Introduction
Lung cancer is the leading cause of cancer-related
death worldwide and accounts for more than one million deaths per
year (1). The morbidity of lung
cancer has continued to increase worldwide, particularly in
developing countries, which is partly due to air pollution. Lung
cancer can be divided into two histological types: small cell lung
cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC can be
further subdivided into: lung adenocarcinoma, large cell carcinoma
and squamous cell carcinoma. These cancer subtypes have distinct
morphologies and molecular profiles, and arise from distinct
locations within the lung. Approximately 40% of lung cancers are
adenocarcinomas. Despite advances in the diagnosis and treatment of
lung adenocarcinoma, the 5-year overall survival rate of patients
with lung adenocarcinoma is still extremely low (2). In recent years, great achievements
have been made in our understanding of lung adenocarcinoma driver
gene mutations and rearrangements, which play critical roles in
tumor development and progression (3). Among them, somatic mutations in the
gene that encodes the epidermal growth factor receptor (EGFR) and
rearrangements that involve the gene that encodes anaplastic
lymphoma kinase (ALK), are the most well-characterized examples
(4,5). Large, prospective randomized trials
have shown that molecularly selected patients with advanced lung
adenocarcinoma can benefit from treatment with EGFR and ALK
tyrosine kinase inhibitors (TKIs). However, despite the success of
such agents in the treatment of genetically defined subsets of lung
cancer, mutations such as those in EGFR and rearrangements within
the ALK gene are identified in only a limited number of patients
(6). Moreover, the majority of
patients who initially respond to EGFR-TKI therapy develop acquired
resistance within 6–12 months (5).
Therefore, further research is needed to identify new therapeutic
targets and tools for the treatment of lung adenocarcinoma.
Histone deacetylases (HDACs) are enzymes that
modulate the acetylation status of histones and other important
cellular proteins (7). Eighteen
HDACs have been identified in humans, and these enzymes are divided
into four classes, as follows, based on sequence phylogeny and
function: class I (HDAC1, 2, 3 and 8), class II (HDAC4, 5, 6, 7, 9
and 10), class III (SIRT1-7) and class IV (HDAC11) (8). HDACs have been recognized as
potentially useful therapeutic targets for a broad range of human
disorders including cancer (8,9). HDACs
are considered to be among the most promising targets in drug
development for cancer therapy. Two HDAC inhibitors, SAHA
(vorinostat) and romidepsin (FK228), have been approved by the US
Food and Drug Administration for the treatment of cutaneous T cell
lymphoma (10,11). However, these inhibitors elicit
profound side-effects since they target several HDAC isoforms
(12,13). Isoform-selective HDAC inhibitors may
offer a therapeutic advantage due to minimal toxicity. Among the 18
HDACs, HDAC6 has recently sparked great interest due to its
functional role in tumor progression. HDAC6 is a key regulator of
many signaling pathways that have been linked to cancer (14,15). A
diverse set of HDAC6 substrates is involved in tumorigenesis, such
as Hsp90, α-tubulin and cortactin. Deacetylation of the Hsp90 core
component by HDAC6 activates the chaperone activity of Hsp90 and
stabilizes Hsp90-associated molecules including EGFR, Akt, c-Raf,
FLT3 and mutant p53 (16). HDAC6
can promote cell migration through the deacetylation of α-tubulin
(17). HDAC6 has been shown to
regulate the cell cycle, apoptosis, and metastasis, among other
cellular processes (18,19). However, unlike the inhibition of
other HDACs, the inhibition of HDAC6 is not believed to be
associated with severe toxicity, which makes HDAC6 a possible
target for the treatment of cancer. However, few studies have been
conducted in regards to the role of HDAC6 in lung
adenocarcinoma.
In the present study, HDAC6 was found to be
overexpressed in lung adenocarcinoma cell lines, and the
overexpression of HDAC6 promoted lung adenocarcinoma proliferation
and conferred resistance to gefitinib, a widely used EGFR-TKI, in a
deacetylase activity-dependent manner. The inhibition of the
deacetylase activity of HDAC6 by CAY10603 (20), a potent and selective HDAC6
inhibitor, inhibited the proliferation and induced th apoptosis of
lung adenocarcinoma cell lines. CAY10603 synergized with gefitinib
to induce the apoptosis of lung adenocarcinoma cells via the
destabilization of EGFR. Our results suggest that inhibition of
HDAC6 may be a promising strategy for the treatment of lung
adenocarcinoma.
Materials and methods
Cell culture
The human lung adenocarcinoma cell lines A549,
HCC827 and H1975 were obtained directly from the American Type
Culture Collection (ATCC; Manassas, VA, USA). The cell lines were
cultured in RPMI-1640 medium (HyClone, Logan, UT, USA) supplemented
with 10% fetal bovine serum (FBS) and 1%
penicillin/streptomycin.
Antibodies and reagents
CAY10603 and gefitinib were purchased from Selleck
(Houston, TX, USA). The anti-PARP (9542) and anti-BID (2002)
antibodies were obtained from Cell Signaling Technology (Danvers,
MA, USA). The anti-caspase 3 (EAP0893) antibody was purchased from
Elabscience (Wuhan, Hubei, China). The anti-p53 (ab179477),
anti-Bax (ab32503), anti-EGFR (ab52894) and anti-p-ERK (ab76299)
antibodies were purchased from Abcam (Cambridge, MA, USA). The
anti-HDAC6 (12834-1-AP), Bcl2 (12789-1-AP), Bcl-xL (10783-1-AP),
Mcl1 (16225-1-AP), p21 (10355-1-AP) and ERK1/2 (16443-1-AP)
antibodies were obtained from Proteintech (Chicago, IL, USA). The
anti-β-actin mouse monoclonal antibody was purchased from Abgent
(San Diego, CA, USA).
Cell proliferation and colony formation
assays
In regards to the cell proliferation assay, cells
that were cultured in 96-well plates were treated with the
indicated compounds, and cell proliferation was measured at the
indicated times by the Cell Counting Kit-8 (CCK-8) (Dojindo, Tokyo,
Japan) assay. In regards to the colony formation assay, cells were
seeded at 1,000/well into 6-well plates and were cultured over a
14-day period. Colonies were fixed in 4% paraformaldehyde, washed
with H2O and stained with 0.1% crystal violet.
Annexin V assay of cell apoptosis
Cells were cultured in 6-well plates and were
treated with the indicated compounds, trypsinized and collected.
The collected cells were washed with PBS, resuspended in binding
buffer, and stained with Annexin V-PE and 7-AAD for 15 min
according to the manufacturer's protocol from Becton-Dickinson (San
Jose, CA, USA). Fluorescence was estimated with a Becton-Dickinson
flow cytometer.
Plasmids and transfection
The plasmids that express wild-type HDAC6 (HDAC6 WT)
and deacetylase-deficient HDAC6 (HDAC6 MT) were kindly provided by
Professor Jun Zhou of Nankai University (21,22).
The plasmids were transfected into cells with TurboFect DNA
transfection reagent (Thermo Scientific, Waltham, MA, USA). Control
siRNA and siRNAs targeting human HDAC6 were purchased from RiboBio
(Guangzhou, China). siRNA was transfected into cells using
Lipofectamine RNAiMAX from Invitrogen (Waltham, MA, USA).
Western blotting
Cells were lysed in lysis buffer (Beyotime, Jiangsu,
China). The samples were electrophoresed by SDS-PAGE and
transferred to polyvinylidene fluoride membranes (Roche). The
membranes were blocked with 5% dried skim milk and then incubated
with the indicated primary antibody followed by incubation with the
appropriate horseradish peroxidase (HRP)-conjugated secondary
antibody (Proteintech). Finally, the bands were visualized with
WesternBright ECL HRP substrate (Advansta, Menlo Park, CA, USA) and
developed with Kodak film.
Statistical analysis
Statistical analysis was performed using the
Student's t-test. Data are expressed as the mean ± standard
deviation (SD). P<0.05 was considered to indicate a
statistically significant difference.
Results
HDAC6 is overexpressed in lung
adenocarcinoma and is negatively correlated with the prognosis of
patients with lung adenocarcinoma
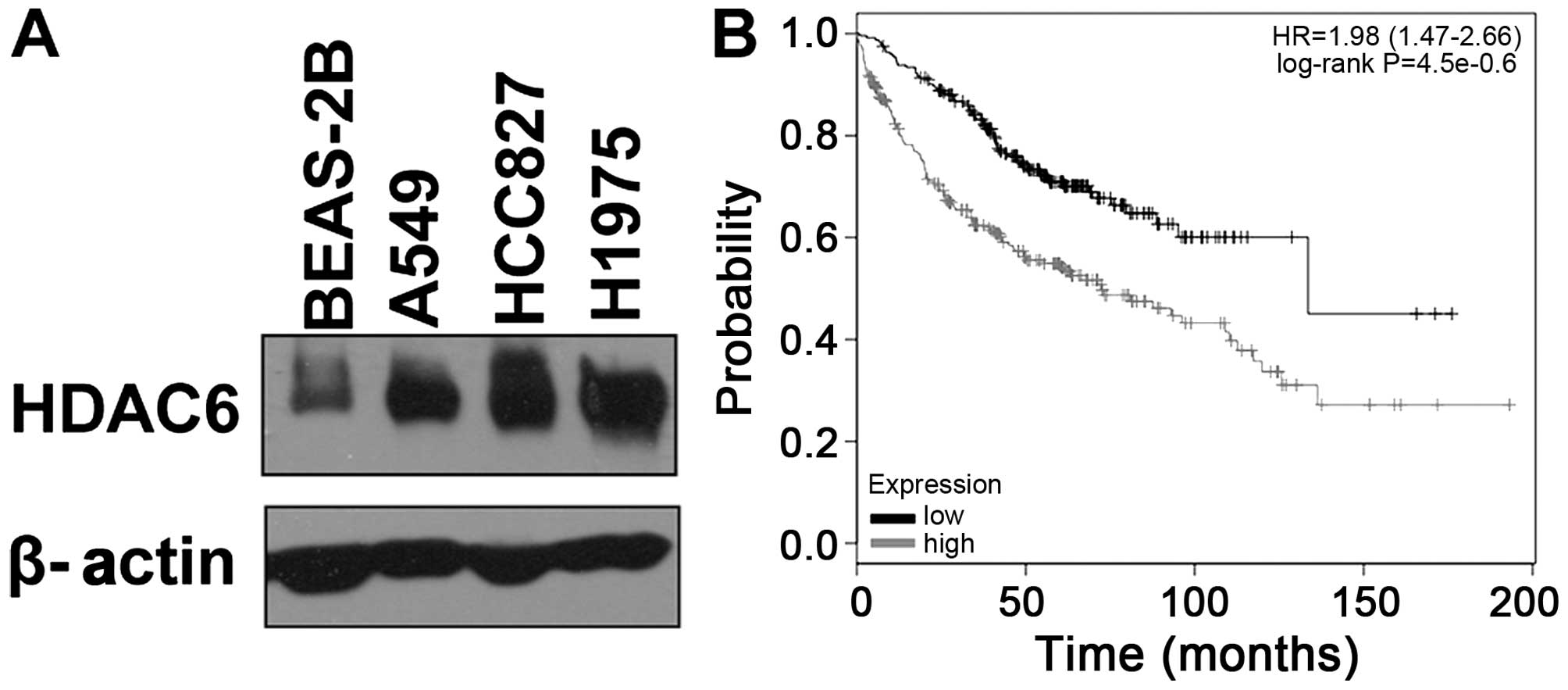
It has been reported that expression of HDAC6
protein is upregulated in several types of human cancers, but the
role of HDAC6 in lung adenocarcinoma tumorigenesis is still unknown
(19). To determine the expression
level of HDAC6 protein in lung adenocarcinoma, we first compared
the expression of HDAC6 between lung adenocarcinoma cell lines and
a normal human lung cell line. Three lung adenocarcinoma cell lines
(A549, HCC827 and H1975) and one normal human lung cell line
(BEAS-2B) were tested. As shown in Fig.
1A, HDAC6 was overexpressed in the lung adenocarcinoma cell
lines compared with the normal lung cell line. Moreover, following
the search of an online Kaplan-Meier plotter database, and analysis
of the prognostic value of biomarkers using transcriptomic data in
NSCLC (23), we found that the
expression of HDAC6 was negatively correlated with the prognosis of
patients with lung adenocarcinoma (P<0.01) (Fig. 1B).
HDAC6 promotes the proliferation of lung
adenocarcinoma cells in a deacetylase activity-dependent
manner
Since HDAC6 is overexpressed in lung adenocarcinoma,
we speculated that HDAC6 may regulate the proliferation of lung
adenocarcinoma cells. To assess the effects of HDAC6 on cell
proliferation and whether this is dependent on its deacetylase
activity, A549 and HCC827 cells were transfected with wild-type
HDAC6 (HDAC6 WT) and the catalytically inactive H216A/H611A mutant
HDAC6 (HDAC6 MT) overexpression plasmids, and cell proliferation
was measured by CCK-8 assay. We found a significant increase in the
cell proliferation rate on day 4 when HDAC6 was upregulated
(Fig. 2A and B). Moreover, the
HDAC6 MT-transfected group showed nearly the same proliferation
rate as the control group (Fig. 2A and
B). Conversely, knockdown of HDAC6 led to impaired cell
proliferation in both cell lines (Fig.
2C and D). These results indicate that HDAC6 promotes cell
proliferation of lung adenocarcinoma in a deacetylase
activity-dependent manner.
HDAC6 confers resistance to
gefitinib-mediated killing of lung adenocarcinoma cells
Dramatic clinical responses to the selective
EGFR-TKIs gefitinib and erlotinib have been observed in patients
with advanced lung adenocarcinoma, particularly in those with
tumors that harbor activating EGFR mutations (1). Gefitinib was the first small-molecule
EGFR-TKI that received FDA approval in 2003. However, almost all
tumors develop acquired resistance to these TKIs within 9–15 months
(24). EGFR-TKIs are used to treat
lung adenocarcinomas that carry activating EGFR mutations, whereas
they exert only modest antitumor activity in lung adenocarcinomas
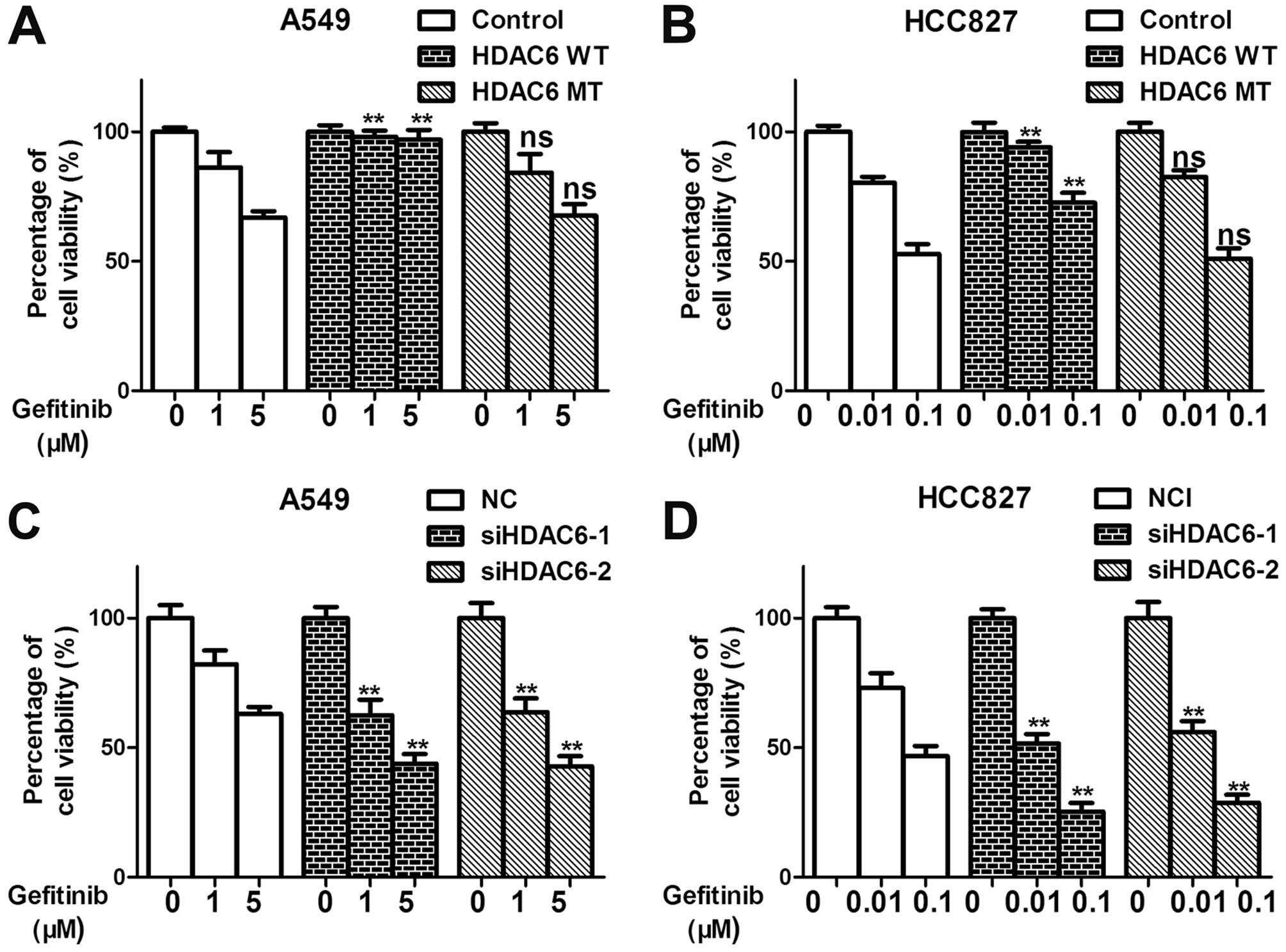
that express wild-type EGFR. In the present study, we showed that
the overexpression of HDAC6 remarkably impaired gefitinib
efficiency with respect to the inhibition of cell proliferation of
the lung adenocarcinoma cell lines A549 (EGFR WT) and HCC827 (EGFR
DelE746A750) in a deacet-ylase activity-dependent manner (Fig. 3A and B). Knockdown of HDAC6 also
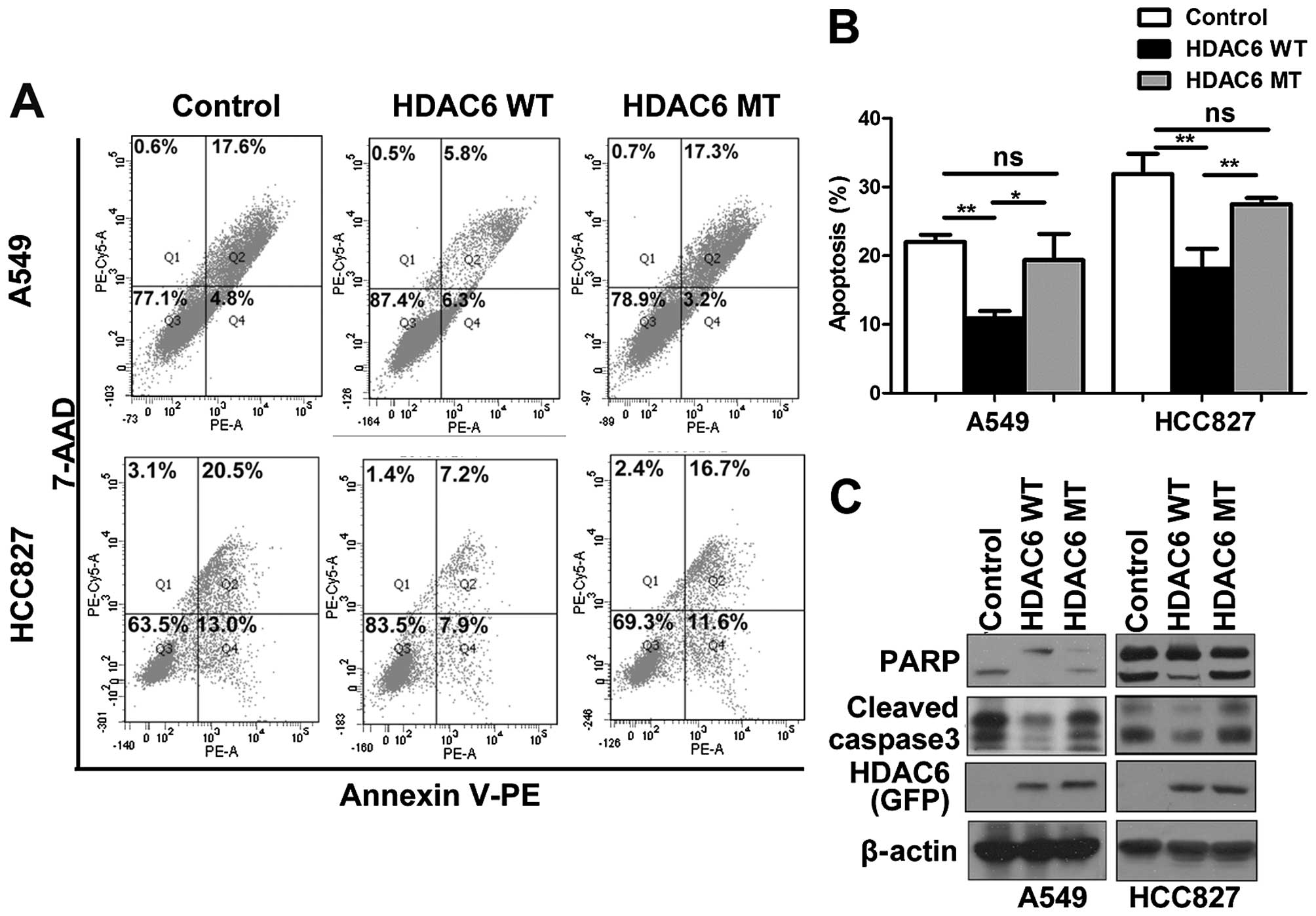
sensitized the A549 and HCC827 cells to geftinib (Fig. 3C and D). Furthermore, by Annexin V
staining and western blotting, the cleavage of PARP and caspase 3
was detected and showed that HDAC6 overexpression conferred
resistance to gefitinib-induced apoptosis, which was dependent on
the deacetylase activity of HDAC6 (Fig.
4A–C). Our results suggest that the overexpression of HDAC6 may
be an intrinsic mechanism that confers resistance to EGFR-TKIs.
Inhibition of HDAC6 deacetylase activity
by CAY10603, a selective HDAC6 inhibitor, inhibits the
proliferation of lung adenocarcinoma cells
The above data indicated that HDAC6 plays a relevant
role in the control of lung adenocarcinoma cell proliferation. To
further assess this possibility, we tested the effect of HDAC6
inhibition on lung adenocarcinoma cell lines using a potent and
selective inhibitor, CAY10603 (chemical structure shown in Fig. 5A). We assessed the growth inhibition
in response to CAY10603 treatment in two human lung adeno-carcinoma
cell lines: A549 and HCC827. A dose-dependent decrease in cell
proliferation was observed for both cell lines tested (Fig. 5B and C). The same conclusion was
drawn from the results of the clone formation assay. We noted a
significant decrease in clone numbers of the lung adenocarcinoma
cell lines after CAY10603 treatment (Fig. 5D).
Inhibition of HDAC6 by CAY10603 notably
induces apoptosis of lung adenocarcinoma cells
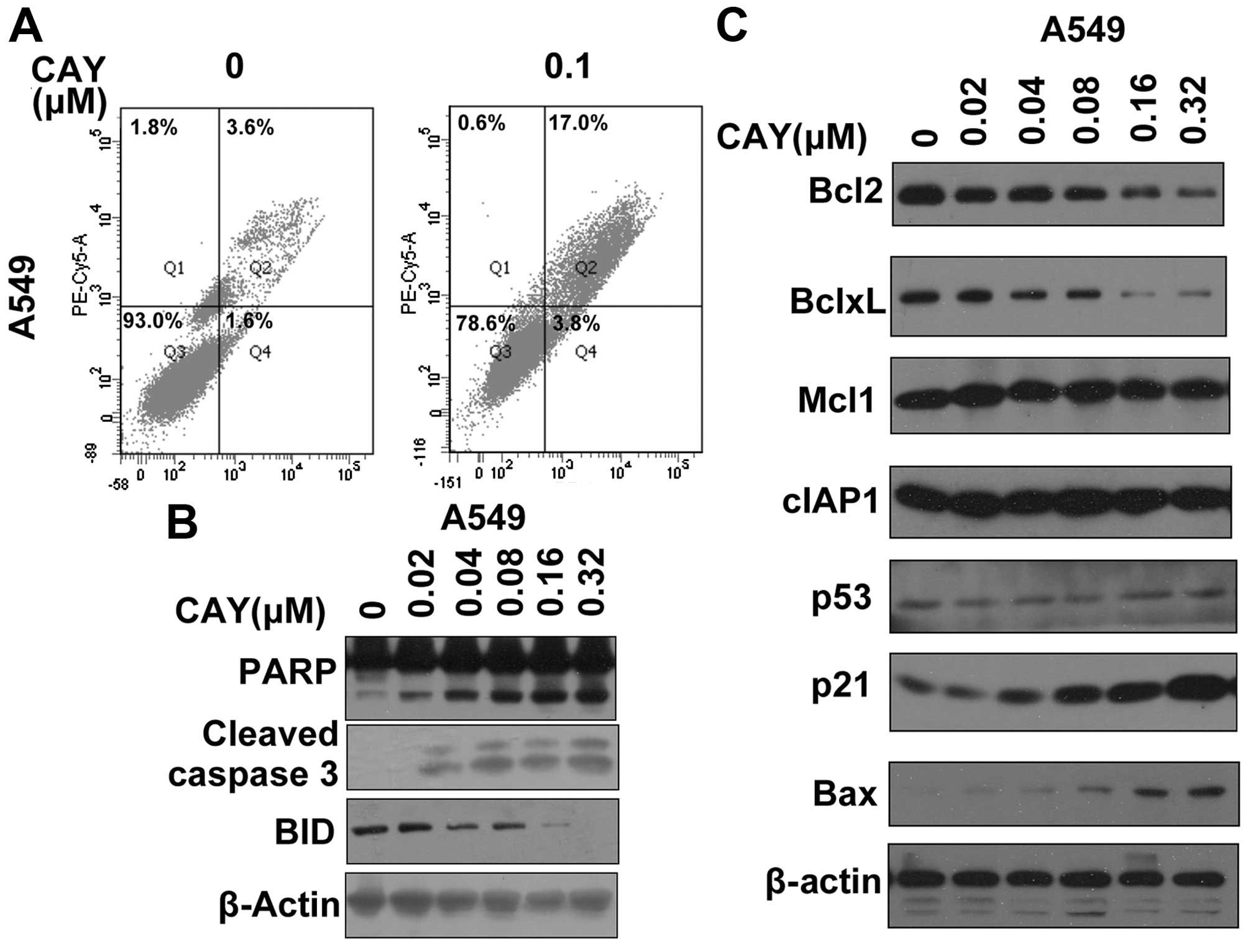
Given that CAY10603 inhibits the proliferation of
lung adenocarcinoma cell lines, we investigated its effect on
apoptosis. CAY10603 clearly induced apoptosis in the lung
adenocarcinoma cell line A549 (Fig.
6A). We determined the effects of CAY10603 on the activation of
caspases and Bid proteins as well as its effect on PARP cleavage,
which serves as a marker of apoptosis. Western blot analysis
demonstrated that PARP, Bid and caspase 3 were cleaved following
CAY10603 treatment in the A549 cells (Fig. 6B). Protein levels of the
apoptosis-related proteins were also detected (Fig. 6C). The inhibition of HDAC6 by
CAY10603 increased the levels of pro-apoptotic proteins such as BAX
and p21 (Fig. 6C). CAY10603 also
led to decreases in the levels of anti-apoptotic proteins including
Bcl2 and Bcl-xL (Fig. 6C).
Inhibition of HDAC6 synergizes with
gefitinib to induce apoptosis in lung adenocarcinoma cell lines,
partly through the destabilization of EGFR and inactivation of the
EGFR pathway
Since HDAC6 overexpression confers resistance to
gefitinib, we speculated that the inhibition of HDAC6 may
contribute to an increase in the efficiency of gefitinib with
respect to lung adenocarcinoma cells. To examine whether a
cooperative effect exists between CAY10603 and gefitinib in the
chemotherapeutic treatment of lung adenocarcinoma, we treated A549
cells with CAY10603 and gefitinib either alone or in combination.
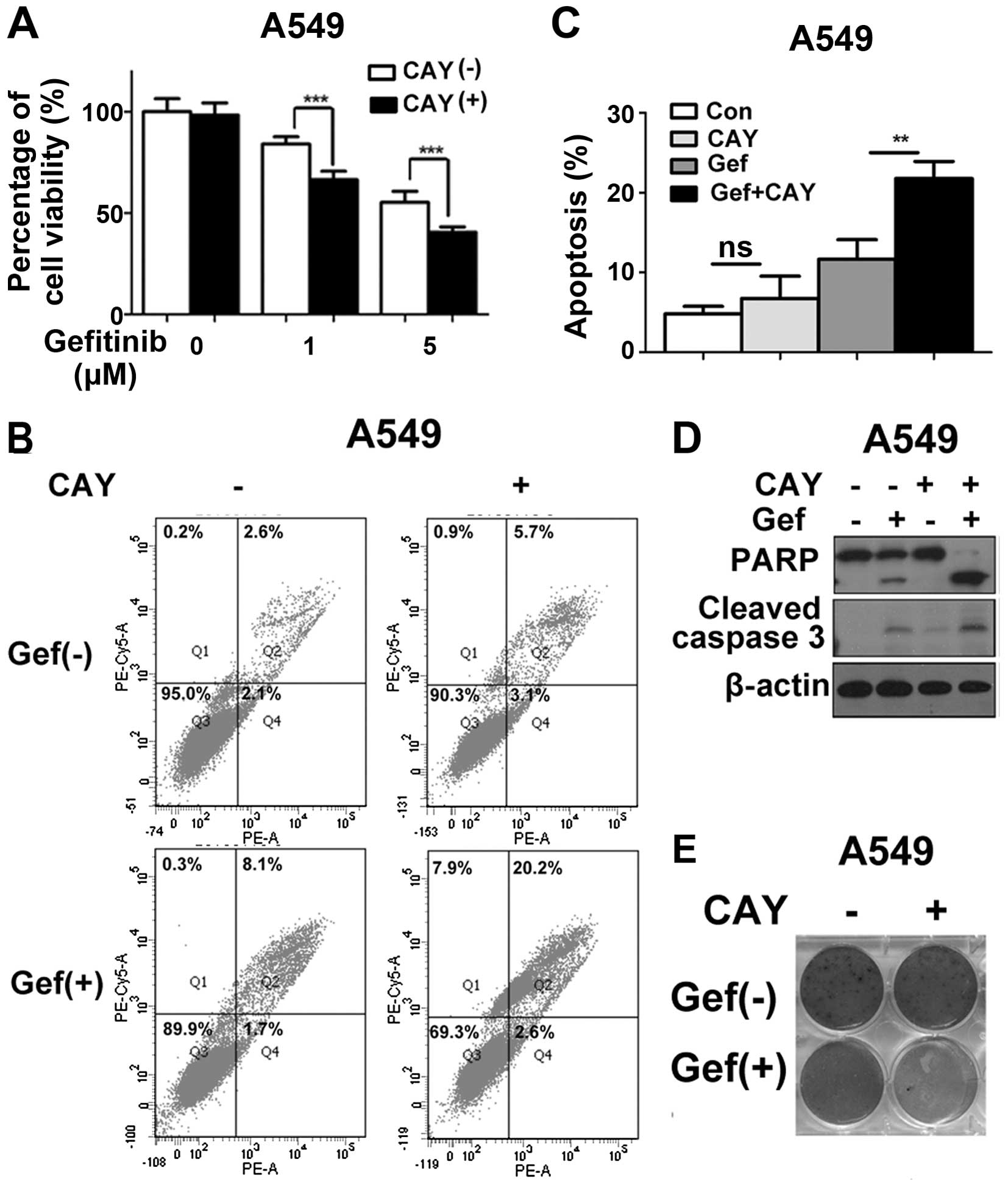
In agreement with our hypothesis, the co-treatment of CAY10603 and
gefitinib significantly reduced the cell viability of the
gefitinib-resistant cell line A549 (Fig. 7A). The rate of apoptosis was also
increased after the co-treatment of these two drugs, as evidenced
by increases in the number of Annexin V-positive cells (Fig. 7B and C). An increase in apoptosis by
this combination drug treatment was further evidenced by the
cleavage of PARP and caspase 3 (Fig.
7D). We also observed that the combination of CAY10603 and
gefitinib remarkably inhibited the clono-genic survival of the A549
cells (Fig. 7E). These data suggest
that the combination treatment of CAY10603 and gefitinib displayed
a significant synergistic therapeutic effect in lung
adenocarcinoma. Next, we aimed to ascertain the mechanism by which
CAY10603 contributes to the gefitinib-induced cell death of lung
adenocarcinoma cells. HDAC6 has been shown to be an important
regulator of EGFR endocytic trafficking and degradation (25,26).
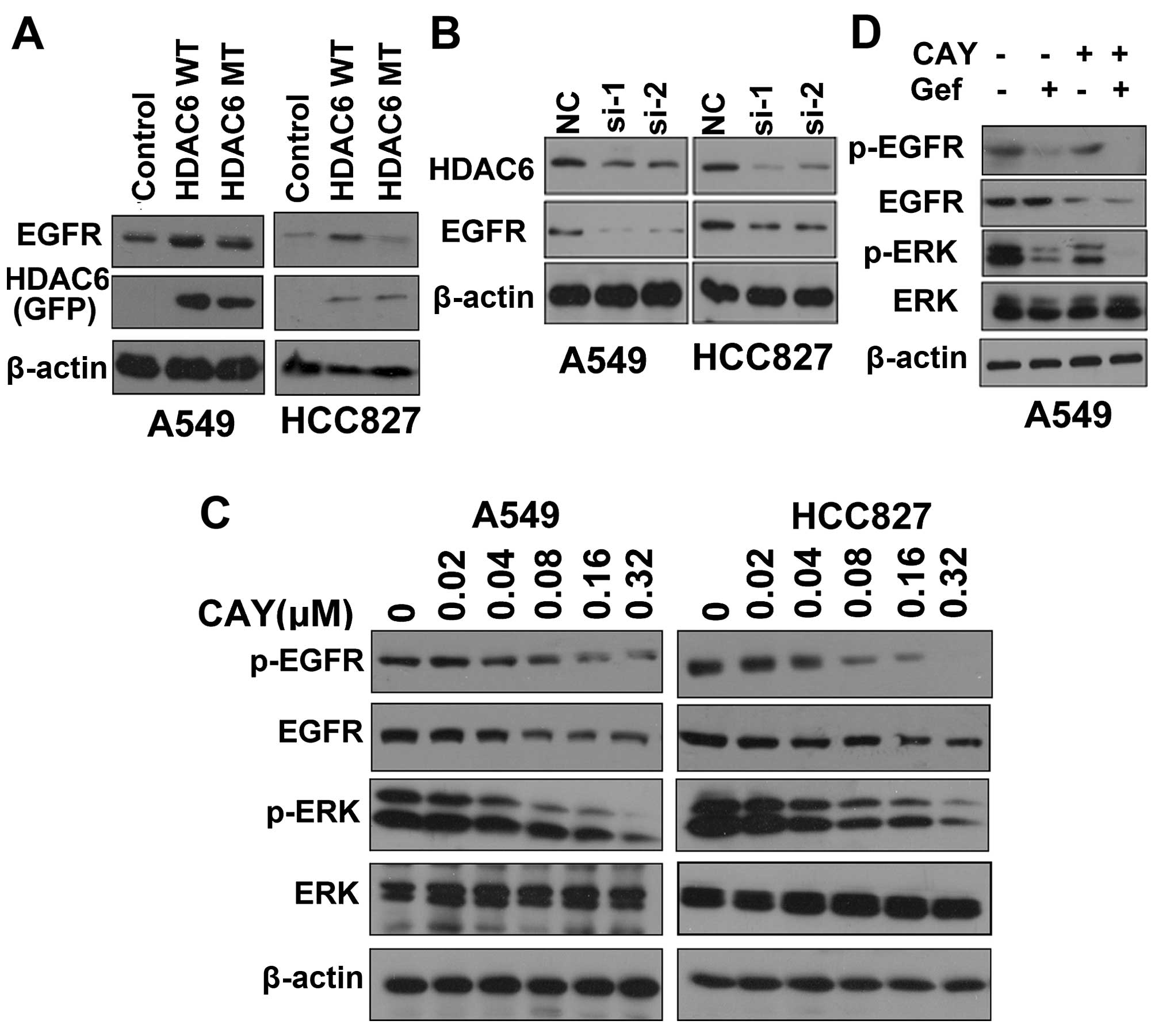
To test whether HDAC6 regulates EGFR stability in lung
adenocarcinoma cells, the cells were transfected with control or
HDAC6 overexpression plasmids for 48 h, and then the cells were
lysed and EGFR protein levels were detected by western blotting. We
found that overexpres-sion of HDAC6 in lung adenocarcinoma cell
lines stabilized EGFR in a deacetylase activity-dependent manner
(Fig. 8A). Conversely, knockdown of
HDAC6 led to destabilization of EGFR (Fig. 8B). Thus, the inhibition of HDAC6 may
also destabilize EGFR and inhibit activation of the EGFR pathway.
CAY10603 treatment destabilized EGFR and inhibited activation of
the EGFR pathway in the A549 and HCC827 cells. ERK, a well-known
EGFR target, was selected as a symbol of EGFR activation (Fig. 8C). We also found that the activation
of ERK was inhibited by the combination of CAY10603 and gefitinib
compared with the administration of either inhibitor alone
(Fig. 8D). Taken together, our
results showed that inhibition of HDAC6 synergized with gefitinib
to induce apoptosis in lung adenocarcinoma via destabilization of
EGFR and inactivation of the EGFR pathway.
Discussion
HDACs are enzymes that are involved in the
regulation of the acetylation of histone and non-histone proteins.
Protein acetylation has emerged as an important post-translational
modification that regulates multiple cellular functions, including
chromatin remodeling and transcriptional regulation, microtubule
dynamics, metabolism, autophagy and apoptosis (7,12).
HDACs are considered to be among the most promising targets for
cancer therapy, and first-generation HDAC inhibitors are currently
being tested in phase I/II clinical trials (9). SAHA and romidepsin have already been
approved by the US FDA for the treatment of cutaneous T cell
lymphoma. As a cytoplasmic HDAC, HDAC6 has been shown to
deacetylate a diverse set of substrates that are involved in
tumorigenesis, including Hsp90, α-tubulin and EGFR (16,25,26).
HDAC6 has also been implicated in the regulation of many
cancer-associated cellular events and signaling pathways, which
makes it an attractive target for cancer therapy (27). HDAC6 has been shown to contribute to
the tumorigenesis of several human cancers, such as
medul-loblastoma, cholangiocarcinoma and prostate cancer (27–29).
HDAC6 inhibitors are being tested both in preclinical settings and
clinical trials as an anticancer agent (ClinicalTrials.gov Identifier: NCT01323751,
NCT02091063, NCT02632071 and NCT02635061). However, the role of
HDAC6 in lung adeno-carcinoma has not been well characterized. In
the present study, we demonstrated that HDAC6 is overexpressed in
lung adenocarcinoma cell lines and promotes the oncogen-esis of
lung adenocarcinoma. HDAC6 also stabilized EGFR in lung
adenocarcinoma cells. The inhibition of HDAC6 by CAY10603, a potent
and selective HDAC6 inhibitor, induced apoptosis and impaired the
proliferation of lung adenocarcinoma cells. Moreover, the
inhibition of HDAC6 synergized with EGFR-TKIs to induce the
apoptosis of lung adenocarcinoma cells via the suppression of EGFR
pathway activation.
Although mounting evidence predicts a therapeutic
benefit for HDAC inhibitors, we still face significant challenges
in their clinical application. Broad spectrum HDAC inhibitors block
multiple HDAC isoforms, particularly HDAC1 and HDAC3 (12,13).
Although HDAC inhibitors have demonstrated significant antitumor
effects, the profound side-effects associated with their use cannot
be ignored. However, unlike the inhibition of other HDACs, the
inhibition of HDAC6 is not believed to be associated with severe
toxicity (30). Moreover, HDAC6
knockout in mice does not lead to embryonic lethality (31). Given that HDAC6 plays an important
role in lung adenocarcinoma, the inhibition of HDAC6 may induce
apoptosis in lung adenocarcinoma cells without the accompaniment of
as many side-effects as broad spectrum HDAC inhibitors. Further
studies that compare the antitumor efficiency and safety of HDAC6
inhibitors with broad spectrum HDAC inhibitors are needed,
particularly in animal models.
EGFR plays a critical role in the control of
cellular proliferation, differentiation and survival of lung
adenocarcinoma cells. Activating mutations in EGFR such as exon 19
deletions and the L858R substitution in exon 21 are important
markers of response to EGFR-TKI therapy in lung adenocar-cinoma
(24). Acquired resistance to
EGFR-TKIs is a major problem that continues to puzzle clinicians
since the majority of patients who initially respond to therapy
develop acquired resistance within 6–12 months. Several mechanisms
that may be responsible for this acquired resistance have been
identified, including a secondary T790M mutation in EGFR, c-Met
amplification and transformation to SCLC (5). In addition to these well characterized
mechanisms, further elucidation of novel mechanisms of acquired
drug resistance is essential to overcome the problem of EGFR-TKI
resistance. Furthermore, patients without EGFR mutations still
manifest acquired resistance to EGFR inhibitors. Therefore, it
remains urgent to explore additional mechanisms of acquired
resistance to EGFR inhibitors. As mentioned above, the
overexpression of HDAC6 stabilizes EGFR, while the inhibition of
HDAC6 destabilizes EGFR. Here, we showed that overexpression of
HDAC6 may partly account for the intrinsic resistance to EGFR-TKIs
due to the activation of the EGFR pathway.
The kinase-independent role of EGFR has attracted
increased attention in recent years. Zhang et al found that
the survival of cancer cells can be maintained by EGFR
independently of its kinase activity through the prevention of
autophagic cell death of cancer cells (32). Thus, EGFR can still promote the
survival of cancer cells when kinase activity is blocked by
EGFR-TKIs. Acceleration of the degradation of EGFR may solve this
problem, and the inhibition of HDAC6 can both destabilize EGFR and
inhibit activation of the EGFR pathway. This may partly explain the
synergistic effects of CAY10603 and gefitinib.
In summary, we report that HDAC6 is upregulated in
lung adenocarcinoma and that inhibition of HDAC6 impairs the
proliferation of lung adenocarcinoma cells. Moreover, the HDAC6
inhibitor CAY10603 synergizes with gefitinib to induce apoptosis in
lung adenocarcinoma cells, which occurs partly through the
destabilization of EGFR and inactivation of the EGFR pathway.
Therefore, the inhibition of HDAC6 may be a potential strategy for
treating lung adenocarcinoma and overcoming resistance to
EGFR-TKIs.
Abbreviations:
|
SCLC
|
small cell lung cancer
|
|
NSCLC
|
non-small cell lung cancer
|
|
HDACs
|
histone deacetylases
|
|
EGFR
|
epidermal growth factor receptor
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
Hsp90
|
heat shock protein 90
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
SIRT
|
silent mating type information
regulation 2 homolog
|
Acknowledgments
We acknowledge Professor Jun Zhou of the Nankai
University for kindly providing the HDAC6 overexpression plasmids.
The present study was supported by the Natural Sciences Foundation
of Hubei Province (no. 2013CFA006).
References
|
1
|
Pao W and Girard N: New driver mutations
in non-small-cell lung cancer. Lancet Oncol. 12:175–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ettinger DS, Wood DE, Akerley W, Bazhenova
LA, Borghaei H, Camidge DR, Cheney RT, Chirieac LR, D'Amico TA,
Demmy TL, et al National comprehensive cancer network: Non-Small
Cell Lung Cancer, Version 6.2015. J Natl Compr Canc Netw.
13:515–524. 2015.PubMed/NCBI
|
|
3
|
Pao W and Chmielecki J: Rational,
biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nat Rev Cancer. 10:760–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Popper HH, Ryska A, Tímár J and Olszewski
W: Molecular testing in lung cancer in the era of precision
medicine. Transl Lung Cancer Res. 3:291–300. 2014.
|
|
5
|
Politi K and Herbst RS: Lung cancer in the
era of precision medicine. Clin Cancer Res. 21:2213–2220. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reck M, Heigener DF, Mok T, Soria JC and
Rabe KF: Management of non-small-cell lung cancer: Recent
developments. Lancet. 382:709–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haberland M, Montgomery RL and Olson EN:
The many roles of histone deacetylases in development and
physiology: Implications for disease and therapy. Nat Rev Genet.
10:32–42. 2009. View
Article : Google Scholar
|
|
9
|
Falkenberg KJ and Johnstone RW: Histone
deacetylases and their inhibitors in cancer, neurological diseases
and immune disorders. Nat Rev Drug Discov. 13:673–691. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duvic M, Olsen EA, Breneman D, Pacheco TR,
Parker S, Vonderheid EC, Abuav R, Ricker JL, Rizvi S, Chen C, et
al: Evaluation of the long-term tolerability and clinical benefit
of vorinostat in patients with advanced cutaneous T-cell lymphoma.
Clin Lymphoma Myeloma. 9:412–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: Development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bruserud Ø, Stapnes C, Ersvaer E, Gjertsen
BT and Ryningen A: Histone deacetylase inhibitors in cancer
treatment: A review of the clinical toxicity and the modulation of
gene expression in cancer cell. Curr Pharm Biotechnol. 8:388–400.
2007. View Article : Google Scholar
|
|
13
|
Marsoni S, Damia G and Camboni G: A work
in progress: The clinical development of histone deacetylase
inhibitors. Epigenetics. 3:164–171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kaliszczak M, Trousil S, Åberg O, Perumal
M, Nguyen QD and Aboagye EO: A novel small molecule hydroxamate
preferentially inhibits HDAC6 activity and tumour growth. Br J
Cancer. 108:342–350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Namdar M, Perez G, Ngo L and Marks PA:
Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA
damage and sensitizes transformed cells to anticancer agents. Proc
Natl Acad Sci USA. 107:20003–20008. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krämer OH, Mahboobi S and Sellmer A:
Drugging the HDAC6-HSP90 interplay in malignanT cells. Trends
Pharmacol Sci. 35:501–509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Li N, Caron C, Matthias G, Hess
D, Khochbin S and Matthias P: HDAC-6 interacts with and
deacetylates tubulin and microtubules in vivo. EMBO J.
22:1168–1179. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park SJ, Kim JK, Bae HJ, Eun JW, Shen Q,
Kim HS, Shin WC, Yang HD, Lee EK, You JS, et al: HDAC6 sustains
growth stimulation by prolonging the activation of EGF receptor
through the inhibition of rabaptin-5-mediated early endosome fusion
in gastric cancer. Cancer Lett. 354:97–106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aldana-Masangkay GI and Sakamoto KM: The
role of HDAC6 in cancer. J Biomed Biotechnol. 2011:8758242011.
View Article : Google Scholar
|
|
20
|
Kozikowski AP, Tapadar S, Luchini DN, Kim
KH and Billadeau DD: Use of the nitrile oxide cycloaddition (NOC)
reaction for molecular probe generation: A new class of enzyme
selective histone deacetylase inhibitors (HDACIs) showing picomolar
activity at HDAC6. J Med Chem. 51:4370–4373. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang Y, Ran J, Liu M, Li D, Li Y, Shi X,
Meng D, Pan J, Ou G, Aneja R, et al: CYLD mediates ciliogenesis in
multiple organs by deubiquitinating Cep70 and inactivating HDAC6.
Cell Res. 24:1342–1353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ran J, Yang Y, Li D, Liu M and Zhou J:
Deacetylation of α-tubulin and cortactin is required for HDAC6 to
trigger ciliary disassembly. Sci Rep. 5:129172015. View Article : Google Scholar
|
|
23
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar
|
|
24
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deribe YL, Wild P, Chandrashaker A, Curak
J, Schmidt MH, Kalaidzidis Y, Milutinovic N, Kratchmarova I,
Buerkle L, Fetchko MJ, et al: Regulation of epidermal growth factor
receptor trafficking by lysine deacetylase HDAC6. Sci Signal.
2:ra842009.PubMed/NCBI
|
|
26
|
Gao YS, Hubbert CC and Yao TP: The
microtubule-associated histone deacetylase 6 (HDAC6) regulates
epidermal growth factor receptor (EGFR) endocytic trafficking and
degradation. J Biol Chem. 285:11219–11226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gradilone SA, Radtke BN, Bogert PS, Huang
BQ, Gajdos GB and LaRusso NF: HDAC6 inhibition restores ciliary
expression and decreases tumor growth. Cancer Res. 73:2259–2270.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dhanyamraju PK, Holz PS, Finkernagel F,
Fendrich V and Lauth M: Histone deacetylase 6 represents a novel
drug target in the oncogenic Hedgehog signaling pathway. Mol Cancer
Ther. 14:727–739. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson
JB and Wang Z: HDAC6 regulates androgen receptor hypersensitivity
and nuclear localization via modulating Hsp90 acetylation in
castration-resistant prostate cancer. Mol Endocrinol. 23:1963–1972.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Haggarty SJ, Koeller KM, Wong JC,
Grozinger CM and Schreiber SL: Domain-selective small-molecule
inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin
deacetylation. Proc Natl Acad Sci USA. 100:4389–4394. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kaluza D, Kroll J, Gesierich S, Yao TP,
Boon RA, Hergenreider E, Tjwa M, Rössig L, Seto E, Augustin HG, et
al: Class IIb HDAC6 regulates endothelial cell migration and
angiogenesis by deacety-lation of cortactin. EMBO J. 30:4142–4156.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH,
Fidler IJ and Hung MC: Survival of cancer cells is maintained by
EGFR independent of its kinase activity. Cancer Cell. 13:385–393.
2008. View Article : Google Scholar : PubMed/NCBI
|