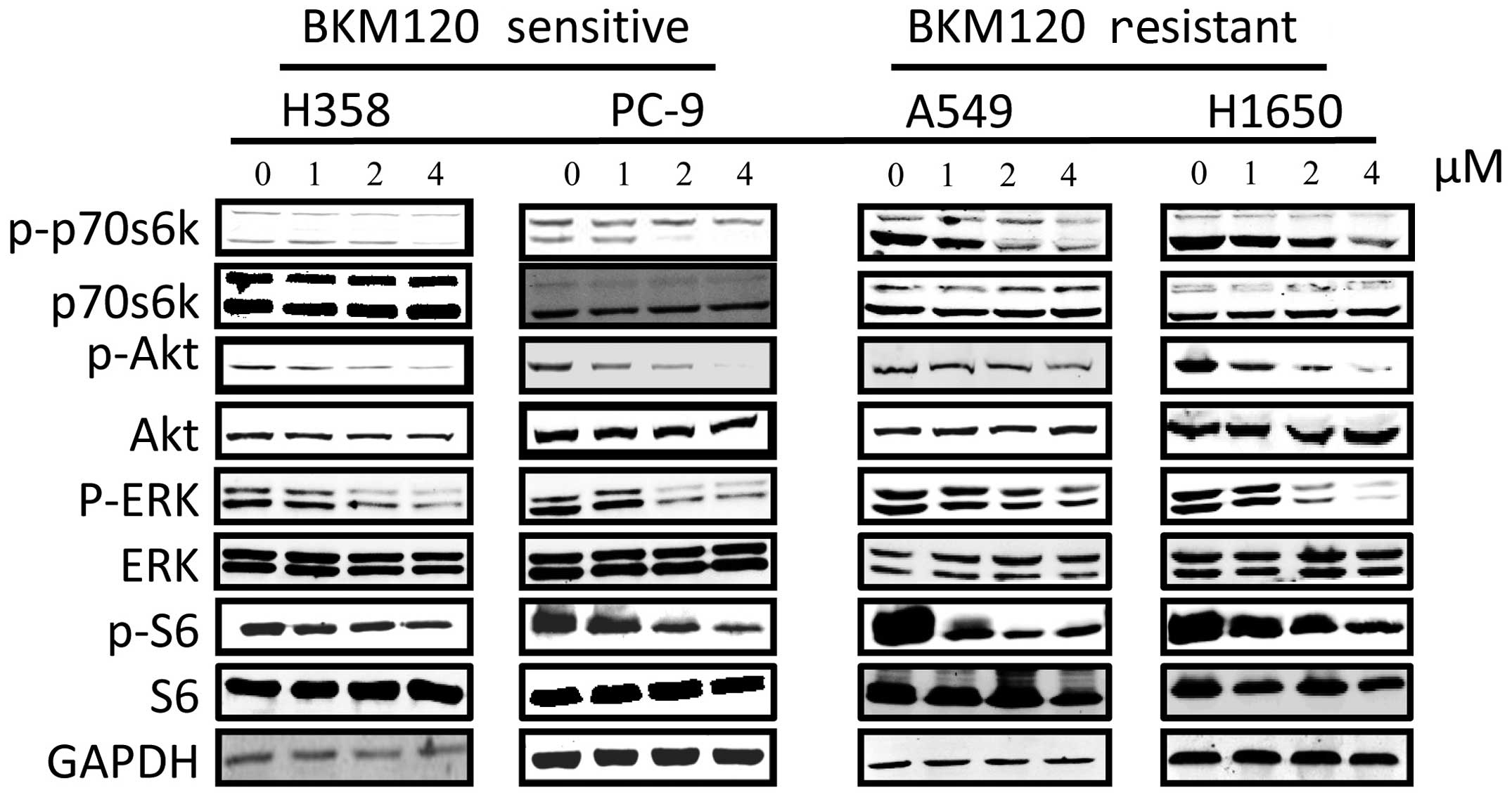 | Figure 5Western blot analysis of the effect
of BKM120 on the mTOR and P13K signalling pathways in different
NSCLC cell lines. The four NCSLC cell lines were treated with
BKM120 for 24 h. The levels of phospho(p)-ERK, p-p70s6k, p-AKT,
p-S6, AKT, ERK, p70s6k, S6, and GAPDH were determined by western
blot analysis. Total ERK1/2 (42/44 kDa), total p70s6k (70/85 kDa),
total S6 (32 kDa) and GAPDH (37 kDa) served as loading controls.
Each experiment was repeated at least three times. |
Combination usage of PI3K and MEK
inhibitors synergistically enhance the apoptosis in
BKM120-resistant A549 cells
Since A549 and H1650 cells are relatively less
sensitive to BKM120 treatment, we further examined whether
co-targeting the PI3K pathway by BKM120 and the MEK pathway by
PD0325901 could synergistically enhance the cancer inhibiting
effect. Previously, PD0325901 was used at a concentration of 100 nM
in combination with BKM120 in the KRAS mutant human and murine CRC
cells (20). Therefore, we took 100
nM as a reference concentration and used it in combination with
BKM120 on A549, PC-9, H358 and H1650 cells. Fig. 6 demonstrates that single use of
either one of these drugs did not induce significant levels of
apoptosis in both the A549 and H1650 cells; however, when both
drugs were used in combination, the levels of apoptosis increased
synergistically in the A549 cells, mildly in the H1650 and H358
cells but apoptosis was not increased in the PC-9 cells.
Discussion
The PI3K pathway has been regarded as an important
drug target for cancer treatment, including NSCLC (8). RAD001 and BEZ235 as well as several
rapamycin analogues are the current mTOR inhibitors undergoing
development designed for cancer with rapamycin resistance (21). RAD001 and BEZ235 both increased AKT
phosphorylation in various lung cancer cell lines such as A549 and
H157 (18). BKM120, similar to
RAD001, is also a PI3K inhibitor, which has been developed and is
also in clinical trial for various types of cancer (22).
In our previous study, we found that BKM120
effectively inhibited the growth of human NSCLC cell lines with
different types of gene mutations (18). It was also suggested that BKM120 was
more sensitive to cell lines with PIK3CA mutation (18). In the present study, we examined the
effect of BKM120 in four NSCLC cell lines with EGFR or
KRAS mutations. We found that H358 and PC-9 cells were
sensitive to BKM120 treatment. Although similar mutations are
present in H358 and PC-9 cells, these cell lines were less
sensitive to BKM120, indicating that the presence of EGFR and KRAS
mutations are not the only criteria to determine whether single
BKM120 treatment is enough. Further mTOR and MEK activation
screening is needed to decide whether combinational therapy should
be used. Similar to RAD001, our data showed that BKM120 decreased
AKT phosphorylation in all four cell lines. Using human NSCLC cell
lines with different mutations, we demonstrated that BKM120
effectively inhibited the growth of NSCLC cell lines with a
different degree of sensitivity. In a recent study, it was
suggested that BKM120 could induce G1 arrest at a low concentration
in A549, H157 and Calu-1 cells, which contain KRAS mutation
(18). In addition, another study
reported that BKM120 could induce G2 phase cell cycle arrest in
T-cell acute lymphoblastic leukemia (23). Here, we further showed that
inhibition of the PI3K pathway resulted in both a decrease in
proliferation in the four lung cancer cell lines and an increase in
apoptosis in the H358 and PC-9 cells, which were both sensitive to
BKM120. It was reported that BKM120 arrests cancer cells at the G1
phase at low concentrations (≤2 µM); our data indicated that
BKM120 also induced G1 arrest but at a lower concentration.
However, after treating the cells with BKM120 at higher
concentrations (>2 µM), it alternatively induced G2
arrest.
A previous study reported that co-treatment of an
MEK inhibitor and BKM120 in NSCLC cell lines with KRAS
mutations significantly inhibited the growth of tumors both in
vitro and in vivo (24).
In the clinic, drug decisions are usually based on a genetic
mutation test to govern the use of drugs common in personalized
therapy (25). For patients with
EGFR mutations and who are less sensitive to gefitinib, the
clinical outcome is poor (26);
therefore, more new drug options are required. In addition,
although KRAS mutations were identified in the 1970s, this
target remains undruggable (27).
Out data indicated that for NSCLC patients with EGFR or
KRAS mutations, inhibition of the PI3K pathway alone or in
combination with the MEK inhibitor may suppress tumor growth, and
was even effective in the gefitinib-resistance H1650 cell line.
Recent studies also suggest that combinational use of PI3K
inhibitor with other agents have demonstrated greater efficacy than
monotherapy (20). When we used
BKM120 alone at a high dose (4 µM), the PC-9 cell line with
EGFR or the H358 cell line with KRAS mutation demonstrated
inhibition of cell growth and induction of apoptosis, while the
gefitinib resistant cancer cell lines H1650 and A549 both were less
sensitive to BKM120 than the other cell lines. However, combined
drug treatment demonstrated good cancer inhibition, indicating the
need for more rational combinational therapy for
gefitinib-resistant patients in the future.
In the present study, we found that following BKM120
treatment, AKT and ERK were activated as shown by the increased
phosphorylation levels in the lung cancer cells. This is further
supported by previous studies on the PI3K pathway which found that
single treatment with a PI3K inhibitor is not sufficient as it
induces at least one signaling mediator in the alternate pathway.
Overall, BKM120 alone or in combination with PD0325901 was
demonstrated to be useful in NSCLC with KRAS and EGFR
mutations. More valid biomarkers must be identified for deciding on
the use of single or combinational therapy in the future.
Acknowledgments
The present study was supported by FDCT grants from
the Science and Technology Development Fund of Macao (Project code:
021/2013/A1 and 005/2014/AMJ) grant to E.L.L.
References
|
1
|
Zardavas D, Fumagalli D and Loi S:
Phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin
pathway inhibition: A breakthrough in the management of luminal
(ER+/HER2−) breast cancers? Curr Opin Oncol.
24:623–634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
She J, Yang P, Hong Q and Bai C: Lung
cancer in China: Challenges and interventions. Chest.
143:1117–1126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Domvri K, Darwiche K, Zarogoulidis P and
Zarogoulidis K: Following the crumbs: From tissue samples, to
pharmacogenomics, to NSCLC therapy. Transl Lung Cancer Res.
2:256–258. 2013.PubMed/NCBI
|
|
6
|
Ma BB, Hui EP and Mok TS: Population-based
differences in treatment outcome following anticancer drug
therapies. Lancet Oncol. 11:75–84. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fumarola C, Bonelli MA, Petronini PG and
Alfieri RR: Targeting PI3K/AKT/mTOR pathway in non small cell lung
cancer. Biochem Pharmacol. 90:197–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vansteenkiste JF, Canon JL, Braud FD,
Grossi F, De Pas T, Gray JE, Su WC, Felip E, Yoshioka H, Gridelli
C, et al: Safety and efficacy of Buparlisib (BKM120) in patients
with PI3K pathway-activated non-small cell lung cancer: Results
from the Phase II BASALT-1 Study. J Thorac Oncol. 10:1319–1327.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mantripragada K and Khurshid H: Targeting
genomic alterations in squamous cell lung cancer. Front Oncol.
3:1952013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maira SM, Pecchi S, Huang A, Burger M,
Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, et al:
Identification and characterization of NVP-BKM120, an orally
available pan-class I PI3-kinase inhibitor. Mol Cancer Ther.
11:317–328. 2012. View Article : Google Scholar
|
|
11
|
Gupta P, Das PK and Ukil A:
Antileishmanial effect of 18β-glycyrrhetinic acid is mediated by
Toll-like receptor-dependent canonical and noncanonical p38
activation. Antimicrob Agents Chemother. 59:2531–2539. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heidegger I, Pircher A, Klocker H and
Massoner P: Targeting the insulin-like growth factor network in
cancer therapy. Cancer Biol Ther. 11:701–707. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu JA, Li H, Meng X, Fullerton DA,
Nemenoff RA, Mitchell JD and Weyant MJ: Group IIa secretory
phospholipase expression correlates with group IIa secretory
phospholipase inhibition-mediated cell death in K-ras mutant lung
cancer cells. J Thorac Cardiovasc Surg. 144:1479–1485. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Simonetti S, Molina MA, Queralt C, de
Aguirre I, Mayo C, Bertran-Alamillo J, Sanchez JJ, Gonzalez-Larriba
JL, Jimenez U, Isla D, et al: Detection of EGFR mutations with
mutation-specific antibodies in stage IV non-small-cell lung
cancer. J Transl Med. 8:1352010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ware KE, Hinz TK, Kleczko E, Singleton KR,
Marek LA, Helfrich BA, Cummings CT, Graham DK, Astling D, Tan AC,
et al: A mechanism of resistance to gefitinib mediated by cellular
reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth
loop. Oncogenesis. 2:e392013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren H, Chen M, Yue P, Tao H, Owonikoko TK,
Ramalingam SS, Khuri FR and Sun SY: The combination of RAD001 and
NVP-BKM120 synergistically inhibits the growth of lung cancer in
vitro and in vivo. Cancer Lett. 325:139–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Stiegler AL, Boggon TJ, Kobayashi
S and Halmos B: EGFR-mutated lung cancer: A paradigm of molecular
oncology. Oncotarget. 1:497–514. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roper J, Sinnamon MJ, Coffee EM, Belmont
P, Keung L, Georgeon-Richard L, Wang WV, Faber AC, Yun J, Yilmaz
ÖH, et al: Combination PI3K/MEK inhibition promotes tumor apoptosis
and regression in PIK3CA wild-type, KRAS mutant colorectal cancer.
Cancer Lett. 347:204–211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Benjamin D, Colombi M, Moroni C and Hall
MN: Rapamycin passes the torch: A new generation of mTOR
inhibitors. Nat Rev Drug Discov. 10:868–880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Slomovitz BM and Coleman RL: The
PI3K/AKT/mTOR pathway as a therapeutic target in endometrial
cancer. Clin Cancer Res. 18:5856–5864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lonetti A, Antunes IL, Chiarini F, Orsini
E, Buontempo F, Ricci F, Tazzari PL, Pagliaro P, Melchionda F,
Pession A, et al: Activity of the pan-class I phosphoinositide
3-kinase inhibitor NVP-BKM120 in T-cell acute lymphoblastic
leukemia. Leukemia. 28:1196–1206. 2014. View Article : Google Scholar
|
|
24
|
Bonelli MA, Cavazzoni A, Saccani F,
Alfieri RR, Quaini F, La Monica S, Galetti M, Cretella D, Caffarra
C, Madeddu D, et al: Inhibition of PI3K pathway reduces
invasiveness and epithelial-to-mesenchymal transition in squamous
lung cancer cell lines harboring PIK3CA gene alterations. Mol
Cancer Ther. 14:1916–1927. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Diamandis M, White NM and Yousef GM:
Personalized medicine: Marking a new epoch in cancer patient
management. Mol Cancer Res. 8:1175–1187. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Antonicelli A, Cafarotti S, Indini A,
Galli A, Russo A, Cesario A, Lococo FM, Russo P, Mainini AF,
Bonifati LG, et al: EGFR-targeted therapy for non-small cell lung
cancer: Focus on EGFR oncogenic mutation. Int J Med Sci.
10:320–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stephen AG, Esposito D, Bagni RK and
McCormick F: Dragging ras back in the ring. Cancer Cell.
25:272–281. 2014. View Article : Google Scholar : PubMed/NCBI
|