Introduction
Prostate cancer (PCa) is the most common male
malignancy in developed countries (1). Androgen deprivation therapy is
considered an effective strategy in the treatment of advanced PCa
patients (2). Castration-resistant
PCa often initially responds to taxane-based chemotherapy, but
eventually acquisition of chemoresistance leads to failure of the
treatment (3,4). Therefore, it is important to find
novel therapeutic targets to improve the treatment of patients with
hormone-refractory, drug-resistant prostate cancer.
S-phase kinase associated protein 2 (Skp2), a
well-characterized F-box protein, is a crucial component of the SCF
(Skp1-Cullin1-F-box) type of E3 ubiqutin ligase complexes (5). SCFSkp2 is an E3 ubiquitin
ligase for androgen receptor (AR), p27, p21, p57, p130, FoxO1,
E-cadherin and other substrate ubiquitination and degradation
(6). Importantly, Skp2 is found to
be upregulated in various types of cancers including prostate
cancer (7). It has been shown that
Skp2 protein expression was correlated with tumor stage,
histological grade, and recurrence in prostate cancer (7). In addition, prostate tissue-specific
overexpression of Skp2 causes hyperplastic and dysplastic changes
and low-grade carcinomas in the mouse prostate gland (8). Skp2 knockout mice suppresses PCa
development induced by loss of either p19ARF or PTEN (9). Skp2 deficiency restricts PCa
development by triggering Arf-p53-independent cellular senescence
through upregulation of p21, p27 and ATF4 in vivo (9). Numerous studies indicate that Skp2
interacts with AR, p27, PTEN, PI3K/Akt and BRCA2 signaling, thus
plays critical roles in prostate tumorigenesis (6).
Emerging evidence suggests that Skp2 is also
involved in the development of drug resistance in human cancers
(10). It has been demonstrated
that overexpression of Skp2 is associated with resistance to
preoperative doxorubicin-based chemotherapy in primary breast
cancer (11). Moreover, Skp2
silencing sensitizes Her2-positive tumors to Herceptin treatment
(12). Recently, it has been
reported that Skp2 is associated with acquisition of
epithelial-mesenchymal transition (EMT) and paclitaxel resistance
in breast cancer cells (10).
However, the role of Skp2 in paclitaxel resistance of PCa remains
unclear.
In this study, we demonstrated that Skp2 is
upregulated in paclitaxel-resistant PCa cells DU145-TxR or
PC-3-TxR. We found that knockdown of Skp2 restored paclitaxel
sensitive in DU145-TxR or PC-3-TxR cells. Importantly, p27, a
cyclin dependent kinase inhibitor, was found to be decreased in
DU145-TxR or PC-3-TxR cells while increased in Skp2 silencing
DU145-TxR or PC-3-TxR cells. We provided evidence that Skp2 is a
promising therapeutic target for treatment of hormone-refractory,
drug-resistant prostate cancer.
Materials and methods
Cell culture and reagents
PC3 and DU145 human PCa cells were purchased from
ATCC and cultured in RPMI-1640 medium supplemented with 10% fetal
bovine serum and penicillin/streptomycin (Invitrogen, Carlsbad, CA,
USA). Paclitaxel-resistant PC-3-TxR and DU145-TxR cells were
generated as previously described (3). PC-3-TxR and DU145-TxR cells were
cultured in RPMI-1640 medium with 10 nM paclitaxel (Abcam,
ab120143) to maintain their drug-resistant phenotypes. Before each
experiment, these cells were grown for at least one day in normal
medium. HEK293 human embryonic kidney cells were obtained from ATCC
and cultured in Dulbecco's modified Eagle's medium supplemented
with 10% fetal bovine serum. Skp2 inhibitor C1 (SKPin C1) was
purchased from MedChem Express and SZL-P1-41 (compound #25) from
TOCRIS.
shRNA-mediated silencing
For lentiviral shRNA infection, 293T cells were
transfected with Skp2 or control shRNA along with packing plasmids
and envelope plasmid using Lipofectamine 2000 reagents according to
the manufacturer's instructions. When 293T cells were cultured to
80–90% confluence in a 10-cm dish, the shuttle plasmid of insertion
sequences and packaging plasmids (pGag/Pol, pRev, pVSV-G) were
transfected and cultured for 72 h, then virus was collected. Skp2
shRNA sequence and negative control sequence was
5′-GGGAGTGACAAAGACTTTG-′3 and 5′-TTCTCCGAACGTGTCACGT-′3,
respectively. Real-time PCR and western blotting were used to
confirm the efficiency of Skp2 interference.
Cell viability assay
Cells were seeded at a density of 3×103
cells/well in 96-well plates containing 200 µl of complete
medium in five replicate wells. Cells were allowed to attach
overnight before treating with the indicated dose of paclitaxel for
24, 48, 72 or 96 h. Subsequently, viable cells were treated with
0.3 mg/ml of MTS for 2 h and MTS conversion was analyzed by
Enzyme-Labeled Instrument (Thermo Fisher Multiskan, GO, USA) at 490
nm.
Real-time quantitative RT-PC
Total RNAs were extracted from cells using TRIzol
reagent (Invitrogen) according to the manufacturer's instructions
and treated with RNase-free DNase. Reverse transcription reaction
was performed on 1 µg of total RNA per sample using the
SuperScript III reverse transcriptase kit (Invitrogen) according to
the manufacturer's protocol. After reverse transcription, the
real-time polymerase chain reaction (PCR) was performed using the
Power SYBR Green PCR MasterMix (Applied Biosystems, Foster City,
CA, USA) on the ABI 7500 thermocycler (Applied Biosystems)
following the instrument manual. The sequences of the primers are
as follows: GAPDH (sense: 5′-TTCATTGACCTCAACTACAT-3′, antisense:
5′-GAGGGGCCATCCACAGTCTT-3′. Skp2 (sense:
5′-GCTGCTAAAGGTCTCTGGTGT-3′, anti-sense:
5′-AGGCTTAGATTCTGCAACTTG-3′). E-cadherin (sense:
5′-ACGCATTGCCACATACA-3′, antisense: 5′-CGTTAGCCTCGTTCTCA-3′)
Vimentin (sense: 5′-ATGGCTCGTCACCTTCG-3′, antisense:
5′-AGTTTCGTTGATAACCTGTCC-3′).
Western blot analysis
Cell lysates were prepared with RIPA buffer (PBS, 1%
Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS and protease
inhibitor cocktail (Roche). The proteins were separated by SDS-PAGE
and then electrotransferred to membranes. Membranes were blocked in
5% non-fat milk in TBST (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1%
Tween-20) for 1 h at room temperature. Membranes were then
incubated with primary antibody overnight at 4°C. After incubation
with a secondary horseradish peroxidase (HRP)-linked antibody,
immunoreactive proteins were detected by using enhanced
chemiluminescence supersignal substrate (Thermo/Pierce, Rockford,
IL, USA). The following antibodies were used for western blot
analysis: p21 (Cell Signaling, 2947), p27 (Cell Signaling, 2552),
E-cadherin (Cell Signaling, 3195), Vimentin (Cell Signaling, 5741),
FoxO1 (Cell Signaling, 2880), FoxO4 (Cell Signaling, 9472),
CyclinB1 (Cell Signaling, 4138), CyclinD1 (Cell Signaling 2926),
CyclinE2 (Cell Signaling, 4132), β-catenin (Cell Signaling, 9562),
Skp2 (Invitrogen-187334), β-actin (Sigma, A1978), Fbxw8
(Sigma,HPA038851), Cul1 (Sigma, C7117), Cul2 (Sigma, SAB4200207),
Cul7 (Sigma, C1743), Cul4A (Sigma, C037), Cul4B (Sigma, C9995).
Statistical analysis
Results are presented as means ± SD. Statistical
analyses were performed using the Statistical Package for Social
Science (SPSS for Windows package release 10.0; SPSS Inc., Chicago,
IL, USA). P<0.05 was considered as statistically
significant.
Results
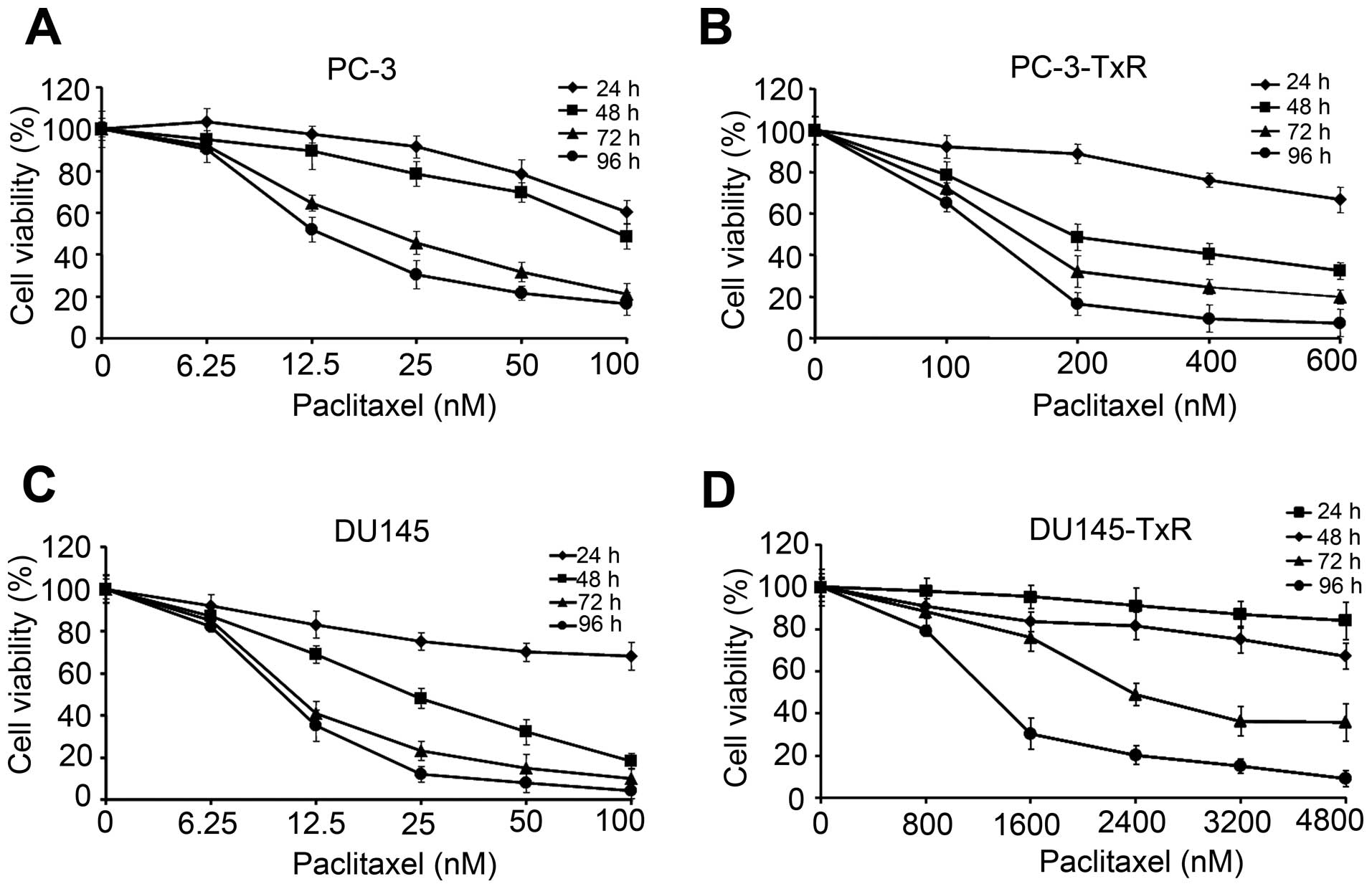
Assessment of paclitaxel sensitivity in
paclitaxel-resistant PCa cells
To investigate the molecular mechanisms underlying
paclitaxel resistance of hormone-refractory prostate cancer, we
first reconfirmed the paclitaxel resistance of DU145-TxR or
PC-3-TxR cells. PC3, PC3-TxR, DU145 and DU145-TxR cells were
treated with different concentration of paclitaxel for 24, 48, 72
and 96 h and cell viability was determined by MTT assay. As shown
in Fig. 1, as related to
vehicle-treated cells, the paclitaxel treatment was statistically
significant at 12.5 nM for the 48 and 72 h treatment in PC-3 cells
(Fig. 1A) and that at 200 nM of
paclitaxel for the 48 and 72 h treatment in PC-3-TxR cells
(Fig. 1B). The paclitaxel treatment
was effective at the dose of 12.5 nM for 48 and 72 h treatment in
DU145 (Fig. 1C) and that at 1600 nM
of paclitaxel for the 48 and 72 h treatment in DU145-TxR cells
(Fig. 1D). The 72 h of
IC50 values of parental cells PC-3 or DU145 were 18.42
and 10.91 nM, respectively. Cell growth inhibition assay
demonstrated that these PC-3-TxR and DU145-TxR cells become
9.04-fold (IC50: 166.47 nM) and 297.49-fold
(IC50: 3245.63 nM) higher than that in both parent
cells, respectively.
Skp2 was significantly elevated in
paclitaxel-resistant PCa cells
Emerging evidence has demonstrated that Skp2 is
involved in drug resistance in human cancer (10,11).
To explore whether Skp2 has a critical role in paclitaxel-resistant
PCa cells, we measured the expression of Skp2 and other Cullin-RING
E3 ligase components in paclitaxel-resistant cells and parental
cells. We found that the expression of Skp2 was significantly
elevated in paclitaxel-resistant cells compared with parental cells
(Fig. 2A and B). Cul4A, Cul4B,
Fbxw8 were also significantly elevated in paclitaxel-resistant
cells compared with parental cells. We found that Skp2 at mRNA
level was significantly elevated in paclitaxel-resistant cells
compared with parental cells (Fig.
2C). On the contrary, p27, one of the Skp2 substrates, was
significantly decreased in paclitaxel-resistant cells compared with
parental cells (Fig. 2D).
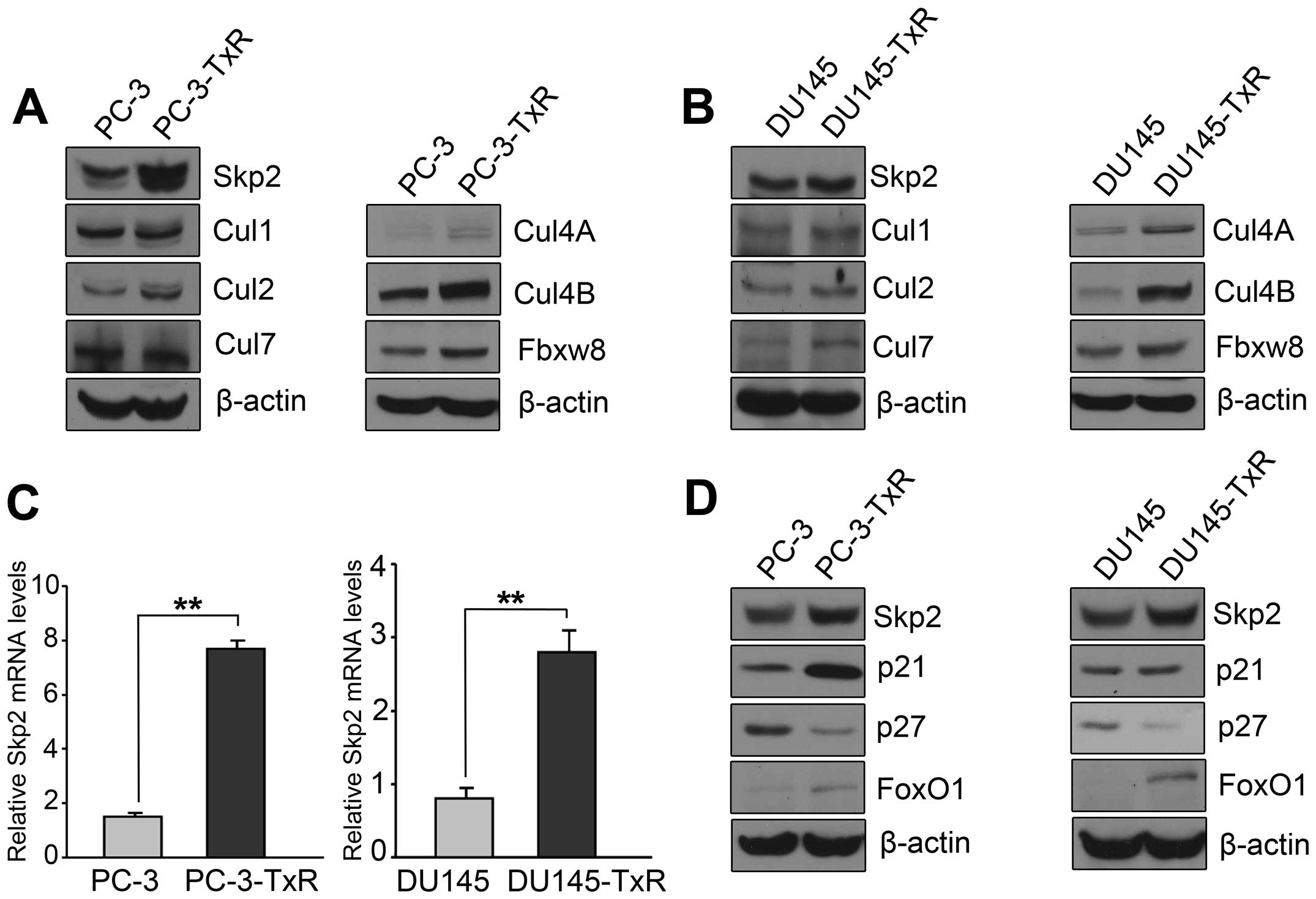 | Figure 2Skp2 is upregulated in paclitaxel
resistant PCa cells. (A) Western blotting was performed to detect
the expression of Skp2, Cul1, Cul2, Cul7, Cul4A, Cul4B, Fbxw8 in
PC-3 and PC-3-TxR cells. (B) Western blotting was performed to
detect the expression of Skp2, Cul1, Cul2, Cul7, Cul4A, Cul4B,
Fbxw8 in DU145 and DU145-TxR cells. (C) Real-time PCR assay was
conducted to detect the expression of Skp2 in parental and
paclitaxel resistant PCa cells. (D) Western blotting was performed
to detect the expression of Skp2, p21, p27 and FoxO1 in PC-3,
PC-3-TxR, DU145 and DU145-TxR cells. β-actin protein served as
loading controls. Bar graphs represent mean ± SD of three
independent experiments. (**P<0.01). |
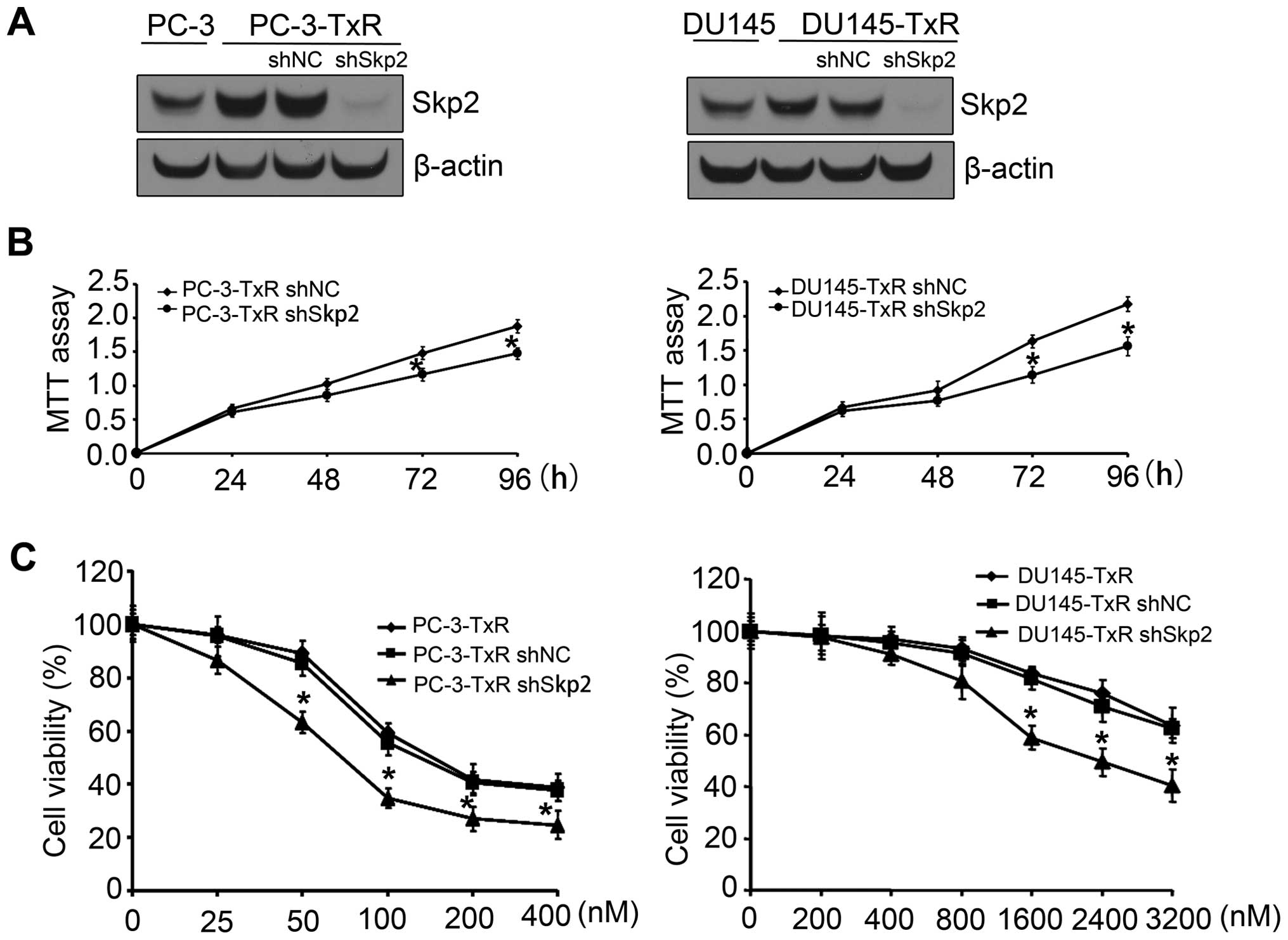
Knockdown of Skp2 enhances sensitivity of
paclitaxel-resistant cells to paclitaxel
We next investigated whether Skp2 is involved in the
development of paclitaxel resistance in PCa cells. As demonstrated
in Fig. 3A, expression of Skp2 was
significantly inhibited by Skp2 shRNA transfection in both PC3-TxR
and DU145-TxR cells. Thus, we determined the effect of
downregulation of Skp2 on paclitaxel sensitivity in PC3-TxR or
DU145-TxR cells. Knockdown of Skp2 induced cell growth inhibition
of PC3-TxR or DU145-TxR cells (Fig.
3B). Moreover, we found that knockdown of Skp2 significantly
enhances paclitaxel-resistant cells to paclitaxel sensitivity
(Fig. 3C).
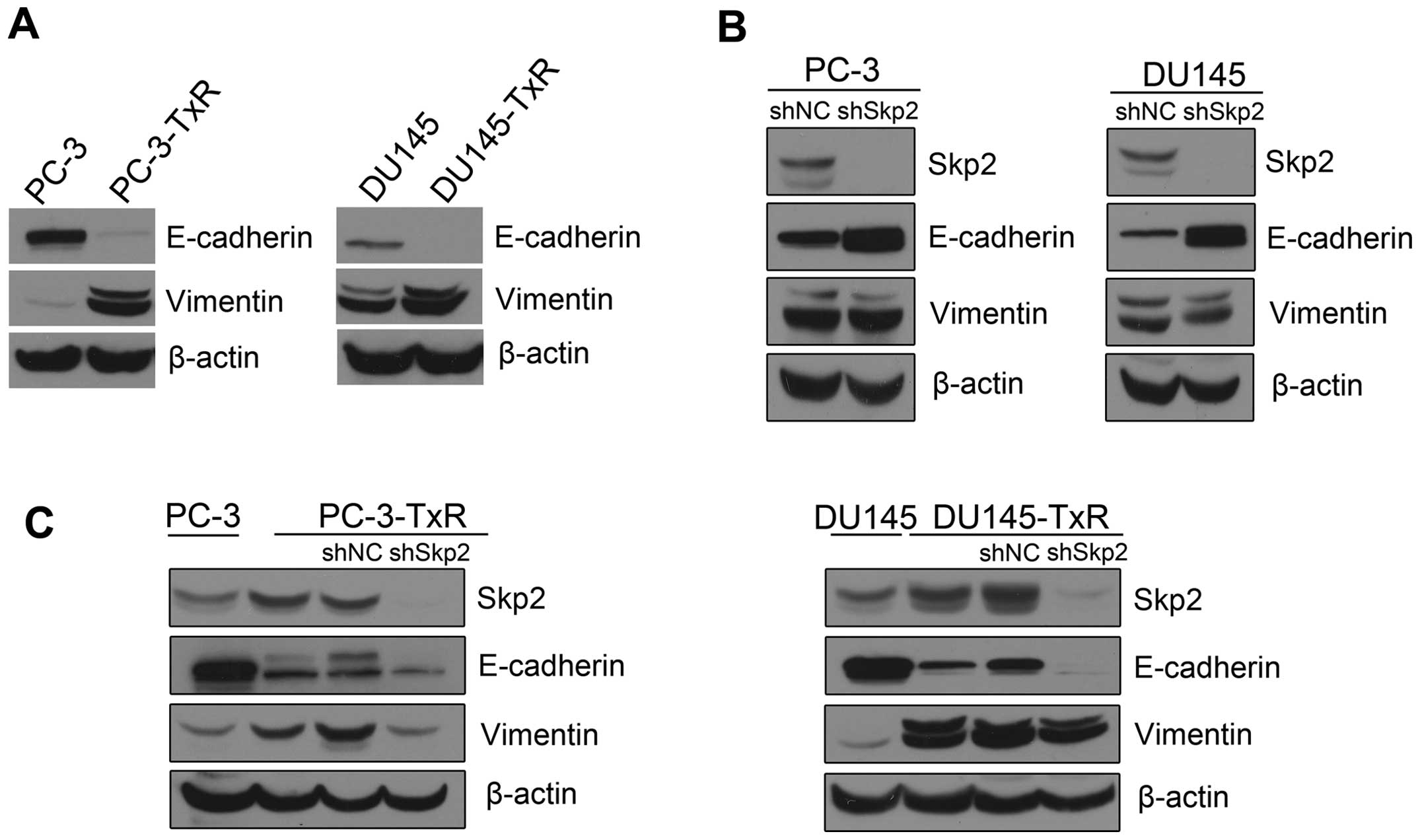
Expression of E-cadherin is not changed
in Skp2 silencing paclitaxel-resistant PCa cells
It has been reported that Skp2 is involved in
regulation of EMT in paclitaxel-resistant breast cancer cells
(10). We found that E-cadherin was
significantly decreased whereas the expression of Vimentin was
highly elevated in paclitaxel-resistant PCa cells (Fig. 4A). To determined whether knockdown
of Skp2 restores E-cadherin expression in paclitaxel-resistant PCa
cells, we examined the expression of E-cadherin and Vimentin in
Skp2 silencing PC-3, DU145, PC3-TxR and DU145-TxR cells. Knockdown
of Skp2 increased E-cadherin expression in PC-3 or DU145 cells
(Fig. 4B). However, E-cadherin
showed no significant change upon knockdown of Skp2 in PC-3-TxR or
DU145-TxR cells (Fig. 4C).
Knockdown of Skp2 restores the expression
of p27 in paclitaxel-resistant PCa cells
To investigate the mechanisms by which upregulated
Skp2 promotes paclitaxel resistance in PCa cells, we examined the
expression of Skp2-related cell cycle proteins by western blot
analysis. We observed that CyclinD1 was increased while CyclinB1
and CyclinE2 decreased in Skp2 silencing DU145-TxR cells, but not
in Skp2 silencing PC-3-TxR cells (Fig.
5A). On the contrary, p27, an inhibitor of cyclin-dependent
kinases, was found to increase in both Skp2 silencing PC-3-TxR and
DU145-TxR cells (Fig. 5B). These
data indicated that Skp2-p27 pathway is associated with the
development of paclitaxel resistance.
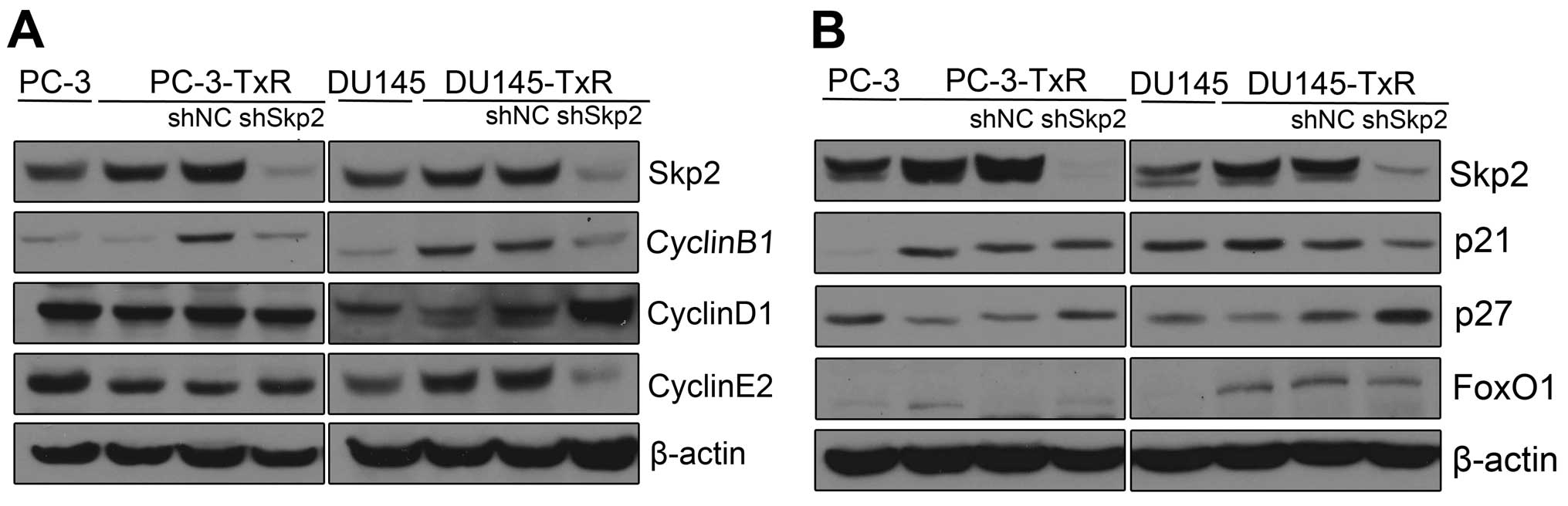 | Figure 5Expression of Skp2-related cell cycle
proteins in Skp2 silencing paclitaxel resistant PCa cells. (A)
Western blotting was performed to detect the expression of Skp2,
CyclinB1, CyclinD1, CyclinE2, p21, p27, FoxO1 in PC-3, PC-3-TxR and
Skp2 silencing PC-3-TxR cells. (B) Western blotting was performed
to detect the expression of Skp2, CyclinB1, CyclinD1, CyclinE2,
p21, p27, FoxO1 in DU145, DU145-TxR and Skp2 silencing DU145-TxR
cells. |
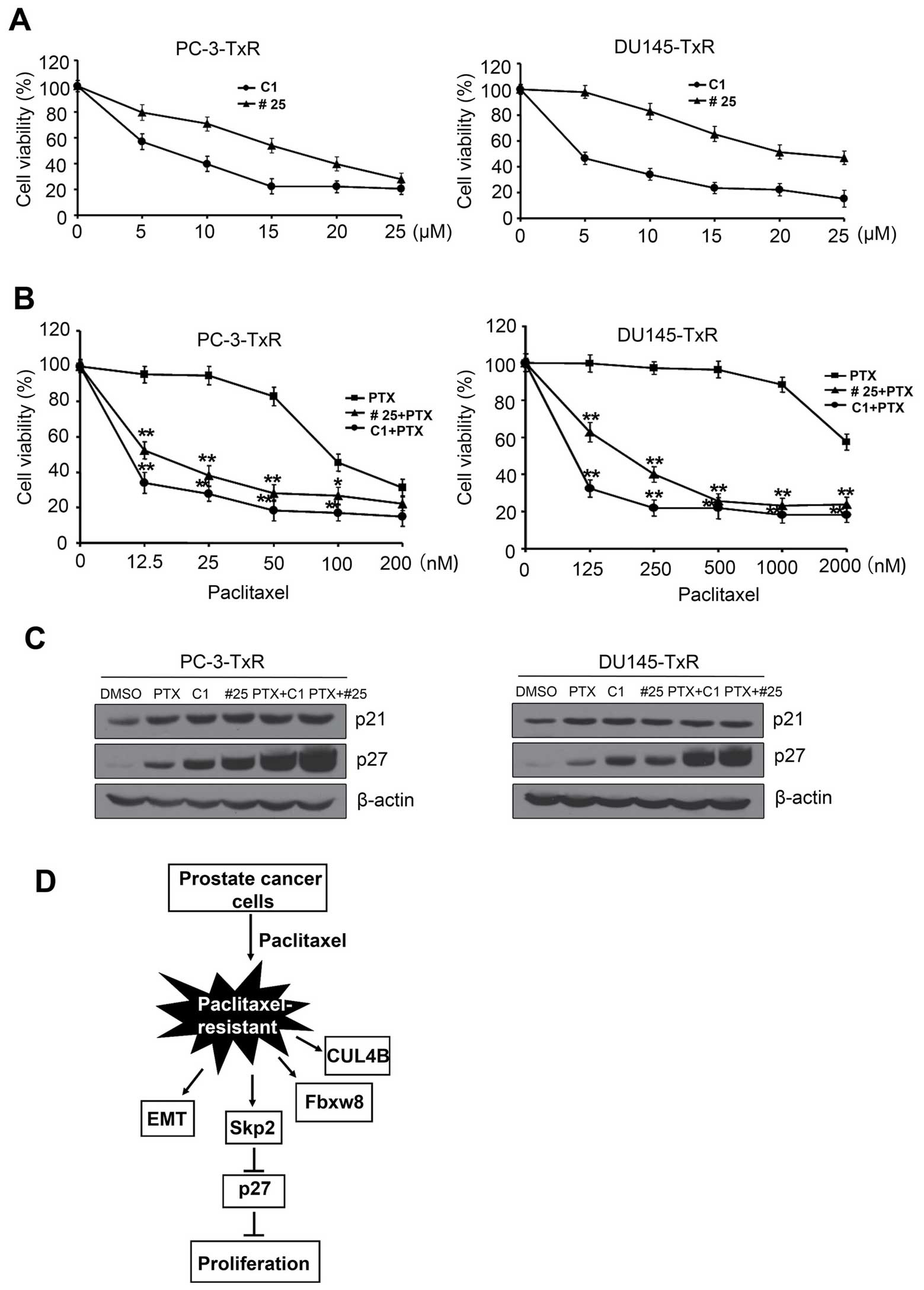
Skp2 inhibitors reverse the resistance of
paclitaxel-resistant PCa cells to paclitaxel
Skp2 inhibitor C1(SKPin C1) or SZL-P1-41 (compound
#25) are potent inhibitors of Skp2 and selectively inhibit
Skp2-mediated p27 degradation (32,33).
To determine the role of Skp2 inhibitors in the development of
paclitaxel resistance in PCa cells, we examined the effects of
SKPin C1 and compound #25 on paclitaxel sensitivity in PC3-TxR or
DU145-TxR cells. As shown in Fig.
6, PC3-TxR or DU145-TxR cells were treated with increasing
concentrations of SKPin C1 or compound #25 for 48 h. Both SKPin C1
and compound #25 suppressed the proliferation of PC3-TxR or
DU145-TxR cells in a dose-dependent manner (Fig. 6A). Next, SKPin C1 (2 µM) or
compound #25 (4 µM) were used to treat PC3-TxR or DU145-TxR
cells with increasing concentrations of paclitaxel. We found that
both SKPin C1 and compound #25 significantly enhances
paclitaxel-resistant cells to paclitaxel sensitivity (Fig. 6B).
Furthermore, paclitaxel or Skp2 inhibitor treatment
alone led to the accumulation of p27 in PC3-TxR or DU145-TxR cells.
Importantly, there is a synergetic effect of the accumulation of
p27 in the combination treatment of paclitaxel with Skp2 inhibitors
(Fig. 6C). A proposed pathway is
shown in (Fig. 6D).
Discussion
Paclitaxel is usually applied for the treatment of
hormone-refractory prostate cancer. However, acquired paclitaxel
resistance is a major limitation to improve the treatment of
advanced prostate cancer (3). In
this study, overexpression of Skp2 was observed in
paclitaxel-resistant PCa cells DU145-TxR or PC-3-TxR. Knockdown of
Skp2 in DU145-TxR or PC-3-TxR cells restored paclitaxel
sensitivity. Moreover, p27 was found to increase in both Skp2
silencing PC-3-TxR and DU145-TxR cells.
Several important molecules and signaling pathways
have been shown to contribute to paclitaxel resistance of PCa
cells. The C-terminal tensin like protein (CTEN, tensin 4) gene was
downregulated 10-fold in PC-3-TxR cells. Downregulation of CTEN
mediates paclitaxel resistance through increasing expression of
EGFR and actin (13).
Overexpression of Sonic Hedgehog (SHH) increases PCa cell
resistance to paclitaxel and leads to increase in ATP-binding
cassette (ABC) transporters expression (14,15).
Recently, it has been reported that Sex determining region Y-box 2
(Sox2) is involved in paclitaxel resistance of PC-3-TxR cells via
the PI3K/Akt pathway (16).
Furthermore, MiR-148a or MiR-34a attenuates paclitaxel resistance
of PC-3-TxR cells by regulating MSK1 or SIRT1 expression,
respectively (4,17). Emerging evidence also showed that
EMT plays an important role in the development of paclitaxel
resistance in various cancer including prostate cancer (10,18).
Nevertheless, the molecular mechanisms responsible for
paclitaxel-resistance has not been fully elucidated.
Cullin-RING ligases are the largest family of E3
ubiquitin ligases. There are seven cullin members, Cul1, Cul2,
Cul3, Cul4A, Cul4B, Cul5, and Cul7 in human (19). In this study, in addition to Skp2,
expression of Cullin-RING ligase components Cul4A, Cul4B, Fbxw8
were also found to be increased in DU145-TxR or PC-3-TxR cells.
Activation of Cullin-RING E3 ligases requires cullin neddylation.
MLN4924, a small molecule inhibitor of the NEDD8-activating enzyme
(NAE), effectively inhibits cullin neddylation and then inactivates
Cullin-RING E3 ligases (20). It
has been demonstrated that neddylation inactivation by MLN4924
inhibited Cullin-RING E3 ligases activity of PCa cells, thus
induced the accumulation of Cullin-RING E3 ligase ubiquitination
substrates, including p21, p27, WEE1, IκBα and DNA replication
licensing proteins CDT1 and ORC1 (20). Therefore, it will be particularly
interesting to determine whether MLN4924 could restore paclitaxel
resistance of PCa cells. Furthermore, targeting the Skp2-SCF
(Skp1-Cul1-F-box) complex can be an effective strategy for the
treatment of cancer. Importantly, Cul1 neddylation is an ideal
target to disrupt the Skp2-SCF complex formation (21). MLN4924 also induced senescence in
PCa cells by inhibiting Cullin-RING E3 ligases activity and causing
p21/p27 accumulation. Moreover, in xenograft tumor models, the
growth of PC3 tumors treated with MLN4924 in vivo was also
suppressed (21). Therefore,
inhibition of neddylation pathway and Cullin-RING E3 ligases could
exert potent anticancer efficacy in PCa cells. Noteworthy, MLN4924
is currently in several phase I clinical trials (20). It is critical to explore the role
and mechanism of MLN4924 in the development of paclitaxel
resistance in PCa cells.
EMT has been implicated in cancer metastasis and
therapeutic resistance in recent years (10,22).
Emerging evidence indicates that EMT plays crucial roles during the
development of castration-resistance and metastasis of prostate
cancer (23). EMT has been shown to
be induced by androgen deprivation therapy in both normal prostate
and prostate cancer (23).
Recently, EMT has also been indicated to be involved in acquiring
drug resistance (10). Accumulating
evidence suggests that EMT was found to be associated with the
resistance of ovarian carcinoma epithelial cells and breast cancer
cells to paclitaxel (10,24). More recently, it has been shown that
DU145-TxR cells exhibited EMT phenotype and became highly invasive
and motile as well as increased tumor growth in mouse xenografts.
E-cadherin, Keratin 8, 18, 19 were downregulated while Vimentin,
ZEB1 and Snail upregulated in DU145-TxR cells compared with DU145
cells (18). Consistent with this
notion, we also found that PC3-TxR and DU145-TxR cells acquired EMT
characteristics. It is well accepted that many transcription
factors such as Snail, Slug, ZEB1, KLF8 can bind to E-cadherin
reporter and repress its expression (10,23).
It has been shown that E-cadherin is destroyed by
ubiquitin-proteasome mediated degradation. Of note, E-cadherin is a
substrate of Skp2-SCF complex (21). Overexpression of Skp2 has been found
to be associated with breast cancer drug resistance and EMT
(10). Knockdown of Skp2 in breast
cancer cells led to partial reversal of EMT phenotype. We observed
that expression of E-cadherin was significantly decreased in
paclitaxel-resistant PCa cells DU145-TxR or PC-3-TxR compared with
their parental cells DU145 or PC-3, respectively. However, there
was no significant change of E-cadherin expression in Skp2 silenced
PC-3-TxR or DU145-TxR cells. These results suggest that Skp2
knockdown did not restore the expression of E-cadherin in PC-3-TxR
or DU145-TxR cells. Our results suggest that Skp2 may not be the
major regulator for E-cadherin expression in paclitaxel-resistant
PCa cells, thus other factors may contribute to EMT phenotype of
PC-3-TxR or DU145-TxR cells. This discrepancy of Skp2 controlled
E-cadherin expression between paclitaxel-resistant PCa cells and
their parental cells is not clear.
The best known Skp2 ubiquitination substrate is p27,
an inhibitor of cyclin-dependent kinases (7). It is well established that Skp2 plays
a critical role in controlling the cell cycle through the G1/S
transition by promoting the destruction of p27 (25). Accumulated evidence also suggests
that Skp2 acts as an oncoprotein in cell proliferation, survival,
and cancer development mainly through its degradation of p27
(6,26). Skp2 knockout mouse embryonic
fibroblast (MEFs) display reduced cell proliferation, accompanied
by enhanced p27 protein expression. Noteworthy, double deficiency
for p27 and Skp2 rescues the cell proliferation defect in Skp2
knockout MEFs and the reduced organ size and body weight observed
in Skp2 knockout mice (27,28). Importantly, Skp2 overexpression is
found in various human cancer samples associated with poor
prognosis and inversely correlated with p27 expression level
(26,29).
It has been demonstrated that Skp2 expression was
inversely correlated with p27 expression in prostate cancer
(29). Moreover, overexpression of
Skp2 in transgenic mice significantly decreased p27 protein
expression level in prostate glands (8). Of note, in addition to functioning as
an inhibitor of cyclin-dependent kinases and a tumor suppressor,
altered regulation of p27 may be involved in resistance to
chemotherapy (30). It has been
shown that p27 is associated with chemotherapeutic drug resistance
in cancer including gastric cancer, lung adenocarcionma and breast
cancer (5,30). In this study, we found that
expression of p27 was significantly decreased while Skp2
overexpressed in paclitaxel-resistant PCa cells DU145-TxR or
PC-3-TxR compared with their parental cells DU145 or PC-3,
respectively. Importantly, expression level of p27 was
significantly reversed by knockdown of Skp2 in both DU145-TxR and
PC-3-TxR cells. We propose that Skp2 promotes PCa proliferation and
paclitaxel resistance by targeting p27. Further investigation is
necessary for elucidating the molecular mechanism of Skp2-p27
pathway in the development of PCa paclitaxel resistance. Taken
together, our results strongly suggested that p27, rather than
E-cadherin, was one of the major substrates of Skp2 in
paclitaxel-resistant PCa cells DU145-TxR or PC-3-TxR.
Pharmacological Skp2 inactivation could restrain
cancer progression in various cancer models including prostate
cancer (32). Given that Skp2 is
involved in the development of paclitaxel resistance in PCa cells,
it would be interesting to test whether Skp2 inhibitors could be
applied for combination treatment of paclitaxel-resistant prostate
cancer. Using high-throughput screening or in silico approaches,
two small molecule inhibitors SKPin C1 or compound #25 targeting
Skp2 mediated p27 ubiquitination have been developed (32). In this study, we observed that SKPin
C1 or compound #25, like Skp2 knockdown, enhances sensitivity of
paclitaxel-resistant prostate cacner cells to paclitaxel and
impairs p27 degradation. Recently, it has been shown that both Skp2
knockdown and compound #25 suppresses cancer stem cell populations
and self-renewal ability (32).
Cancer stem cells are known to develop resistance to chemotherapy
and considered as one major cause of treatment failure (33). Targeting Skp2 by SKPin C1 or
compound #25 and combined with paclitaxel could be a novel approach
for achieving more effective treatment outcome of PCa patients.
In conclusion, we showed that Skp2 was upregulated
in paclitaxel-resistant PCa cells. Skp2 inhibition enhanced
paclitaxel-resistant DU145-TxR as well as PC-3-TxR cells to
paclitaxel sensitivity and reversed the protein expression level of
p27. Our results strongly suggests that inactivation of Skp2 could
be a promising systemic therapy strategy for restoring sensitivity
to paclitaxel. Thus, Skp2 may be a potential molecular target for
the treatment of advanced prostate cancers with acquired resistance
to paclitaxel.
Abbreviations:
|
PCa
|
prostate cancer
|
|
Skp2
|
S-phase kinase associated protein
2
|
|
SCF
|
Skp1-Cullin1-F-box
|
|
TxR
|
paclitaxel resistant
|
|
AR
|
androgen receptor
|
|
SKPin C1
|
Skp2 inhibitor C1
|
|
EMT
|
epithelial-mesenchymal transition
|
|
CTEN
|
C-terminal tensin like protein
|
|
MEF
|
mouse embryonic fibroblast
|
Acknowledgments
This study was supported by grants from National
Natural Science Foundation of China (81130046, 81201669, 81171993,
81272415, 81560483) and Natural Science Foundation of Guangxi
(2013GXNSFAA019211, 2014GXNSFCA118009, 2 013GX NSF E A0530 0 4, 2
012GX NSFCB0530 0 4, 2013GXNSFBA019177, 1355004-5, 201201ZD004,
GZPT13-35, 14122008-22, 11-031-05-K2, KY2015YB057, 14-045-12-K2) as
well as grants from Guangxi Educational Committee (201106LX087,
201203YB034). The authors would like to thank Drs Yong Wan and
Chunlin Zou for helpful discussions and Xin Huang for editing.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pernicová Z, Slabáková E, Kharaishvili G,
Bouchal J, Král M, Kunická Z, Machala M, Kozubík A and Souček K:
Androgen depletion induces senescence in prostate cancer cells
through down-regulation of Skp2. Neoplasia. 13:526–536. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takeda M, Mizokami A, Mamiya K, Li YQ,
Zhang J, Keller ET and Namiki M: The establishment of two
paclitaxel-resistant prostate cancer cell lines and the mechanisms
of paclitaxel resistance with two cell lines. Prostate. 67:955–967.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fujita Y, Kojima K, Ohhashi R, Hamada N,
Nozawa Y, Kitamoto A, Sato A, Kondo S, Kojima T, Deguchi T, et al:
MiR-148a attenuates paclitaxel resistance of hormone-refractory,
drug-resistant prostate cancer PC3 cells by regulating MSK1
expression. J Biol Chem. 285:19076–19084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner
MW and Kaelin WG Jr: Degradation of the SCF component Skp2 in
cell-cycle phase G1 by the anaphase-promoting complex. Nature.
428:194–198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Z, Gao D, Fukushima H, Inuzuka H, Liu
P, Wan L, Sarkar FH and Wei W: Skp2: A novel potential therapeutic
target for prostate cancer. Biochim Biophys Acta. 1825:11–17.
2012.
|
|
7
|
Drobnjak M, Melamed J, Taneja S, Melzer K,
Wieczorek R, Levinson B, Zeleniuch-Jacquotte A, Polsky D, Ferrara
J, Perez-Soler R, et al: Altered expression of p27 and Skp2
proteins in prostate cancer of African-American patients. Clin
Cancer Res. 9:2613–2619. 2003.PubMed/NCBI
|
|
8
|
Shim EH, Johnson L, Noh HL, Kim YJ, Sun H,
Zeiss C and Zhang H: Expression of the F-box protein SKP2 induces
hyperplasia, dysplasia, and low-grade carcinoma in the mouse
prostate. Cancer Res. 63:1583–1588. 2003.PubMed/NCBI
|
|
9
|
Lin HK, Chen Z, Wang G, Nardella C, Lee
SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, et al: Skp2
targeting suppresses tumorigenesis by Arf-p53-independent cellular
senescence. Nature. 464:374–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Q, Huang J, Wu Q, Cai Y, Zhu L, Lu X,
Chen S, Chen C and Wang Z: Acquisition of epithelial-mesenchymal
transition is associated with Skp2 expression in
paclitaxel-resistant breast cancer cells. Br J Cancer.
110:1958–1967. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Davidovich S, Ben-Izhak O, Shapira M,
Futerman B and Hershko DD: Over-expression of Skp2 is associated
with resistance to preoperative doxorubicin-based chemotherapy in
primary breast cancer. Breast Cancer Res. 10:R632008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chan CH, Li CF, Yang WL, Gao Y, Lee SW,
Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, et al: The Skp2-SCF
E3 ligase regulates Akt ubiquitination, glycolysis, herceptin
sensitivity, and tumorigenesis. Cell. 149:1098–1111. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y, Mizokami A, Izumi K, Narimoto K,
Shima T, Zhang J, Dai J, Keller ET and Namiki M: CTEN/tensin 4
expression induces sensitivity to paclitaxel in prostate cancer.
Prostate. 70:48–60. 2010. View Article : Google Scholar
|
|
14
|
Statkiewicz M, Maryan N, Lipiec A, Grecka
E, Grygorowicz MA, Omiotek M, Gorska A, Mikula M and Malecki M: The
role of the SHH gene in prostate cancer cell resistance to
paclitaxel. Prostate. 74:1142–1152. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh S, Chitkara D, Mehrazin R, Behrman
SW, Wake RW and Mahato RI: Chemoresistance in prostate cancer cells
is regulated by miRNAs and Hedgehog pathway. PLoS One.
7:e400212012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li D, Zhao LN, Zheng XL, Lin P, Lin F, Li
Y, Zou HF, Cui RJ, Chen H and Yu XG: Sox2 is involved in paclitaxel
resistance of the prostate cancer cell line PC-3 via the PI3K/Akt
pathway. Mol Med Rep. 10:3169–3176. 2014.PubMed/NCBI
|
|
17
|
Kojima K, Fujita Y, Nozawa Y, Deguchi T
and Ito M: MiR-34a attenuates paclitaxel-resistance of
hormone-refractory prostate cancer PC3 cells through direct and
indirect mechanisms. Prostate. 70:1501–1512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JJ, Yin B, Christudass CS, Terada N,
Rajagopalan K, Fabry B, Lee DY, Shiraishi T, Getzenberg RH, Veltri
RW, et al: Acquisition of paclitaxel resistance is associated with
a more aggressive and invasive phenotype in prostate cancer. J Cell
Biochem. 114:1286–1293. 2013. View Article : Google Scholar
|
|
19
|
Sarikas A, Hartmann T and Pan ZQ: The
cullin protein family. Genome Biol. 12:220–232. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang X, Li L, Liang Y, Li C, Zhao H, Ye D,
Sun M, Jeong LS, Feng Y, Fu S, et al: Targeting the neddylation
pathway to suppress the growth of prostate cancer cells:
Therapeutic implication for the men's cancer. Biomed Res Int.
2014:9743092014.PubMed/NCBI
|
|
21
|
Inuzuka H, Gao D, Finley LW, Yang W, Wan
L, Fukushima H, Chin YR, Zhai B, Shaik S, Lau AW, et al:
Acetylation-dependent regulation of Skp2 function. Cell.
150:179–193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li P, Yang R and Gao WQ: Contributions of
epithelial-mesenchymal transition and cancer stem cells to the
development of castration resistance of prostate cancer. Mol
Cancer. 13:552014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun Y, Wang BE, Leong KG, Yue P, Li L,
Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, et al: Androgen
deprivation causes epithelial-mesenchymal transition in the
prostate: Implications for androgen-deprivation therapy. Cancer
Res. 72:527–536. 2012. View Article : Google Scholar
|
|
24
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.PubMed/NCBI
|
|
25
|
Zhao H, Bauzon F, Fu H, Lu Z, Cui J,
Nakayama K, Nakayama KI, Locker J and Zhu L: Skp2 deletion unmasks
a p27 safeguard that blocks tumorigenesis in the absence of pRb and
p53 tumor suppressors. Cancer Cell. 24:645–659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng XY, Ding W, Xie LP and Chen ZD:
Correlation of Skp2 and P27kip1 protein expression and
clinicopathological features of prostate cancer. Ai Zheng.
23:215–218. 2004.In Chinese. PubMed/NCBI
|
|
27
|
Nakayama K, Nagahama H, Minamishima YA,
Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R,
Tsukiyama T, Ishida N, et al: Targeted disruption of Skp2 results
in accumulation of cyclin E and p27(Kip1), polyploidy and
centrosome overduplication. EMBO J. 19:2069–2081. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu L: Skp2 knockout reduces cell
proliferation and mouse body size: And prevents cancer? Cell Res.
20:605–607. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ben-Izhak O, Lahav-Baratz S, Meretyk S,
Ben-Eliezer S, Sabo E, Dirnfeld M, Cohen S and Ciechanover A:
Inverse relationship between Skp2 ubiquitin ligase and the cyclin
dependent kinase inhibitor p27Kip1 in prostate cancer. J Urol.
170:241–245. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Le TV, Seo Y, Ryu CJ, Lee HR and Park HJ:
Increased expression of p27 is associated with the cisplatin
resistance in gastric cancer cell line YCC-3. Arch Pharm Res.
33:1127–1132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao YF, Zhao JY, Yue H, Hu KS, Shen H,
Guo ZG and Su XJ: FOXD1 promotes breast cancer proliferation and
chemotherapeutic drug resistance by targeting p27. Biochem Biophys
Res Commun. 456:232–237. 2015. View Article : Google Scholar
|
|
32
|
Chan CH, Morrow JK, Li CF, Gao Y, Jin G,
Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, et al: Pharmacological
inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem
cell traits and cancer progression. Cell. 154:556–568. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chan CH, Morrow JK, Zhang S and Lin HK:
Skp2: A dream target in the coming age of cancer therapy. Cell
Cycle. 13:679–680. 2014. View Article : Google Scholar : PubMed/NCBI
|