Introduction
Colorectal cancer (CRC) is presently the third most
frequently diagnosed cancer and the fourth leading cause of
cancer-related deaths worldwide (1). The incidence of CRC is now rapidly
increasing in previously known low-risk areas, such as Asia, Latin
America and Africa (2). Despite
considerable progress in clarifying the critical factors involved
in the prevention of different types of CRCs, this mortality is
primarily attributed to the high incidence of metastasis due to its
acquired resistance to anticancer drugs (3). Therefore, the development of novel
therapeutic strategies with increased selectivity and reduced
toxicity for CRC treatment is required.
One of the hallmarks of cancer is the evasion of
tumor cells by apoptosis, a highly regulated process of programmed
cell death. The disruption of this process is a major contributing
factor in the pathology of cancer (4). It has been well established that the
apoptotic process triggers extrinsic [death receptor (DR)-mediated]
or intrinsic (mitochondria-dependent) caspase-dependent pathways
(5,6). In addition, the integrity of the
mitochondrial membrane is regulated by the interactions of Bcl-2
family proteins. Reactive oxygen species (ROS) derived from
mitochondrial oxidative metabolism are also an important mediator
of apoptosis induction (7). Among
several cellular signaling pathways, the
phosphatidylinositol-3-kinase (PI3K)/Akt signal transduction
pathway integrates the survival signals provided by extracellular
and intracellular stimuli to promote cell growth and to inhibit
cell death, which is mediated by intracellular ROS generation
(7,8). Moreover, the PI3K/Akt pathway is
highly activated in most human cancers, including CRC (9,10).
Current research is highly interested in exploiting this pathway
for the discovery of potential cancer treatment. Therefore, agents
that target this apoptosis pathway without affecting normal cells
play crucial roles as potential drug targets in cancer
treatment.
For thousands of years, herbal medicines have been
used with apparent safety and efficacy to alleviate and treat
various diseases, including cancer, in numerous countries (11,12).
In particular, typical traditional herbal medicinal prescriptions
consist of several components that are combined to minimize
side-effects, maximize therapeutic efficacy by facilitating
synergistic actions between the drugs while preventing potential
adverse effects, and improve the quality of life of patients
(13). Among such components,
Hwang-Heuk-San (HHS) is an aqueous polyherbal formulation that
consists of crude ingredients extracted from four medicinal herbs
(Table I), which are described in
the Donguibogam, an ancient Korean medical book (14). HHS has been used for hundreds of
years in Korea for the clinical treatment of intestinal abscesses
and carbuncles on the abdominal wall. However, despite its valuable
clinical effects on patients, few attempts have been made to
investigate the molecular mechanism and pharmacological action of
HHS in gastrointestinal malignancy.
 | Table IHerbal components and amount of HHS
decoction. |
Table I
Herbal components and amount of HHS
decoction.
| Herbal medicine
(pharmacognostic nomenclature) | Raw material amount
(g/%) |
|---|
| Rheum
palmatum L. (Rhei Radix et Rhizoma) | 36.0 (47.4) |
| Psoralea
corylifolia L. (Psoraleae Fructus) | 16.0 (21.0) |
| Pharbitis
nil Chois. (Pharbitidis Semen) | 16.0 (21.0) |
| Arctium
lappa L. (Arctii Fructus) | 8.0 (10.5) |
| Total amounts | 76.0 (100.0) |
The aim of the present study was to discover novel
biologically active substances in traditional medicinal resources
for the prevention and treatment of cancers. We aimed to determine
whether HHS could inhibit the growth of and trigger apoptosis in
HCT-116 CRC cells. The results showed that HHS triggered
caspase-dependent apoptosis through the activation of both
intrinsic and extrinsic pathways. The present study is the first to
investigate the potential roles and underlying mechanisms
connecting ROS generation and the PI3K/Akt pathway in mediating
HHS-induced apoptosis in HCT-116 cells.
Materials and methods
Reagents and antibodies
RPMI-1640 medium, fetal bovine serum (FBS),
penicillin and streptomycin were purchased from WelGENE Inc.
(Daegu, Korea);
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT), 4′,6-diamidino-2-phenylindole (DAPI), phenol-chloroform,
isoamyl alcohol, RNase A, dithiothreitol (DTT), propidium iodide
(PI), paraformaldehyde,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolyl
carbocyanine iodide (JC-1) and N-acetylcysteine (NAC), a ROS
scavenger were purchased from Sigma-Aldrich Chemicals Co. (St.
Louis, MO, USA). z-Val-Ala-Asp-fluoromethylketone (z-VAD-fmk), a
pan-caspase inhibitor and LY294002, a selective inhibitor of PI3K,
were obtained from Calbiochem (San Diego, CA, USA) and Cell
Signaling Technology, Inc. (Danvers, MA, USA), respectively.
2′,7′-Dichlorodihydrofluorescein diacetate (DCF-DA) and the
enhanced chemiluminescence (ECL) kit were purchased from Molecular
Probes (Leiden, The Netherlands) and Amersham Pharmacia Biotech
(Arlington Heights, IL, USA), respectively. Primary antibodies
(Table II) were purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), Cell
Signaling Technology, Inc., Bioworld Technology, Inc. (St. Louis
Park, MN, USA) and Abcam (Cambridge, UK). Peroxidase-labeled donkey
anti-rabbit and sheep anti-mouse immunoglobulin were purchased from
Amersham Pharmacia Biotech. All other chemicals not mentioned in
the present study were purchased from Sigma-Aldrich Chemical
Co.
| Table IIAntibodies used in the present
study. |
Table II
Antibodies used in the present
study.
| Antibody | Dilution | Product no. | Species of origin
and supplier |
|---|
| Actin | 1:50000 | BS6007M | Mouse polyclonal,
Bioworld Technology, Inc. |
| Caspase-3 | 1:1000 | sc-7272 | Mouse polyclonal,
Santa Cruz Biotechnology, Inc. |
| Caspase-8 | 1:1000 | sc-7890 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| Caspase-9 | 1:1000 | sc-7885 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| PARP | 1:1000 | sc-7150 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| XIAP | 1:1000 | sc-11426 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| cIAP-1 | 1:1000 | sc-7943 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| cIAP-2 | 1:1000 | sc-7944 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| survivin | 1:1000 | sc-17779 | Mouse polyclonal,
Santa Cruz Biotechnology, Inc. |
| TRAIL | 1:1000 | sc-7877 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| DR4 | 1:1000 | sc-7863 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| DR5 | 1:1000 | sc-65314 | Mouse polyclonal,
Santa Cruz Biotechnology, Inc. |
| Fas | 1:1000 | sc-715 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| FasL | 1:1000 | sc-957 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| Bcl-2 | 1:1000 | sc-783 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| Bax | 1:1000 | sc-493 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| Bid | 1:1000 | sc-11423 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| Cytochrome c | 1:1000 | sc-7159 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| COX IV | 1:500 | 20E8C12 | Mouse monoclonal,
Abcam |
| PI3K | 1:1000 | 4257P | Rabbit polyclonal,
Cell Signaling Technology, Inc. |
| p-PI3K | 1:1000 | 4228P | Rabbit polyclonal,
Cell Signaling Technology, Inc. |
| Akt | 1:1000 | sc-8312 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
| p-Akt | 1:1000 | sc-101629 | Rabbit polyclonal,
Santa Cruz Biotechnology, Inc. |
Preparation of the HHS
The four medicinal herbs forming HHS were obtained
from Dongeui Oriental Hospital, Dongeui University College of
Korean Medicine (Busan, Korea). The origin of the medicinal herbs
was taxonomically confirmed by Professor Su Hyun Hong at the
Dongeui University College of Korean Medicine (Busan). Each of the
four herbs in HHS was cut into small pieces, which were mixed
together to obtain a total amount of 76 g in the ratios shown in
Table I. The mixture was boiled in
distilled water (76 g/500 ml) for 3 h. The extract solution was
filtered using a 0.45 μM filter to remove insoluble
materials, and the blended supernatants were lyophilized and and
then crushed into a thin powder (yield, 21% w/w, dried
extract/crude herb). The dried extract (HHS) was dissolved in a 100
mg/ml concentration with distilled water, and the stock solution
was then diluted with medium to the desired concentration prior to
use.
Cell culture and cell viability
assay
CRC HCT116 cells were purchased from the American
Type Culture Collection (ATCC; Manassas, MD, USA) and maintained at
37°C in an incubator with a humidified atmosphere of 5%
CO2 in air. The cells were cultured in RPMI-1640 medium
containing 10% FBS, 100 U/ml of penicillin and 100 mg/ml of
streptomycin in the presence or absence of HHS. Cell viability was
determined using the MTT assay, which is based on the reduction of
a tetrazolium salt by mitochondrial dehydrogenases in viable cells.
In brief, HCT116 cells were seeded into a 96-well plate at a
density of 1×105 cells/well and treated with HHS or
reagents. After incubating for 48 h, the medium was discarded, and
the cells were washed with phosphate-buffered saline (PBS),
followed by incubation with MTT (0.5 mg/ml) at 37°C. After
incubating for a further 3 h, the supernatants were aspirated, and
the formazan crystals in each well were dissolved in dimethyl
sulfoxide (DMSO). Absorbance at 540 nm was read on an enzyme-linked
immunosorbent assay (ELISA) reader (Molecular Devices, Sunnyvale,
CA, USA).
Nuclear morphology analysis of
apoptosis
For the assessment of apoptosis, the cells were
washed with PBS, and the cell suspension was cytospun onto clean
fat-free glass slides using a cytocentrifuge. The cells were fixed
with 3.7% para-formaldehyde in PBS and then stained with 2.5
μg/ml of DAPI solution for 10 min at room temperature. The
cells were washed twice with PBS, and the images were then captured
using a fluorescence microscope (Carl Zeiss, Jena, Germany).
DNA flow cytometric detection of
apoptosis
To analyze apoptotic sub-G1 hypodiploid cells, the
cells were harvested, washed twice with ice-cold PBS, and fixed
with 75% ethanol at 4°C for 30 min. The cells were then incubated
with 1 U/ml of RNase A (DNase-free) and 10 μg/ml of PI
overnight at room temperature in the dark. A flow cytometric
analysis was performed using a flow cytometer (FACSCalibur;
Becton-Dickinson, San Jose, CA, USA). The sub-G1 hypodiploid cells
were assessed based on histograms generated by the CellQuest and
ModFit computer programs (Becton-Dickinson). The level of apoptotic
cells containing sub-G1 DNA content was determined as a percentage
of the total number of cells. The cells were also stained with 5
μl of Annexin V-fluorescein isothiocyanate (FITC) (R&D
Systems, Minneapolis, MN, USA) and 5 μl of PI. Following
incubation for 15 min at room temperature in the dark, the degree
of apoptosis was quantified as a percentage of the Annexin
V-positive and PI-negative (Annexin V+/PI−
cells) cells using a flow cytometer (15).
Protein extraction and western blot
analysis
The cells were harvested and lysed with lysis buffer
[40 mM Tris (pH 8.0), 120 mM NaCl, 0.5% Nonidet P-40, 0.1 mM sodium
orthovanadate, 2 μg/ml aprotinin, 2 μg/ml leupeptin
and 100 μg/ml phenymethylsulfonyl fluoride] for 30 min. In a
parallel experiment, the mitochondrial and cytosolic fractions were
isolated using a mitochondrial fractionation kit (Active Motif,
Carlsbad, CA, USA) according to the manufacturer's instructions.
The protein concentration was measured using a Bio-Rad protein
assay (Bio-Rad, Hercules, CA, USA). For the western blot analysis,
equal amounts of protein (30–50 μg/lane) were fractionated
in sodium dodecyl sulphate (SDS)-polyacrylamide gels prior to
transfer to the nitrocellulose membranes (Schleicher & Schuell,
Keene, NH, USA). The membranes were blocked with Tris-buffered
saline (10 mM of Tris-Cl, pH 7.4) containing 0.5% Tween-20 and 5%
non-fat dry milk for 1 h at room temperature and then probed with
the desired antibodies for 1 h. The membranes were then washed with
PBS and incubated with the secondary antibody conjugated to
horseradish peroxidase for 1 h at room temperature.
Immunoreactivity was detected using the ECL western blotting
detection system according to the manufacturer's instructions.
Measurement of mitochondrial membrane
potential (MMP, Δψm)
The MMP values were determined using the lipophilic
cationic probe JC-1, a radiometric dual-emission fluorescent dye
that is absorbed and concentrated by respiring mitochondria and can
reflect MMP changes in cells. Briefly, the cells were fixed and
permeabilized with 0.2% Triton X-100 in PBS for 10 min at room
temperature and then incubated with 10 μM of JC-1 for 30 min
at 37°C in the dark. Subsequently, the cells were washed with PBS
to remove unbound dye, and the amount of JC-1 retained by 10,000
cells/sample was measured at 488 and 575 nm using a flow cytometer
(16).
Determination of caspase activity
The enzymatic activity of the caspases induced by
HHS was assayed using colorimetric assay kits (R&D Systems).
Briefly, the cells were lysed in the supplied lysis buffer
according to the manufacturer's instructions. The supernatants were
collected, and equal amounts of protein were incubated with the
supplied reaction buffer containing colorimetric tetrapeptides,
Asp-Glu-Val-Asp (DEAD)-p-nitroaniline (pNA) for caspase-3,
Ile-Glu-Thr-Asp (IETD)-pNA for caspase-8 and Leu-Glu-His-Asp
(LEHD)-pNA for caspase-9. The caspase activity was determined by
measuring changes in absorbance at 405 nm using the ELISA
reader.
Measurement of intracellular ROS
The fluorescent probe DCF-DA method was used to
measure the level of intracellular ROS in the cells as previously
described (17). Twenty-four hours
after plating, the cells were treated with 1 mM NAC for 1 h; HHS
was then added, and the cells were incubated for another 48 h.
After the media were changed, 10 μM DCF-DA was added to each
well, and the cells were incubated for an additional 30 min at
37°C. After washing with PBS, the cells were trypsinized and
resuspended in PBS. The fluorescence was measured at specific time
intervals using a flow cytometer.
Statistical analysis
The data derived from at least three independent
experiments are expressed as mean ± standard deviation (SD).
Statistical comparisons between different groups were performed
using a one-way ANOVA, which was followed by Student's t-tests
after each treated group was compared to the negative control.
Values of p<0.05 were considered to indicate a statistically
significant result.
Results
HHS suppresses cell viability and induces
apoptosis of the HCT116 cells
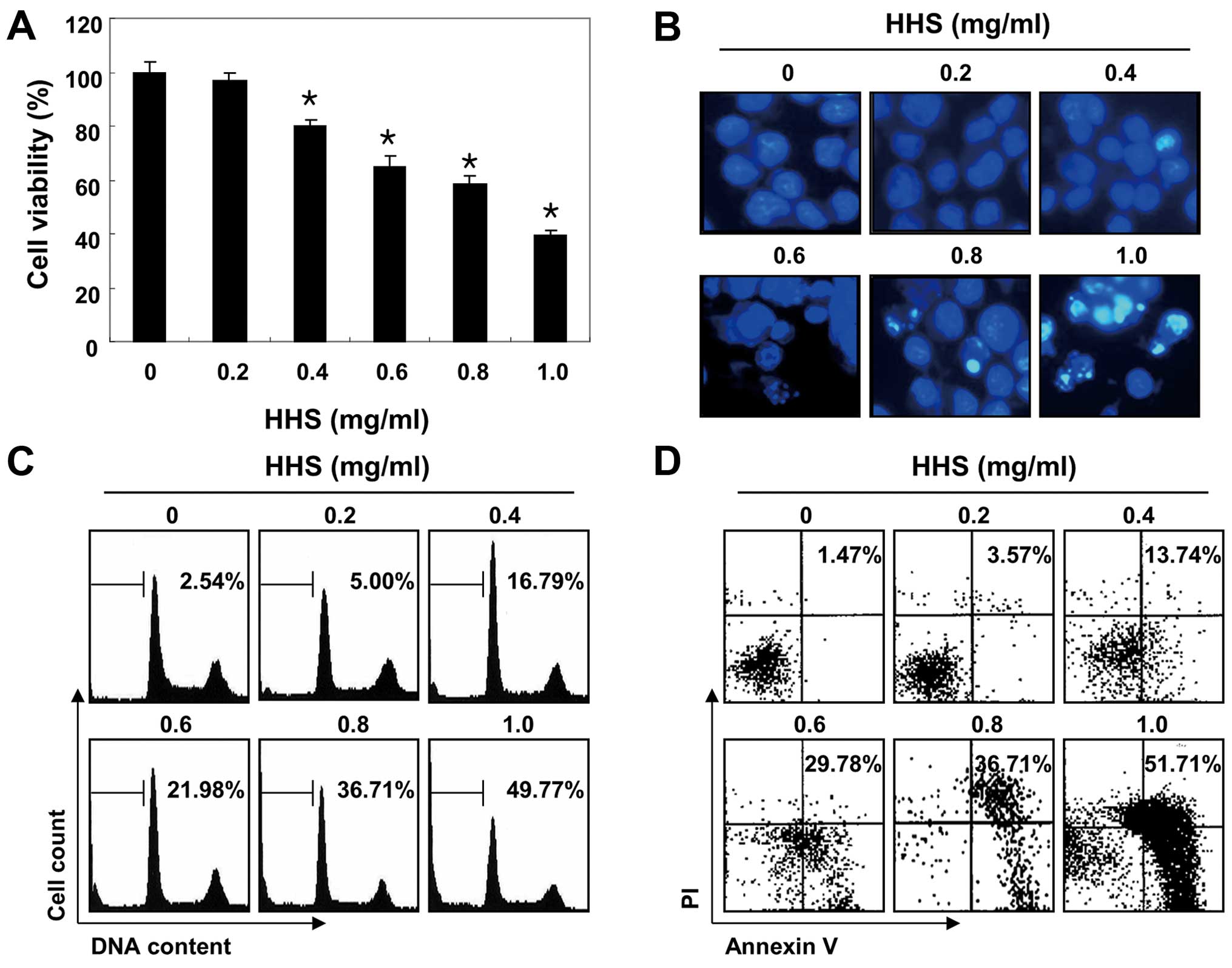
To evaluate the effect of HHS on cell viability, the
cells were treated with various concentrations of HHS for 48 h, and
an MTT assay was performed. As shown in Fig. 1A, the results indicated that HHS
exhibited cytotoxic effects in a concentration-dependent manner. In
addition, DAPI staining revealed that the apparent morphological
changes, such as nuclear condensation and fragmentation, were
concentration-dependently increased in the cells treated with HHS,
indicating an increasing number of apoptotic cells, whereas these
features were not observed in the control cells (Fig. 1B). To measure apoptotic cell death
upon HHS treatment, we stained the cells with PI, and the cell
cycle profiles were analyzed using a flow cytometer. The results
showed that HHS increased the percentage of cells in the apoptotic
hypodiploid sub-G1 phase in a dose-dependent manner (Fig. 1C). Annexin V-FITC/PI double staining
was also performed to assess the rate of apoptotic cell death, and
a significant degree of apoptosis was detected in the HHS-treated
cells (Fig. 1D).
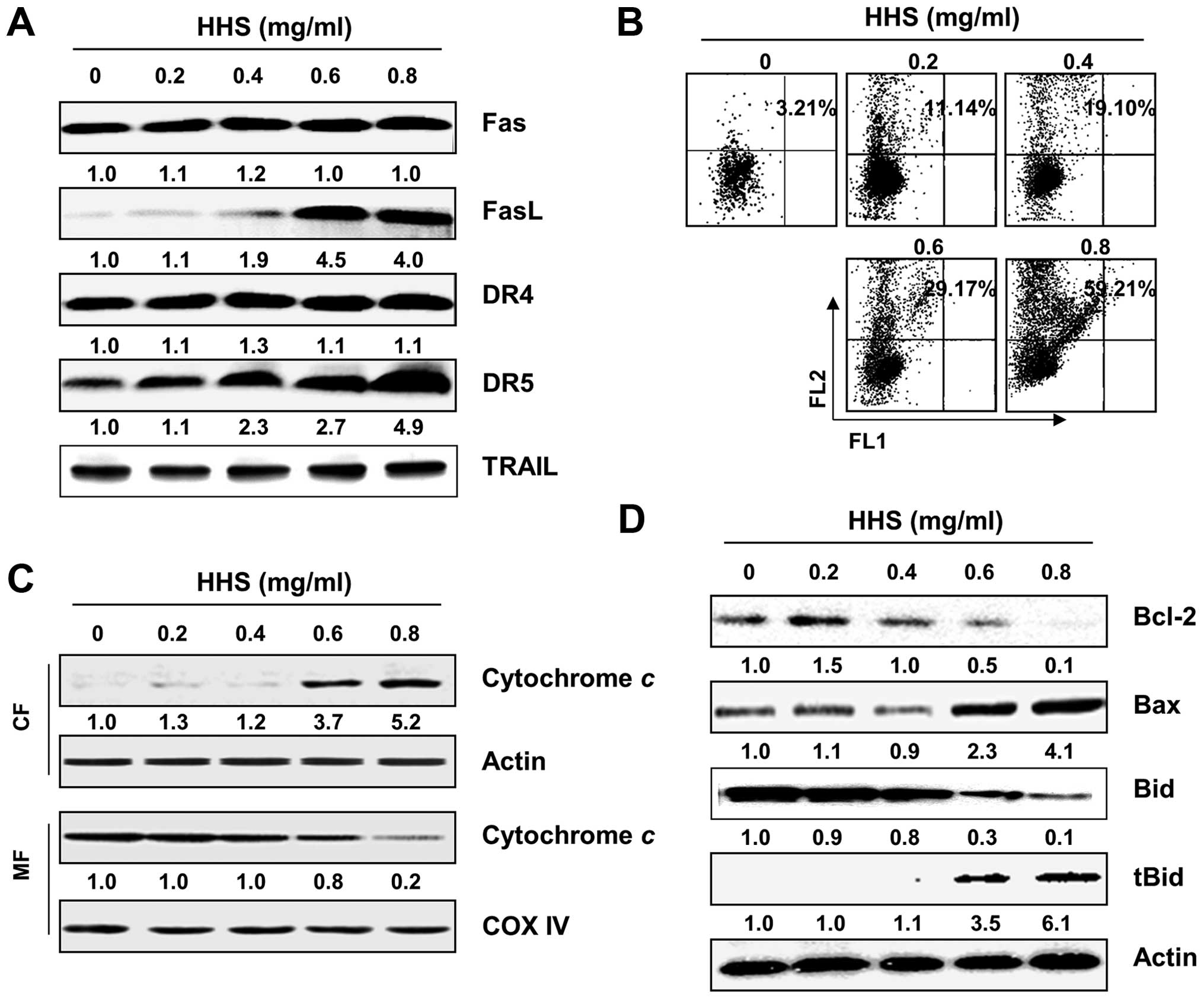
HHS induces DR-mediated apoptosis in the
HCT116 cells
To elucidate the mechanism of HHS-induced apoptosis
in the HCT116 cells, we determined whether HHS affects the
expression of DR-related proteins. As shown in Fig. 2A, the treatment of HCT116 cells with
HHS increased the expression of the Fas ligand (FasL), DR4 and DR5
in a concentration-dependent manner, whereas the levels of Fas and
tumor necrosis factor- related apoptosis-inducing ligand (TRAIL)
were relatively unchanged in response to the HHS treatment. The
western blot analysis also revealed that the exposure of HCT116
cells to HHS caused the progressive accumulation of truncated Bid
(tBid), a BH3 interacting domain death agonist, presumably
resulting from truncation by the activated caspase-8, in the
HHS-treated cells (Fig. 2D).
HHS induces mitochondria-mediated
apoptosis in HCT116 cells
To confirm whether HHS induced intrinsic
mitochondrial-mediated apoptosis, we determined the effect of HHS
on MMP, a hallmark of intrinsic apoptosis, by staining with JC-1
dye. The flow cytometric results demonstrated that HHS induced a
significant reduction in the MMP levels with increased
concentrations of HHS (Fig. 2B),
suggesting the depolarization of the mitochondria by HHS. Since the
loss of MMP promotes the release of pro-apoptotic proteins, such as
cytochrome c into the cytosol (18,19),
we determined the levels of cytochrome c in the cytosolic
and mitochondrial fractions of HCT116 cells that were treated with
different concentrations of HHS. As shown in Fig. 2C, HHS promoted a
concentration-dependent increase in the release of cytochrome
c from the mitochondria into the cytosol. Since the Bcl-2
family proteins play a crucial role in mitochondrial homeostasis
(20,21), the effect of HHS on the levels of
Bcl-2 family proteins was also monitored. Our results showed that
the incubation of the HCT116 cells with HHS caused a marked
reduction in the level of the anti-apoptotic Bcl-2 protein and
increased the level of the pro-apoptotic Bax protein (Fig. 2D).
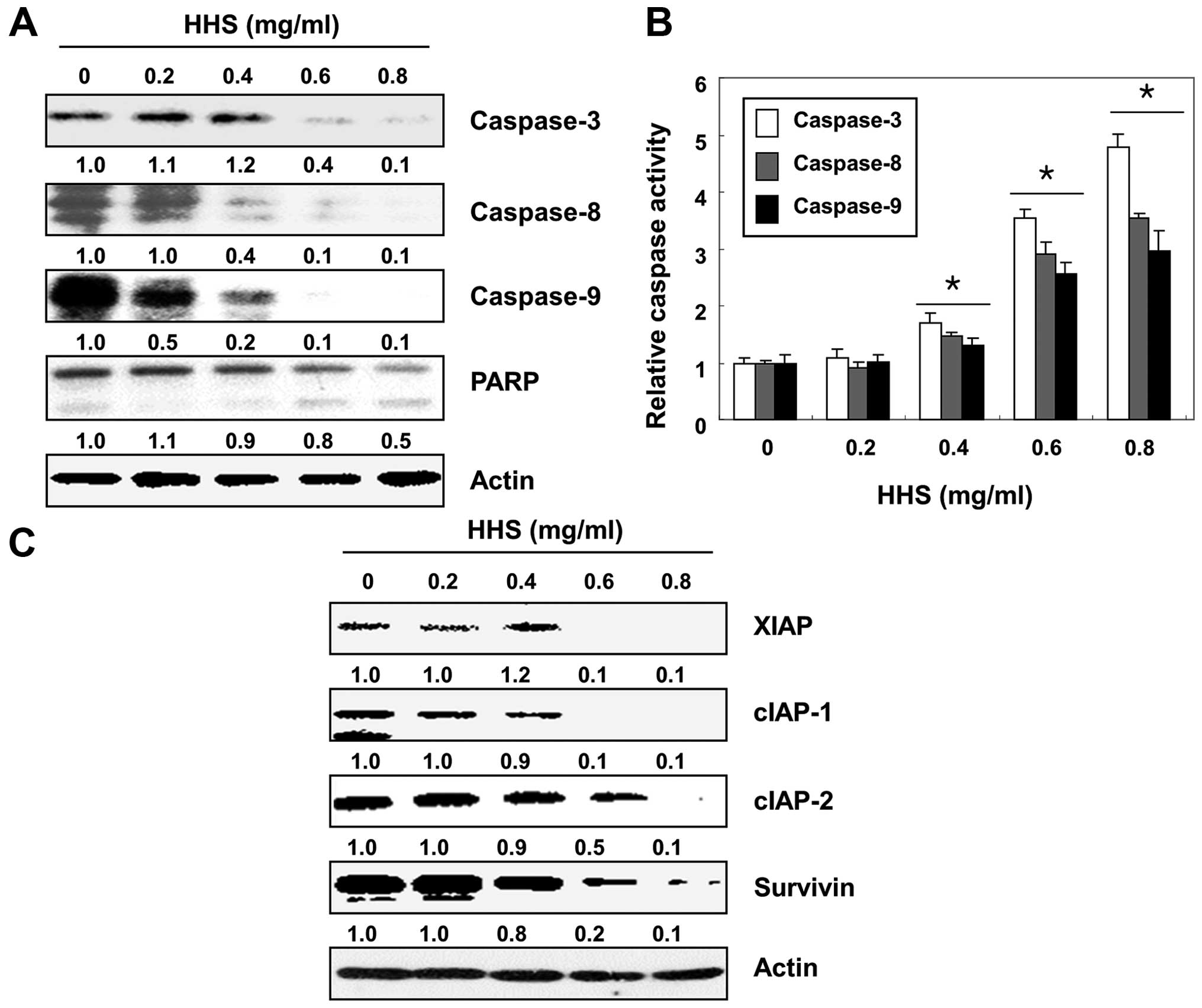
HHS-induced apoptosis is
caspase-dependent in the HCT116 cells
We then examined the expression and activity of
caspases in the HHS-treated HCT116 cells to determine whether
HHS-induced apoptosis was associated with the activation of the
caspases. The results showed that HHS treatment downregulated the
levels of pro-caspase-8 and -9, which are the initiator caspases of
the extrinsic and intrinsic pathway, respectively, in a
concentration-dependent manner (Fig.
3A). The subsequent immunoblotting analysis showed that the
levels of pro-caspase-3, an effector caspase, were also
downregulated in the HHS-treated cells. Correspondingly, the HHS
treatment led to the progressive proteolytic cleavage of
poly-(ADP-ribose) polymerase (PARP, 113-kDa), a known caspase-3
substrate (22), which yielded an
85-kDa cleaved fragment. For the further quantification of the
proteolytic activation of caspases, the lysates equalized by the
protein from the cells treated without or with HHS were assayed for
their enzymatic activities. As shown in Fig. 3B, the HHS treatment resulted in the
significantly increased activity of caspase-8, -9 and -3. In
addition, we examined the expression of inhibitor of the apoptosis
protein (IAP) family proteins, which bind to caspases and lead to
their inactivation (23,24). As shown in Fig. 3C, HHS treatment reduced the
expression of the IAP family proteins, such as XIAP, cIAP-1, cIAP-2
and survivin, indicating that the HHS-induced activation of
caspases was associated with the reduction in the IAP family
proteins.
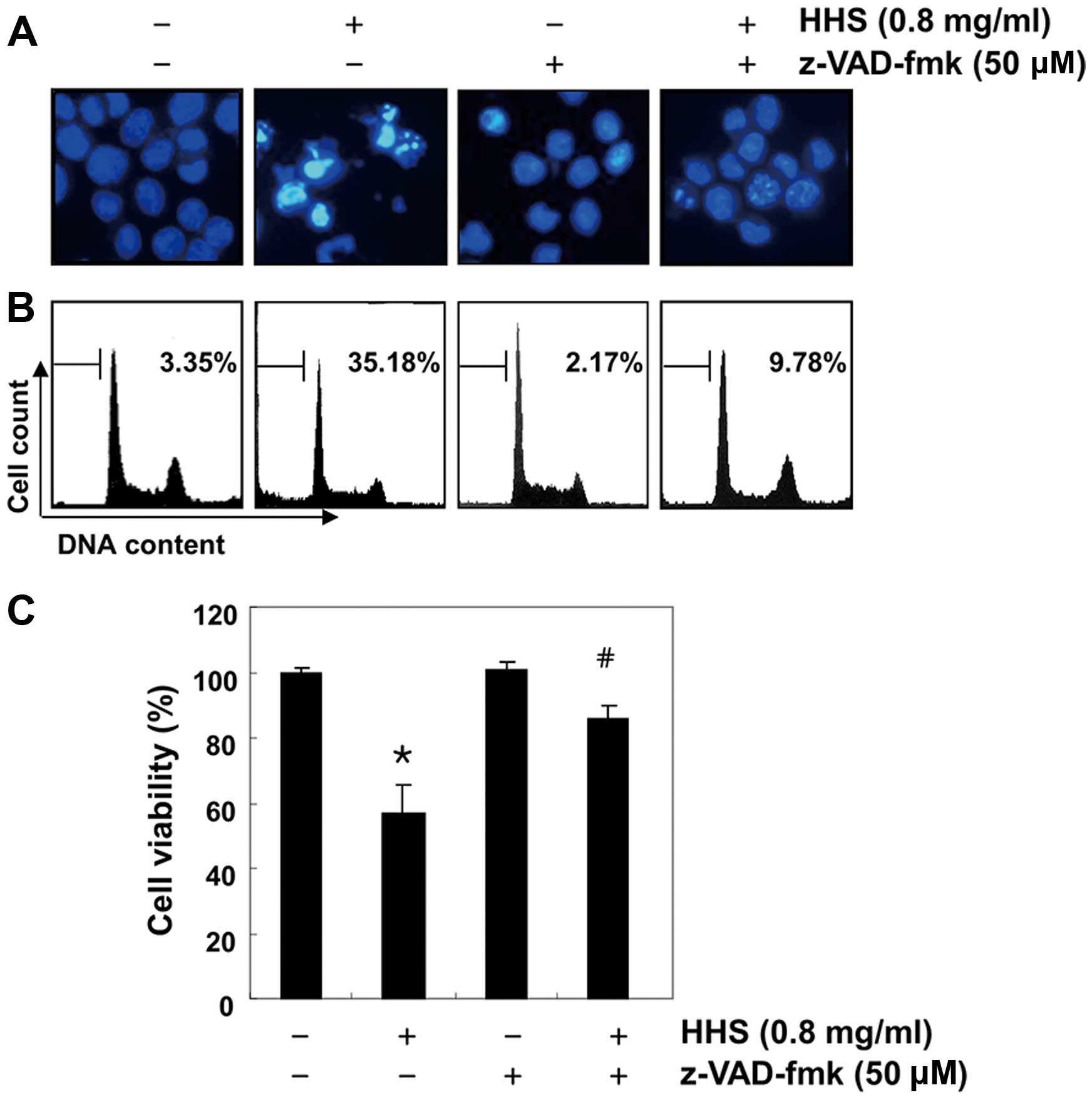
To confirm the involvement of caspase activation in
the apoptosis induced by HHS, we conducted a test to determine
whether the caspase inhibitor could attenuate the HHS-induced
apoptosis and viability reduction. As shown in Fig. 4A and B, pre-treatment with
z-VAD-fmk, a pan-caspase inhibitor, resulted in the significant
prevention of apoptotic features and the accumulation of
hypodiploid cells. The MTT assay also revealed that cells that were
co-treated with HHS and z-VAD-fmk had significantly greater cell
viability than those that were treated with HHS only (Fig. 4C).
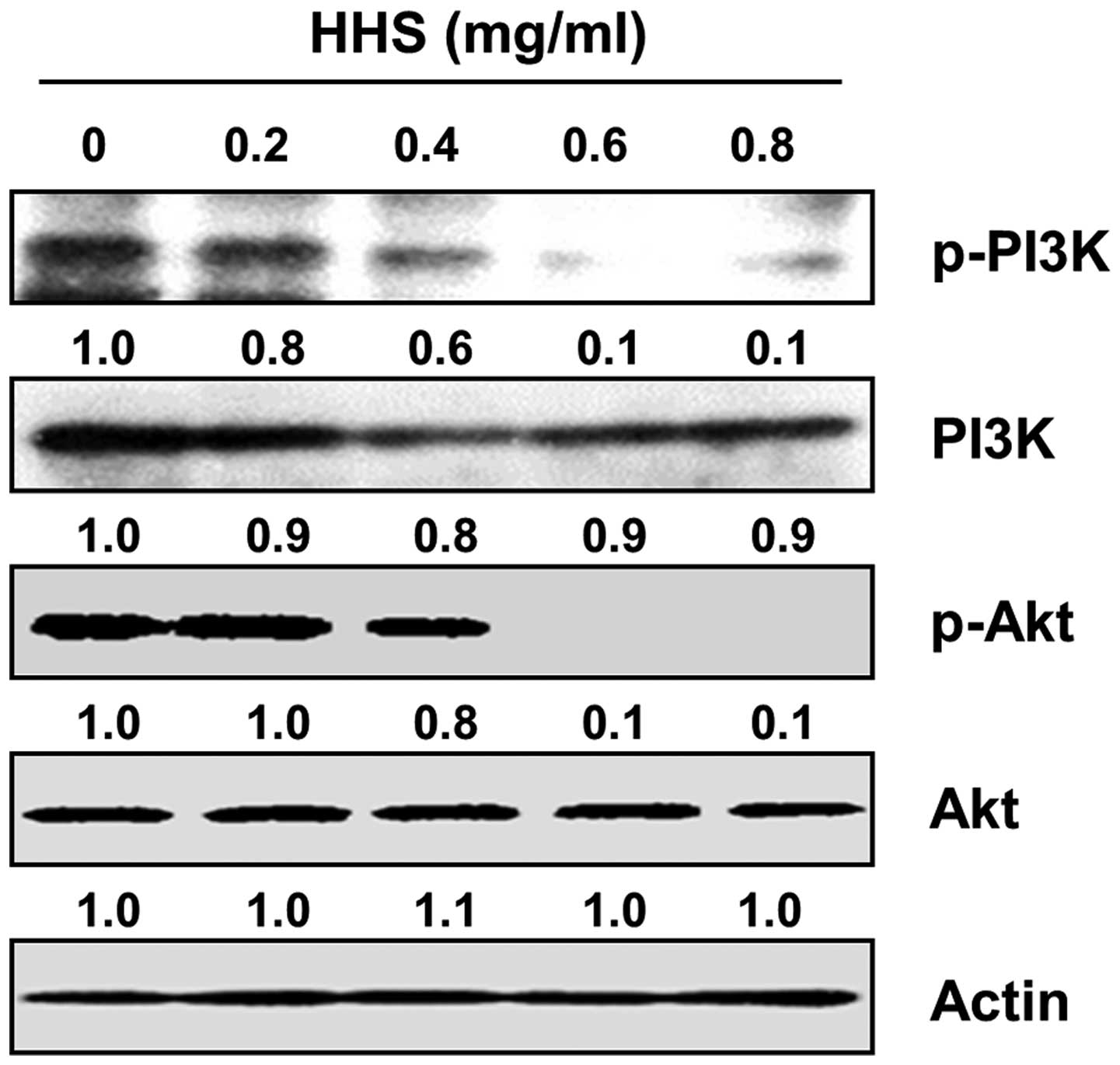
HHS inhibits the activation of PI3K/Akt
signaling in the HCT116 cells
The PI3K/Akt pathway has been known to transduce
anti-apoptotic signals that confer a survival advantage and
resistance of cancer cells against various chemotherapeutic agents
(7,9). We therefore sought to determine the
involvement of the PI3K/Akt signaling pathway in the HHS-induced
apoptosis in HCT116 cells. The constitutive activation of both PI3K
and AKT, one of the main downstream effectors of PI3K, was observed
in the control cells, whereas HHS treatment significantly
suppressed their phosphorylation in a concentration-dependent
manner (Fig. 5). The levels of
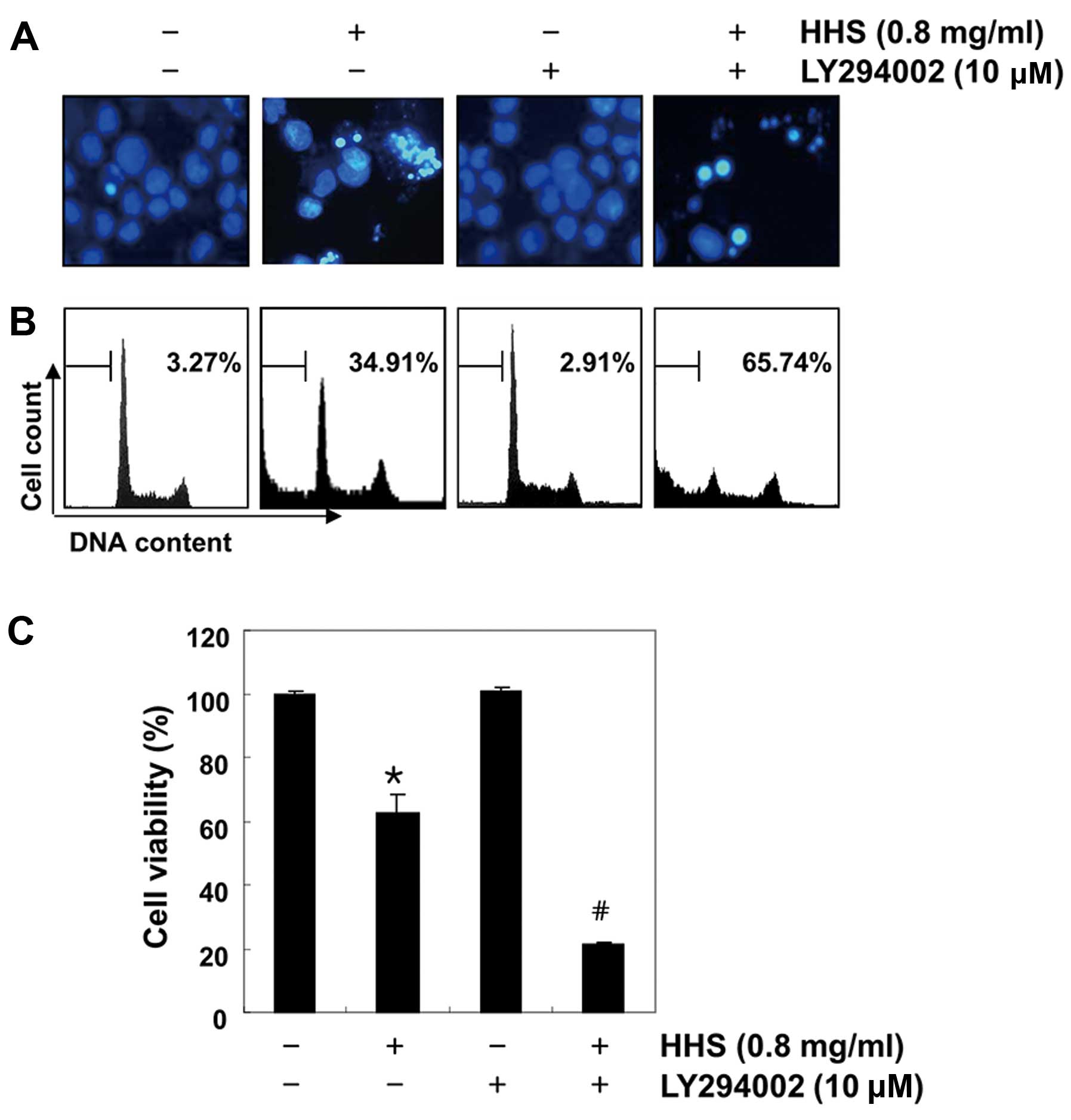
total PI3K and Akt were unaffected by HHS treatment. To determine
whether the mechanism in PI3K/Akt signaling is involved in
HHS-induced apoptosis, the cells were treated with HHS in the
presence or absence of LY294002, a representative PI3K inhibitor.
As shown in Fig. 6A and B, the
treatment of LY294002 markedly increased the HHS-induced apoptosis,
which was demonstrated by nuclear morphological changes and the
flow cytometric analysis. In addition, in combination with
LY294002, HHS strongly inhibited cell viability (Fig. 6C), indicating that the inactivation
of the PI3K/Akt signaling pathway is involved in HHS-induced HCT116
cell apoptosis.
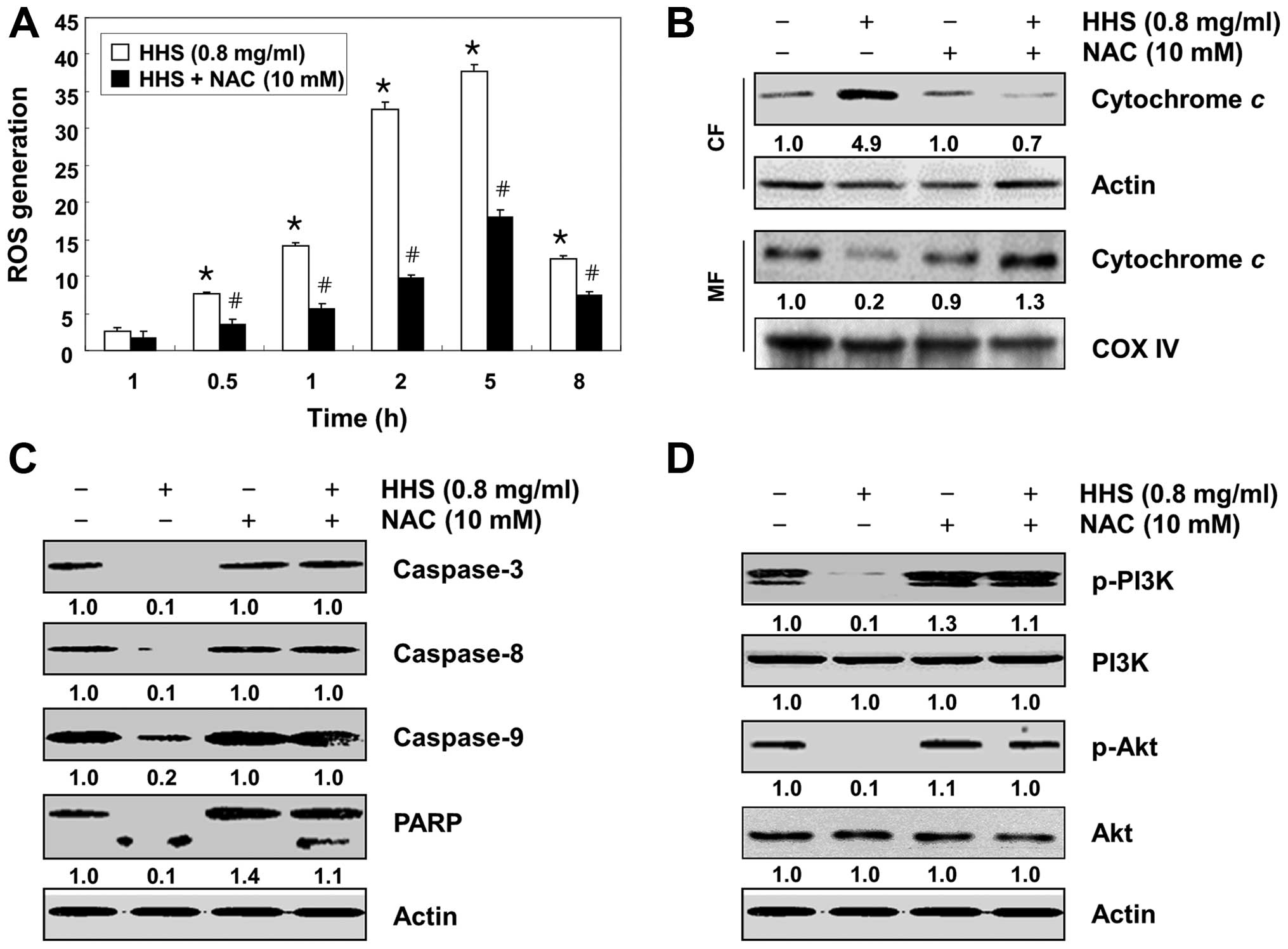
HHS-induced apoptosis is mediated by
intracellular ROS generation in the HCT116 cells
Since the increase of intracellular ROS generation
is proposed to be one of the early events in the activation of
apoptotic signaling, which alters the cellular redox state and MMP
(8,25), we determined whether the generation
of ROS is responsible for the apoptosis induced by HHS in HCT116
cells. The flow cytometric analysis revealed that the generation of
ROS was observed at 0.5 h, and the levels continued to increase up
to 5 h in response to the HHS treatment. In contrast, the
pretreatment of cells with NAC, a commonly used reactive oxygen
intermediate scavenger, significantly blocked the HHS-induced
increase in ROS generation (Fig.
7A).
In order to show that the generation of ROS is a key
step in HHS-induced mitochondrial dysfunction, we compared the
levels of cytochrome c in the cytosolic and mitochondrial
fractions. As shown in Fig. 7B, the
blocking of ROS generation by the pretreatment of cells with NAC
effectively prevented the HHS-induced cytochrome c release
into the cytosol, suggesting that the increment in the
intracellular ROS level by HHS was linked to mitochondrial
dysfunction. NAC pretreatment also markedly prevented the
HHS-induced cleavage of caspases (caspase-8, -9 and -3) and the
degradation of PARP (Fig. 7C).
Next, we confirmed whether the HCC-induced ROS generation had any
functional role in the inactivation of the PI3K/Akt pathway. The
results indicated that the pretreatment of HCT116 cells with NAC
effectively attenuated the HHS-induced phosphorylation of both PI3K
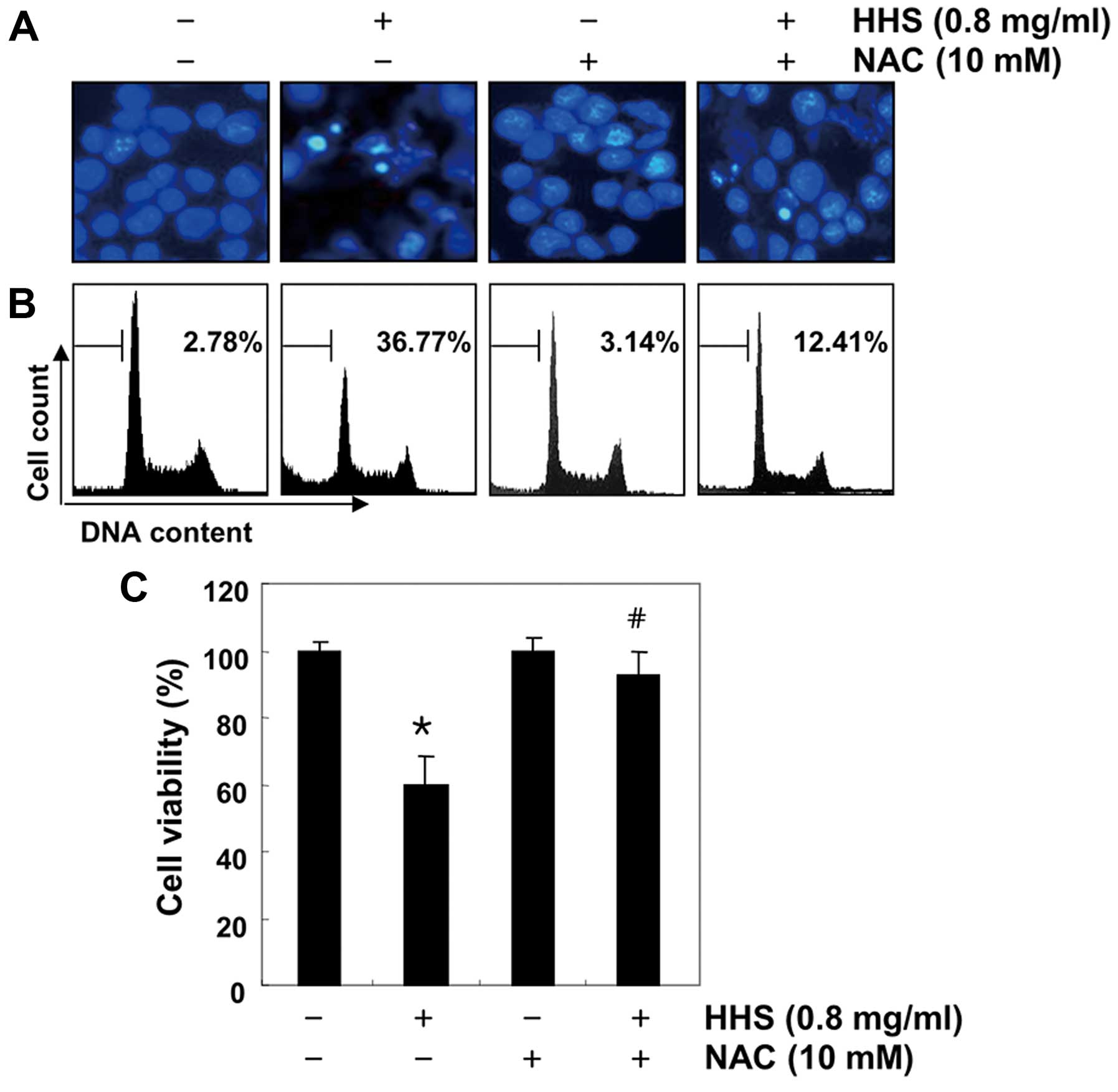
and Akt (Fig. 7D). Furthermore, the
apoptotic events in the HHS-treated HCT116 cells were prevented to
a considerable extent by the blocking of ROS generation (Fig. 8A and B), which was associated with
recovered cell viability, which was revealed by the MTT assay
(Fig. 8C), indicating that the
HHS-induced inactivation of the PI3K/Akt pathway was mediated by
ROS generation.
Discussion
In the present study, the HHS-induced inhibition of
cell proliferation was associated with the induction of apoptotic
cell death in HCT116 cells through both the intrinsic and extrinsic
pathways. We also demonstrated that exposure of the HCT116 cells to
HHS resulted in the induction of apoptosis, which was mediated by
ROS generation and the downregulation of the PI3K/Akt signaling
cascades.
Since apoptosis plays a crucial role in determining
the fate of a cell, apoptosis-inducing agents have been
investigated as tools for the management of cancer treatment. In
general, apoptosis is initiated by two cellular mechanisms:
extrinsic and intrinsic signaling pathways (5,6). In
the extrinsic pathway, membrane-bound DRs face the exterior of the
plasma membranes to receive stimuli from pro-apoptotic ligands and
transmit signals by activating downstream initiators, including
caspase-8, by the formation of a death-induced signaling complex
(26,27). In contrast, one of the key events in
the initiation of intrinsic apoptosis is the disintegration of MMP,
which induces the release of cytochrome c from the
mitochondria to the cytosol and elicits caspase-9 activation. The
activated caspase-8 also directly cleaves Bid to tBid, leading to
the activation of the intrinsic pathway through mitochondrial
dysfunction, which links the extrinsic pathway to the intrinsic
pathway (5,28). Activated caspase-8 and -9 then
promote the cleavage of the effector caspases, including caspase-3
and -7, into their active forms, which causes the proteolysis of
their substrates, eventually leading to apoptotic cell death
(27,29). In the present study, HHS enhanced
the expression of FasL, DR4 and DR5, the activation of caspase-8
and -3, and the concomitant PARP cleavage with a rise in tBid. We
also found that the HHS treatment resulted in the loss of MMP, the
release of cytochrome c, and the activation of caspase-9.
However, pretreatment with the pan-caspase inhibitor, z-VAD-fmk,
strongly attenuated HHS-induced cell death and afforded significant
protection against HHS-induced growth reduction. Therefore, the
results indicate that HHS appears to induce apoptosis primarily
through the caspase-dependent, intrinsic and extrinsic apoptotic
pathways in HCT116 cells.
In addition, activation of the intrinsic apoptosis
pathway is tightly regulated by Bcl-2 family proteins, which are
key regulators of mitochondrial membrane function (26,30).
In mammalian cells, the anti-apoptotic proteins of the Bcl-2
family, such as Bcl-2 and Bcl-xL, inhibit apoptosis and promote
cell survival, which is thought to be involved in the resistance to
conventional cancer treatment, including CRC (31,32).
However, pro-apoptotic proteins from the same gene family,
including Bax and Bad, can critically induce apoptotic cell death.
In particular, Bax, in combination with tBid, can induce the
release of cytochrome c by creating pore channels in the
process of oligomerization in the outer mitochondrial membrane or
by opening other channels (18,19).
Therefore, apoptosis largely depends on the expression ratio
between anti-apoptotic and pro-apoptotic protein levels. Here we
found that HHS treatment resulted in a concentration-dependent
inhibition of the expression of Bcl-2 protein and a concomitant
increase in the expression of Bax. These results suggest that Bax
was oligomerized to anchor onto the outer mitochondrial membrane,
forming mitochondrial permeability transition pores that disrupted
the MMP in the HCT116 cells. Therefore, the disturbance of the
Bcl-2/Bax ratio could contribute to the mitochondrial response to
the generation of tBid, leading to mitochondrial disturbance and
finally releasing cytochrome c and activating caspase-9 to
intensify the initial apoptotic response.
Recently, the activation of the PI3K/Akt signaling
pathway and its downstream transcription factors has been shown to
play a critical role in the control of cellular proliferation
(7,33). A growing amount of evidence suggests
that PI3K/Akt is activated in most cancer tissues, including CRC,
compared to normal tissue. Several studies have shown that the
dysregulation of the PI3K/Akt signaling pathway leads to cancer
progression (9,10). Moreover, there is much scientific
interest in finding molecular pathways and novel compounds that
target PI3K/Akt signaling as a new potential therapeutic option for
cancer treatment (10,34). Therefore, we further clarified the
regulatory role of PI3K/Akt signaling in HHS-mediated HCT116 cell
apoptosis. We found that the treatment with HHS had an inhibitory
effect on the levels of phosphorylation of AMPK and its downstream
target, Akt, in HCT116 cells without the reduction in the
steady-state levels of the total PI3K and Akt. To rule out the
possibility that the pro-apoptotic effect of HHS was caused by the
inactivation of PI3K/Akt after HHS treatment, we pretreated the
cells with a synthetic PI3K inhibitor, LY294002. The results
indicated that the inhibition of PI3K/Akt signaling in the HCT116
cells effectively protected them from HHS-induced apoptosis and
cell viability inhibition. Thus, the results clearly indicate that
HHS has anticancer effects with respect to its ability to inhibit
the PI3K/Akt signaling pathway, which may be one of the factors
leading to apoptosis.
Accumulating evidence indicates that moderate levels
of ROS can promote cell proliferation and survival; however,
excessive ROS production can induce apoptosis (25). Consistent with this finding, many
chemotherapeutic drugs exert their cytotoxic effects through the
generation of ROS by causing oxidative stress, while many
inhibitors of apoptosis show antioxidant activity (35,36).
Indeed, the mitochondrial dysfunction induced by excessive ROS
generation leads to MMP loss and apoptotic processes (37–39).
In the present study, we also found that incubation with HHS
significantly increased the ROS level, which was abolished by the
pretreatment of cells with the antioxidant NAC. The NAC treatment
also rescued cells from HHS-induced apoptosis and growth reduction
by blocking the release of cytochrome c and the cleavage of
caspases and PARP. Furthermore, it is noteworthy that blocking ROS
generation prevented the HHS-induced phosphorylation of PI3K and
Akt, demonstrating that HHS stimulated the production of ROS, which
subsequently inactivated the PI3K/Akt signaling pathway.
In conclusion, the results indicate that HCT116
cells undergo apoptosis in response to treatment with HHS, which
occurs through the activation of the intrinsic and extrinsic
pathways, and activation of caspases is responsible for the
mediation of HHS-induced apoptosis. In addition, we proposed that
the HHS-mediated apoptosis is caused by generation of ROS, which
may contribute to the inactivation of PI3K/Akt signaling and the
induction of caspase-dependent HCT116 cell apoptosis. Although
these findings indicate that ROS play a pivotal role in the
regulation of HHS-induced apoptosis in HCT116 cells, further
studies are needed to identify the active compounds of HHS.
Moreover, elucidation of additional molecular mechanisms and the
biological efficacy of HHS may lead to the development of
therapeutic approaches for the attenuation of CRC.
Acknowledgments
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) grant funded by the Korea government (2012046358 and
2015R1A2A2A01004633).
References
|
1
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hohla F, Winder T, Greil R, Rick FG, Block
NL and Schally AV: Targeted therapy in advanced metastatic
colorectal cancer: Current concepts and perspectives. World J
Gastroenterol. 20:6102–6112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Walczak H and Krammer PH: The CD95
(APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res.
256:58–66. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lavrik IN: Systems biology of apoptosis
signaling networks. Curr Opin Biotechnol. 21:551–555. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li ZY, Yang Y, Ming M and Liu B:
Mitochondrial ROS generation for regulation of autophagic pathways
in cancer. Biochem Biophys Res Commun. 414:5–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakanishi A, Wada Y, Kitagishi Y and
Matsuda S: Link between PI3K/AKT/PTEN pathway and NOX protein in
diseases. Aging Dis. 5:203–211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pandurangan AK: Potential targets for
prevention of colorectal cancer: A focus on PI3K/Akt/mTOR and Wnt
pathways. Asian Pac J Cancer Prev. 14:2201–2205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Danielsen SA, Eide PW, Nesbakken A, Guren
T, Leithe E and Lothe RA: Portrait of the PI3K/AKT pathway in
colorectal cancer. Biochim Biophys Acta. 1855:104–121. 2015.
|
|
11
|
Yang G, Li X, Li X, Wang L, Li J, Song X,
Chen J, Guo Y, Sun X, Wang S, et al: Traditional chinese medicine
in cancer care: A review of case series published in the Chinese
literature. Evid Based Complement Alternat Med. 2012:7510462012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X, Feng Y, Wang N, Cheung F, Tan HY,
Zhong S, Li C and Kobayashi S: Chinese medicines induce cell death:
The molecular and cellular mechanisms for cancer therapy. Biomed
Res Int. 2014:5303422014.PubMed/NCBI
|
|
13
|
Li J, Lu C, Jiang M, Niu X, Guo H, Li L,
Bian Z, Lin N and Lu A: Traditional chinese medicine-based network
pharmacology could lead to new multicompound drug discovery. Evid
Based Complement Alternat Med. 2012:1497622012.
|
|
14
|
Cha WS, Oh JH, Park HJ, Ahn SW, Hong SY
and Kim NI: Historical difference between traditional Korean
medicine and traditional Chinese medicine. Neurol Res. 29(Suppl 1):
S5–S9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwon T, Rho JK, Lee JC, Park YH, Shin HJ,
Cho S, Kang YK, Kim BY, Yoon DY and Yu DY: An important role for
peroxiredoxin II in survival of A549 lung cancer cells resistant to
gefitinib. Exp Mol Med. 47:e1652015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim YS, Li XF, Kang KH, Ryu B and Kim SK:
Stigmasterol isolated from marine microalgae Navicula incerta
induces apoptosis in human hepatoma HepG2 cells. BMB Rep.
47:433–438. 2014. View Article : Google Scholar :
|
|
17
|
Song JL, Choi JH, Seo JH, Kil JH and Park
KY: Antioxidative effects of fermented sesame sauce against
hydrogen peroxide-induced oxidative damage in LLC-PK1 porcine renal
tubule cells. Nutr Res Pract. 8:138–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kadenbach B, Arnold S, Lee I and Hüttemann
M: The possible role of cytochrome c oxidase in stress-induced
apoptosis and degenerative diseases. Biochim Biophys Acta.
1655:400–408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scorrano L and Korsmeyer SJ: Mechanisms of
cytochrome c release by proapoptotic BCL-2 family members. Biochem
Biophys Res Commun. 304:437–444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Degli Esposti M and Dive C: Mitochondrial
membrane permeabilisation by Bax/Bak. Biochem Biophys Res Commun.
304:455–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karbowski M, Norris KL, Cleland MM, Jeong
SY and Youle RJ: Role of Bax and Bak in mitochondrial
morphogenesis. Nature. 443:658–662. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Duriez PJ and Shah GM: Cleavage of
poly(ADP-ribose) polymerase: A sensitive parameter to study cell
death. Biochem Cell Biol. 75:337–349. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Danson S, Dean E, Dive C and Ranson M:
IAPs as a target for anticancer therapy. Curr Cancer Drug Targets.
7:785–794. 2007. View Article : Google Scholar
|
|
24
|
de Graaf AO, de Witte T and Jansen JH:
Inhibitor of apoptosis proteins: New therapeutic targets in
hematological cancer? Leukemia. 18:1751–1759. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hensley P, Mishra M and Kyprianou N:
Targeting caspases in cancer therapeutics. Biol Chem. 394:831–843.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fiandalo MV and Kyprianou N: Caspase
control: Protagonists of cancer cell apoptosis. Exp Oncol.
34:165–175. 2012.PubMed/NCBI
|
|
30
|
Tomek M, Akiyama T and Dass CR: Role of
Bcl-2 in tumour cell survival and implications for pharmacotherapy.
J Pharm Pharmacol. 64:1695–1702. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koehler BC, Jäger D and Schulze-Bergkamen
H: Targeting cell death signaling in colorectal cancer: Current
strategies and future perspectives. World J Gastroenterol.
20:1923–1934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qiao L and Wong BC: Targeting apoptosis as
an approach for gastrointestinal cancer therapy. Drug Resist Updat.
12:55–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marone R, Cmiljanovic V, Giese B and
Wymann MP: Targeting phosphoinositide 3-kinase: Moving towards
therapy. Biochim Biophys Acta. 1784:159–185. 2008. View Article : Google Scholar
|
|
34
|
Moral M and Paramio JM: Akt pathway as a
target for therapeutic intervention in HNSCC. Histol Histopathol.
23:1269–1278. 2008.PubMed/NCBI
|
|
35
|
Kardeh S, Ashkani-Esfahani S and Alizadeh
AM: Paradoxical action of reactive oxygen species in creation and
therapy of cancer. Eur J Pharmacol. 735:150–168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Matés JM, Segura JA, Alonso FJ and Márquez
J: Oxidative stress in apoptosis and cancer: An update. Arch
Toxicol. 86:1649–1665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jourdain A and Martinou JC: Mitochondrial
outer-membrane permeabilization and remodelling in apoptosis. Int J
Biochem Cell Biol. 41:1884–1889. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seo K, Ki SH and Shin SM: Methylglyoxal
induces mitochondrial dysfunction and cell death in liver. Toxicol
Res. 30:193–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Asakura T and Ohkawa K: Chemotherapeutic
agents that induce mitochondrial apoptosis. Curr Cancer Drug
Targets. 4:577–590. 2004. View Article : Google Scholar : PubMed/NCBI
|