Introduction
Hepatocellular carcinoma (HCC) is one of the most
malignant cancers, which accounts for almost 80% of all primary
liver tumors (1). Despite effective
surgical and drug therapy for HCC, such as surgical resection,
partial melting and intervention, chemoradiotherapy, and other
comprehensive treatment, the 5-year survival rate for HCC still
remains very low. This high mortality rate is attributable mainly
to tumor recurrence and metastases (2). Even with liver transplantation, the
prognosis remains poor, owing to persistent tumor recurrence. Thus,
there is a compelling need for a novel and specific therapeutic
target.
Human tissue inhibitor of metalloproteinase-3
(TIMP-3), located on chromosome 22q12 (3) is an endogenous inhibitor of matrix
metalloproteinases (MMPs), including metalloproteinases that have a
disintegrin and metalloproteinase (ADAM) domain, and ADAMs with
thrombospondin (TS)-like domains. These enzymes play important
roles in degrading extracellular matrix (ECM) substrates (4). Unlike other TIMP variants (TIMP-1, 2,
and 4), TIMP-3 has poor aqueous solubility and is a component of
the ECM (5). Studies have
demonstrated that TIMP-3 might function as a potential tumor
suppressor gene through induction of tumor cell apoptosis,
prevention of tumor ECM remodeling, and inhibition of tumor-derived
angiogenic activity (5,6)
The expression of TIMP-3 is silenced in various
types of malignant carcinoma (7).
Regulation of TIMP-3 expression may be influenced by many upstream
and downstream transcription factors. The type II nuclear receptor,
peroxisome proliferator-activated receptor gamma, can upregulate
TIMP-3 expression, which can be detected by cDNA microarray and
chromatin immunoprecipitation coupled to PCR (8). Transcriptional reprogramming of tumors
by overexpression of dormancy-associated microRNAs (DmiRs),
particularly DmiR-580, 588, or 190, led to downregulation of TIMP-3
(9). Many other miRs can
downregulate TIMP-3 expression, such as miR-21 (10), miR-103 (11), miR-181b (12), miR-181a (13), and miR-221/222 (14). These miRs all regulate TIMP-3
expression by targeting mRNAs at the 3′-untranslated regions (UTRs)
for cleavage or translational repression. Thus, miR inhibition can
significantly upregulate TIMP-3 protein expression in cancer cells
(11).
Another mechanism by which TIMP-3 expression is
regulated is the methylation of CpG islands in the TIMP-3 promoter,
which results in a dramatic decrease in TIMP-3 mRNA levels
(15,16). Ten-eleven translocation
methylcytosine dioxygenase 1, encoded by the gene, TET1, has been
shown to suppress tumor growth by downregulating TIMP-3 methylation
(17). Masson et al showed
that loss of TIMP-3 expression in clear cell renal cell carcinoma
results from multiple mechanisms, including promoter
hypermethylation. Methylation-associated inactivation of TIMP-3 has
been reported in bladder cancer and in esophageal and gastric
adenocarcinomas as well as renal cancer, and it is associated with
poor tumor differentiation, high incidence of tumor invasiveness,
and poor clinical outcome (18–20).
Invasion and metastasis of tumor cells are the key
characteristics of malignant tumors. By binding to MMPs, in tumor
cells, TIMP-3 inhibits their ECM-degrading activity, and thus
TIMP-3 is crucially involved in the balance between synthesis and
degradation of the ECM, a process which is closely related to tumor
cell invasion and metastasis (21).
Furthermore, the balance between MMPs and TIMPs is strongly linked
to cellular proliferation, apoptosis, and cell cycle arrest as well
as invasion and metastasis (22).
The activities of both MMP-2, which can promote αvβ3
integrin-mediated adhesion and migration by cleaving fibronectin
(23), and MMP-9, which can induce
cleavage of the cell-cell/cell-matrix adhesion molecule, CD44
(24), plays an important role in
increasing the migration and/or invasion potential of several kinds
of cancer cells. Downregulation of the gene that codes for a
proliferation-inducing ligand (APRIL), which is typically
overexpressed in most tumor cells, leads to reduced expression of
MMP-2 and MMP-9 and enhancement of TIMP-3 and TIMP-4 expression
(4).
It has been reported that suppression of MMP-9 can
inhibit tumor invasion and migration via activating transforming
growth factor-β (25), and loss of
TIMP-3 expression can result from inactivation of transforming
growth factor-β receptor II (18).
Recent studies have also shown that TIMP-3 suppresses tumor
invasion via the TNFα/NF-κB/IL-6 pathway (16). We have previously found that the
transcription factor, peroxisome proliferator activated receptor
gamma, partially inhibits HCC cell invasiveness and metastasis via
binding directly to the TIMP-3 promoter and upregulating
TIMP-3 protein expression (8).
Although TIMP-3 has been shown to inhibit tumor
growth, invasion, and metastasis (26), the effects of this physiological
inhibitor in HCC have not been fully characterized. In this study,
our aim was to assess the methylation status of the TIMP-3 promoter
and expression of TIMP-3 in HCC and elucidate the biological
function of TIMP-3 in HCC by exogenously inducing TIMP-3 expression
in liver cancer cells.
Materials and methods
Patient tissue samples
Tissue samples embedded in paraffin blocks were
obtained from the livers of 80 HCC patients and 60 cancer-free
patients (control subjects). None of these patients had received
any other therapeutic intervention prior to surgery. Pertinent
demographic and clinicopathological data, such as gender, age,
tumor size, hepatitis B virus (HBV) infection, liver cirrhosis,
tumor pathological grade, lymph node metastasis, and distant
metastasis were collected for further analyses. The study protocol
was approved by the Clinical Research Ethics Committee of Guangzhou
Medical University.
Immunohistochemistry (IHC)
IHC staining was performed on 4-µm-thick
paraffin sections using the TIMP-3 primary antibody (1:100
dilution; Santa Cruz Biotechnology, Dallas, TX, USA). Sections were
washed in Tris-buffered saline and stained in a two-step process
involving incubation with a horseradish peroxidase-conjugated
antibody kit (1:2000 dilution, Santa Cruz Biotechnology), followed
by incubation with the chromagen, 3,3′-diaminobenzidine (Dako,
Glostrup, Denmark). TIMP-3 immunostaining was defined according to
the intensity and percentage of TIMP-3- positive tumor cells. The
staining intensity was scored as follows: 0 (negative), 1 (weakly
positive), 2 (moderately positive), and 3 (strongly positive). The
percentage of TIMP-3 positive cells was classified into 4
categories: score of 1 for 0–10%, 2 for 11–50%, 3 for 51–80%, and 4
for 81–100%. The degree of TIMP-3 staining was quantified by using
a two-level grading system (with the intensity score added to the
percentage score to produce a final score), as follows: <3,
negative expression; and 3–9, positive expression.
Human HCC cell lines and culture
The human HCC HepG2 cell line was a gift from AiMing
Li, Department of Digestive System, Southern Medical University.
The human HCC SMMC-7721 cell line was purchased from the
Experimental Animal Center of Sun Yat-Sen University. Cells were
cultured in Dulbecco's modified Eagle's medium (DMEM) with 10%
fetal bovine serum in 5% CO2 at 37°C.
Bisulfite conversion and
methylation-specific PCR (MSP)
MSP was performed as previously described (16) and was used to examine the DNA
methylation status of the TIMP-3 promoter. Genomic DNA was
extracted from 8 different HCC cell lines with NucleoSpin Tissue
mini spin columns (Macherey-Nagel GmbH & Co. KG, Düren,
Germany). Sodium bisulfite modification was performed with the
EpiTect Bisulfie kit (Qiagen, Hilden, Germany). The primers used in
MSP were designed via the MethPrimer web site (http://www.urogene.org/methprimer.html)
and synthesized by Invitrogen Custom DNA Oligos (Life Technologies,
Grand Island, NY, USA). The sequences of the forward and reverse
primers were: MSP forward methylation primer,
5′-TCGAGGATTTAGCGGTAAGTATC-3′; MSP reverse methylation primer,
5′-GAAAACAAAAAATAACGAAACGAA-3′; MSP forward non-methylation primer,
5′-TTGAGGATTTAGTGGTAAGTATTGG-3′; MSP reverse non-methylation
primer, 5′-CAAAAACAAAAAATAACAAAACAAA-3′. The MSP was carried out in
the T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA) under the
following conditions: 30 sec at 94°C, 30 sec at 54°C, and 30 sec at
72°C for 40 cycles, with a final 7 min extension at 72°C. The PCR
products were stained with ethidium bromide, followed by
electrophoresis on 2% agarose gel and visualization under UV light.
TIMP-3 mRNA was quantified by real-time PCR. Real-time
amplification of 1 µg of cDNA was performed with 12.5
µl of Master SYBR Green Supermix in the CFX96 Touch
Real-Time PCR Detection System (Bio-Rad). Amplification was
performed, with the target gene mRNA levels being normalized to the
expression of GAPDH. Results were analyzed by the 2−ΔCt
method.
Demethylation treatment
Selected methylation-positive HCC cells were
cultured with the DNA methyltransferase inhibitor, 5-Aza-CdR, at a
concentration of 5 µM or 0 µM (control).
Pyrosequencing analysis was performed to examine the effects of
5-Aza-CdR on the methylation level of the TIMP-3 promoter
CpG islands in HCC cells. Pyrosequencing was performed by the
PyroMark Q96 ID System (Qiagen) as previously described (27).
Plasmids and transfection
HepG2 and SMMC-7721 cells were transfected with the
pCMV6-TIMP-3 plasmid, which strongly expresses human TIMP-3 under a
CMV promoter. The pCMV6-AC-GFP plasmid was used as the vector
control. The open reading frames cloned in this vector are
expressed in mammalian cells as a tagged protein (C-terminal tGFP),
which can be visualized as green fluorescence under a fluorescence
microscope. Since it has been shown that methylation of TIMP-3 in
HepG2 is closely associated with TIMP-3 silencing in HCC and
because the pCMV6-TIMP-3 plasmid has been successfully transfected
into SMMC-7721 cells (28), we
selected these cell lines for the transfection experiments. The
cells were transfected using MegaTran 1.0 transfection reagent
(OriGene, Rockville, MD, USA) according to the manufacturer's
instructions. Cells were cultured for 2 weeks with the
aminoglycoside, G418 (Thermo Fisher Scientific, Shanghai, China),
to select the gene-transfected cells. Most cells are not
transfected with the expression plasmid and thus are killed by
G418. Those cells that remain growing in this selective medium have
retained the expression plasmid, which stably integrates into the
genome of the targeted cells. The group of cells transfected with
the pCMV6-TIMP-3 plasmid were labeled as TIMP-3, and the group of
cells not transfected with this plasmid were labeled as NC
(control). The cultures were observed under fluorescence microscopy
to confirm the presence of tGFP+ transfected cells, and
the transfection efficiency was determined by counting the number
of tGFP+ cells.
Western blot assay
Total protein was extracted and protein
concentration was measured by the BCA protein assay (Bioworld,
Minneapolis, MN, USA). Protein (40 µg) from each sample was
used for western blotting as previously described (29). Bands were observed using the Pierce
Enhanced Chemiluminescence kit (Thermo Fisher Scientific).
Cell viability assay
Cell proliferation was determined by the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay (Promega, Madison, WI, USA). Briefly, cells (3000/well)
were placed in 96-well plates. After 24 h, 20 µl of MTS
reaction solution was added to cultured cells in 100 µl
medium and incubated at 37°C for 1 h. The optical density was
measured at 490 nm in the Sunrise microplate absorbance reader
(Tecan Group Ltd., Männedorf, Switzerland).
Cell apoptosis assay
Cell apoptosis was assessed using the Annexin V-FITC
Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA)
according to the manufacturer's instructions. Cells were washed
twice with cold phosphate-buffered saline (PBS) and resuspended in
1X binding buffer. Next, 5 µl of Annexin V-FITC and 5
µl of propidium iodide (PI) staining solution were added,
followed by incubation for 15 min in the dark at room temperature
(25°C). Finally, cells were suspended in 400 µl 1X binding
buffer and analyzed within 1 h on the FACSCalibur Flow Cytometer
(BD Biosciences, San Jose, CA, USA). Experiments were conducted in
triplicate.
Cell cycle assay
Cells were trypsinized, washed twice with cold PBS,
and fixed for 1 h in ice-cold 70% ethanol. The cells were
resuspended in cold PBS, and 20 µl of RNase A solution were
then added, followed by incubation for 30 min at 37°C. Finally,
cells were labeled with PI and analyzed on the FACSCalibur Flow
Cytometer. Experiments were conducted in triplicate.
Invasion assay
The invasion assay was performed using transwell
chambers (BD Biosciences) with polycarbonate membrane inserts
(8-µm pore size). Diluted Matrigel (BD Biosciences), which
acts as a chemoattractant, was added to the upper chamber surface.
After 1 h of incubation at 37°C, the cells (2×104 per
well) were collected in serum-free medium and added to the upper
chambers. The lower chamber contained 600 µl DMEM containing
10% fetal bovine serum. After 24 h, cells that invaded through the
Matrigel membrane were fixed in 10% formalin, stained with crystal
violet, and counted under a microscope (4 high-power fields at ×100
magnification). Experiments were conducted in triplicate.
Migration assay
In the migration assay, transwell chambers were also
used (2×104 cells per well), but no Matrigel was used.
Instead, the cells were collected and diluted in serum-free medium,
and placed in the upper chambers. The lower chamber contained 600
µl DMEM containing 10% fetal bovine serum. After 24 h, cells
that had moved to the bottom surface of the filter membrane were
fixed in 10% formalin, stained with crystal violet, and counted
under a microscope (4 high-power fields at ×100 magnification).
Experiments were conducted in triplicate.
Statistical analyses
Data are presented as the mean ± standard deviation
(SD). Differences between two groups were analyzed by Student's
t-test. The correlation between TIMP-3 positive-expression and
liver cancer was analyzed by the Mann-Whitney test. Chi-square
tests were performed to evaluate the relationship between TIMP-3
expression and clinicopathological parameters. Statistical analyses
were performed in the SPSS package for Windows (version 16.0; SPSS
Inc., Chicago, IL, USA). Differences with a P-value <0.05 were
considered statistically significant.
Results
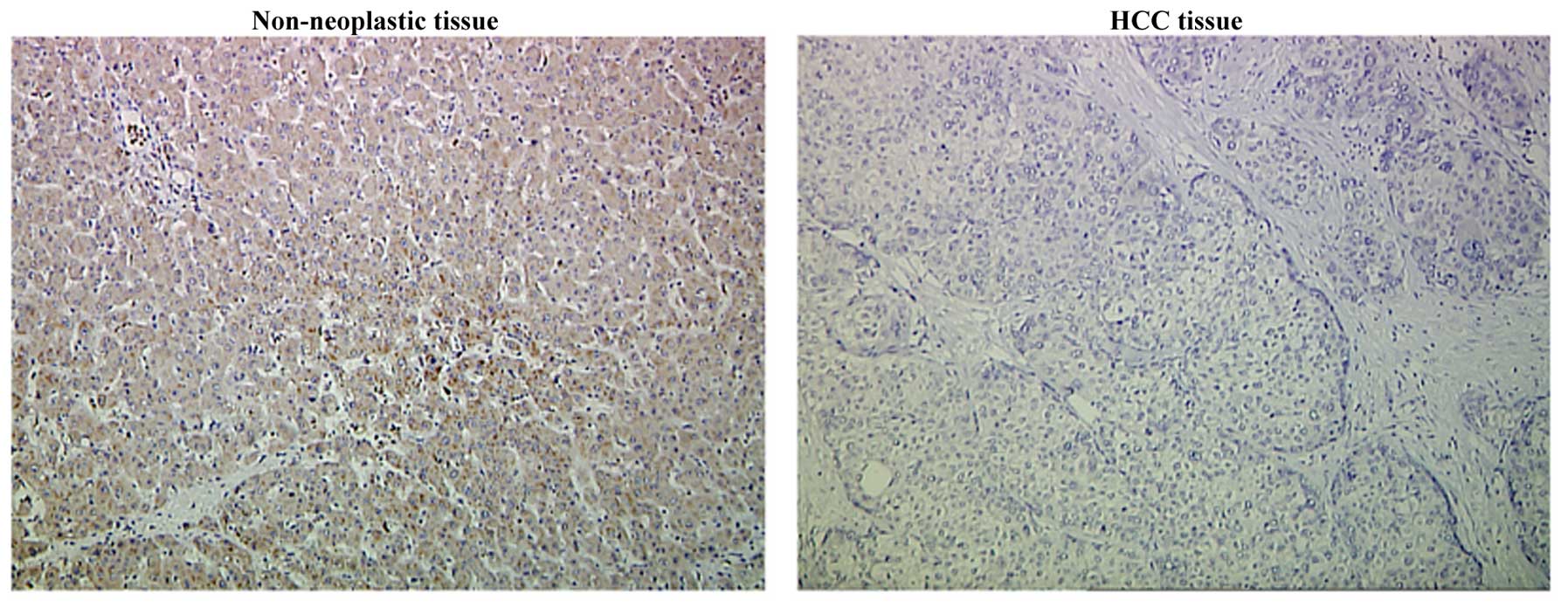
TIMP-3 expression in HCC tissues
As shown in Table I
and Fig. 1, TIMP-3 was expressed in
only 32 of 80 (40%) HCC tissue samples, whereas 37 of 60 tumor-free
samples (61.67%) exhibited TIMP-3 expression (P=0.011). Tissue from
76.67% of younger patients (<50 years) with HCC showed negative
TIMP-3 expression, while only 50% of tissues from older patients
exhibited negative TIMP-3 expression (P=0.018). Reduced expression
of TIMP-3 was correlated with higher frequencies of poorly
differentiated carcinomas and metastasis. No significant
correlations were observed between TIMP-3 expression and the
remaining clinicopathological parameters (Table II).
 | Table IComparison of TIMP-3 expression
between HCC and control subjects. |
Table I
Comparison of TIMP-3 expression
between HCC and control subjects.
| Subjects |
TIMP-3-positivea |
TIMP-3-negativea | χ2 | P-value |
|---|
| All cases | 69 (49.29) | 71 (50.71) | | |
| HCC | 32 (40.00) | 48 (60.00) | 6.439 | 0.011 |
| Control | 37 (61.67) | 23 (38.33) | | |
| Table IICorrelation of TIMP-3 expression with
clinicopathological characteristics in HCC patients. |
Table II
Correlation of TIMP-3 expression with
clinicopathological characteristics in HCC patients.
|
Characteristics | TIMP-3-positive
(%) | TIMP-3-negative
(%) | P-value |
|---|
| Gender | | | 0.598 |
| Male | 25 (41.67) | 35 (58.33) | |
| Female | 7 (35.00) | 13 (65.00) | |
| Age (years) | | | 0.018 |
| <50 | 7 (23.33) | 23 (76.67) | |
| ≥50 | 25 (50.00) | 25 (50.00) | |
| Tumor size | | | 0.144 |
| <5 cm | 20 (47.62) | 22 (52.38) | |
| ≥5 cm | 12 (31.58) | 26 (68.42) | |
|
Differentiation | | | 0.003 |
| Mod-well | 23 (56.10) | 18 (43.90) | |
| Low | 9 (23.08) | 30 (76.92) | |
| Metastasis | | | 0.005 |
| Yes | 11 (25.59) | 32 (74.41) | |
| No | 21 (56.76) | 16 (43.24) | |
| HBV infection | | | 0.205 |
| Yes | 19 (35.19) | 35 (64.81) | |
| No | 13 (50.00) | 13 (50.00) | |
| Fatty liver | | | 0.059 |
| Yes | 4 (80.00) | 1 (20.00) | |
| No | 28 (37.33) | 47 (62.67) | |
| Cirrhosis | | | 0.923 |
| Yes | 21 (39.62) | 32 (60.38) | |
| No | 11 (40.74) | 16 (59.26) | |
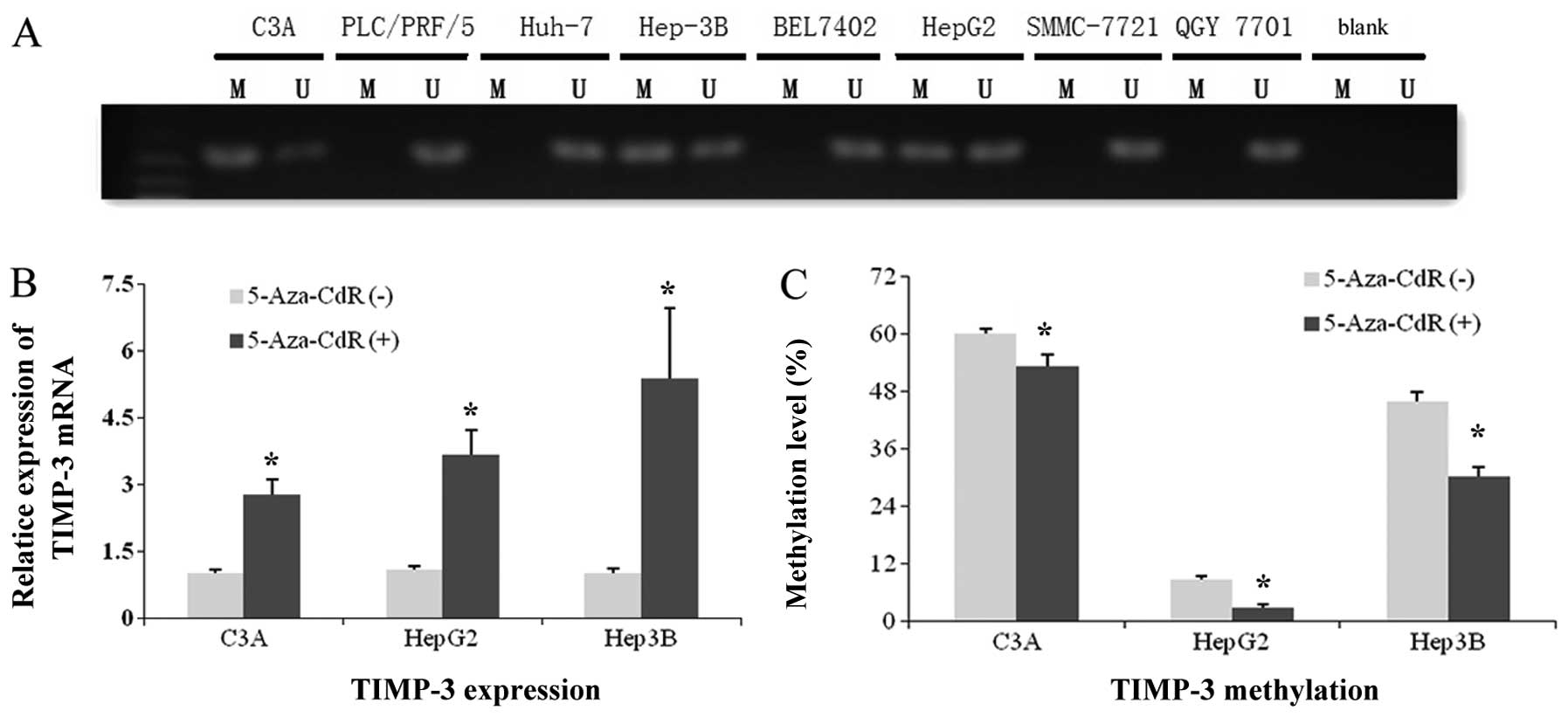
TIMP-3 gene expression and methylation
status in HCC cells
Results of MSP analysis were positive for
TIMP-3 promoter methylation in 3 of 8 HCC cell lines: C3A,
HepG2, and Hep-3B (Fig. 2A). Thus,
we selected these cell lines and treated them with 5-Aza-CdR. As
shown in Fig. 2B, TIMP-3 mRNA in
all three cell lines was upregulated by demethylation treatment
(P<0.05). Results of the pyrosequencing analyses (Fig. 2C) revealed decreased methylation
level in all three cell lines after 5-Aza-CdR treatment
(P<0.05).
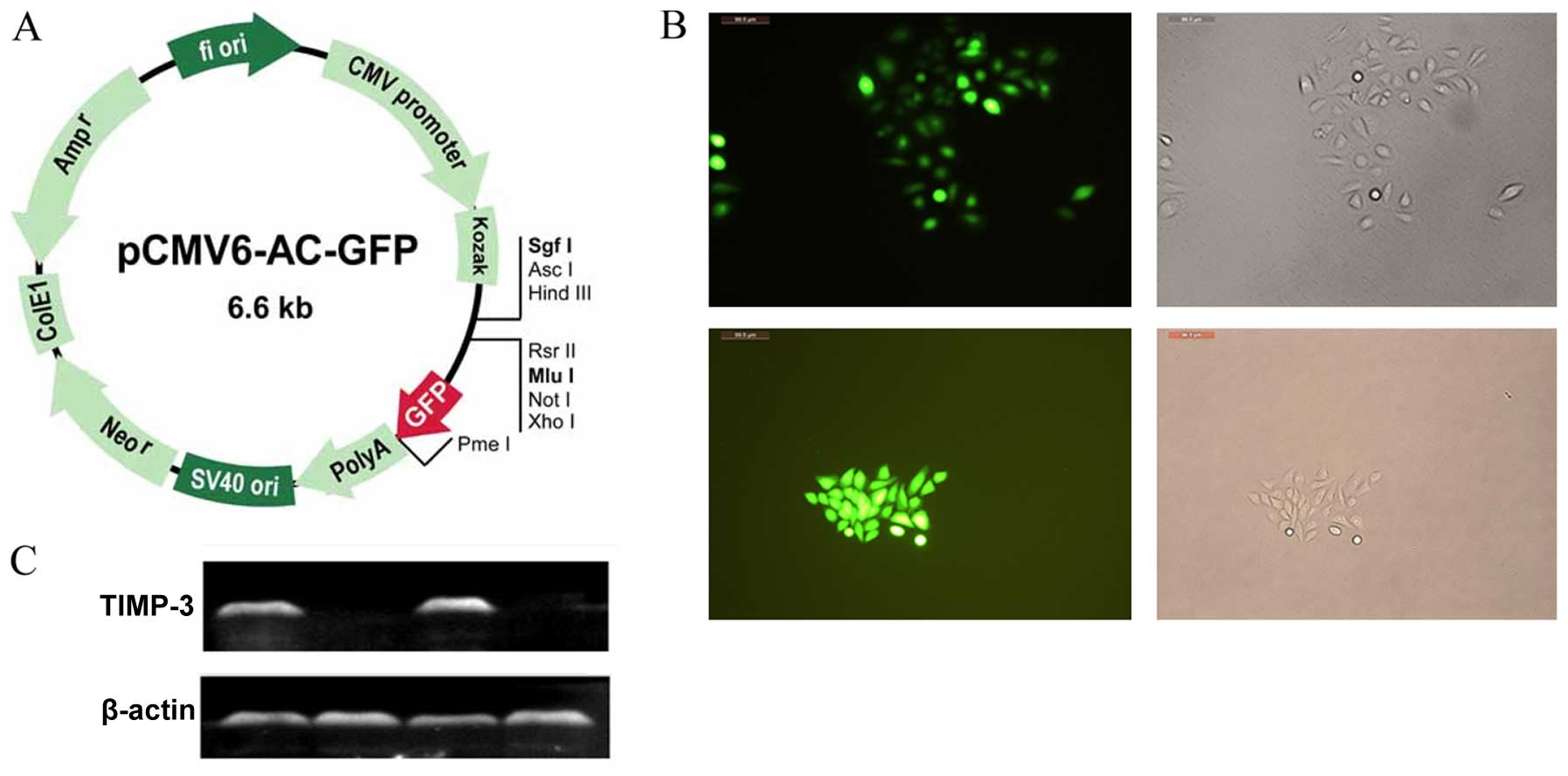
Expression of TIMP-3 plasmid in
transfected HCC cells
An illustration of the GFP expression vector is
shown in Fig. 3A, and the extent of
GFP labeling in SMMC-7721 and HepG2 cells is shown in Fig. 3B. Nearly all the G418-selected cells
exhibited green fluorescence. The western blots revealed robust
TIMP-3 protein expression in the TIMP-3 group, whereas no protein
expression was observed in the NC group (Fig. 3C).
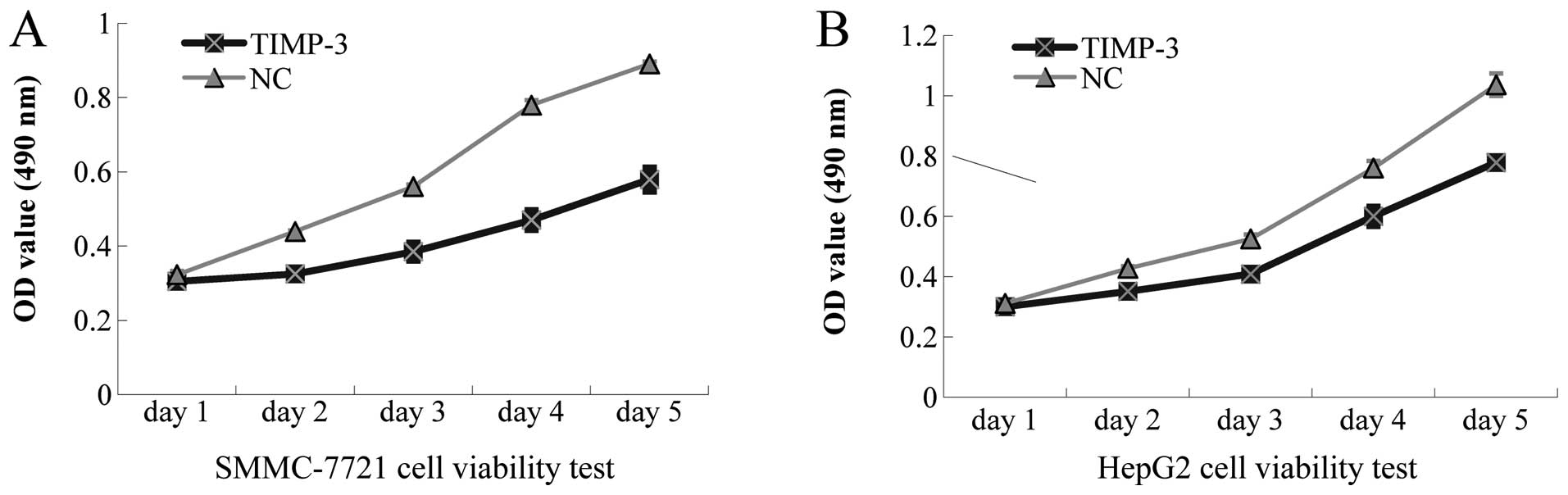
Effects of TIMP-3 overexpression on HCC
cell viability, apoptosis, and cell cycle arrest
The MTS assay revealed suppressed growth of
SMMC-7721 and HepG2 cells, stably transfected with TIMP-3, in a
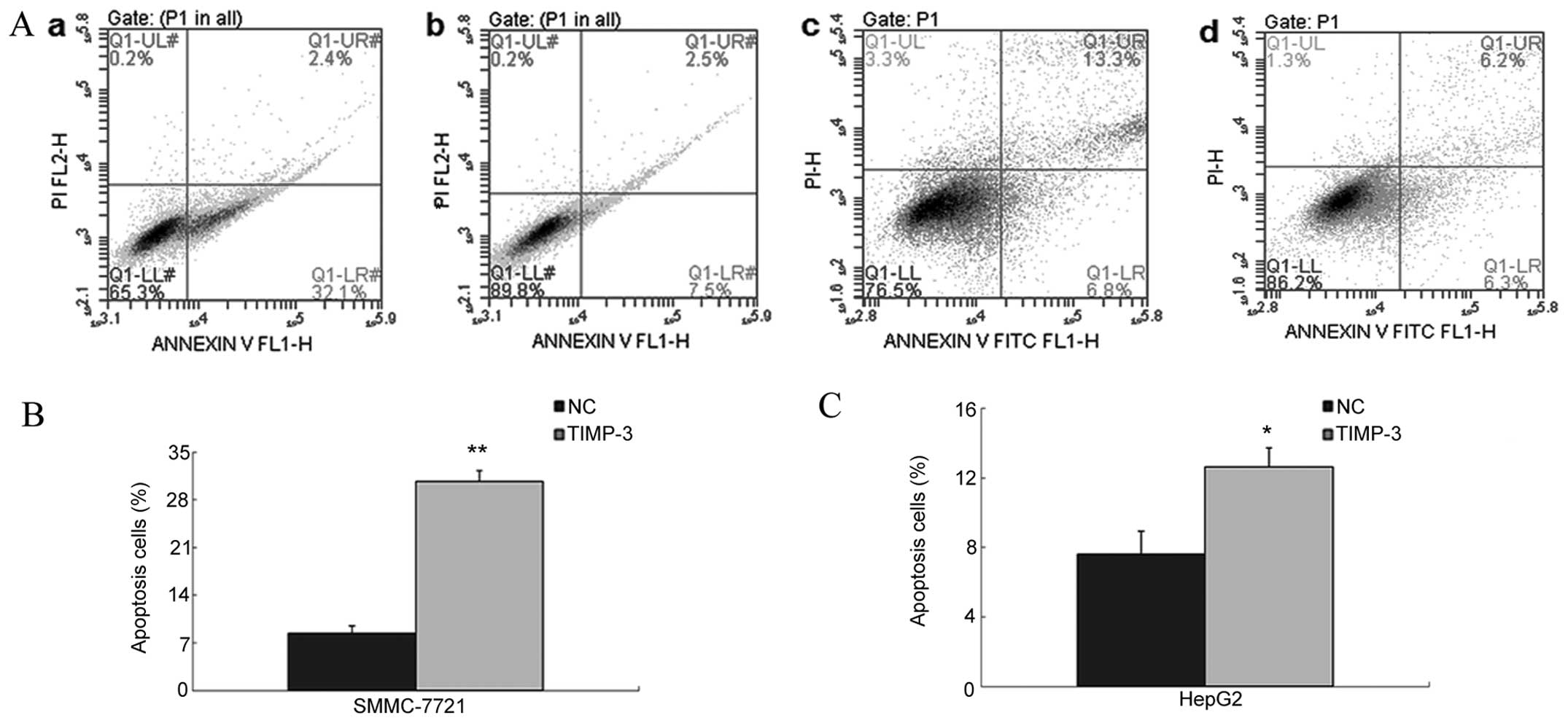
significant, time-dependent manner (Fig. 4). As shown in Fig. 5A–C, Flow cytometric analysis of
Annexin V-FITC/PI revealed an increased mean number of apoptotic
cells in the TIMP-3 group compared to the NC group (SMMC-7721:
30.7±1.6% vs. 8.4±1.1%, P<0.01; HepG2: 12.6±1.1% vs. 7.6±1.3%,
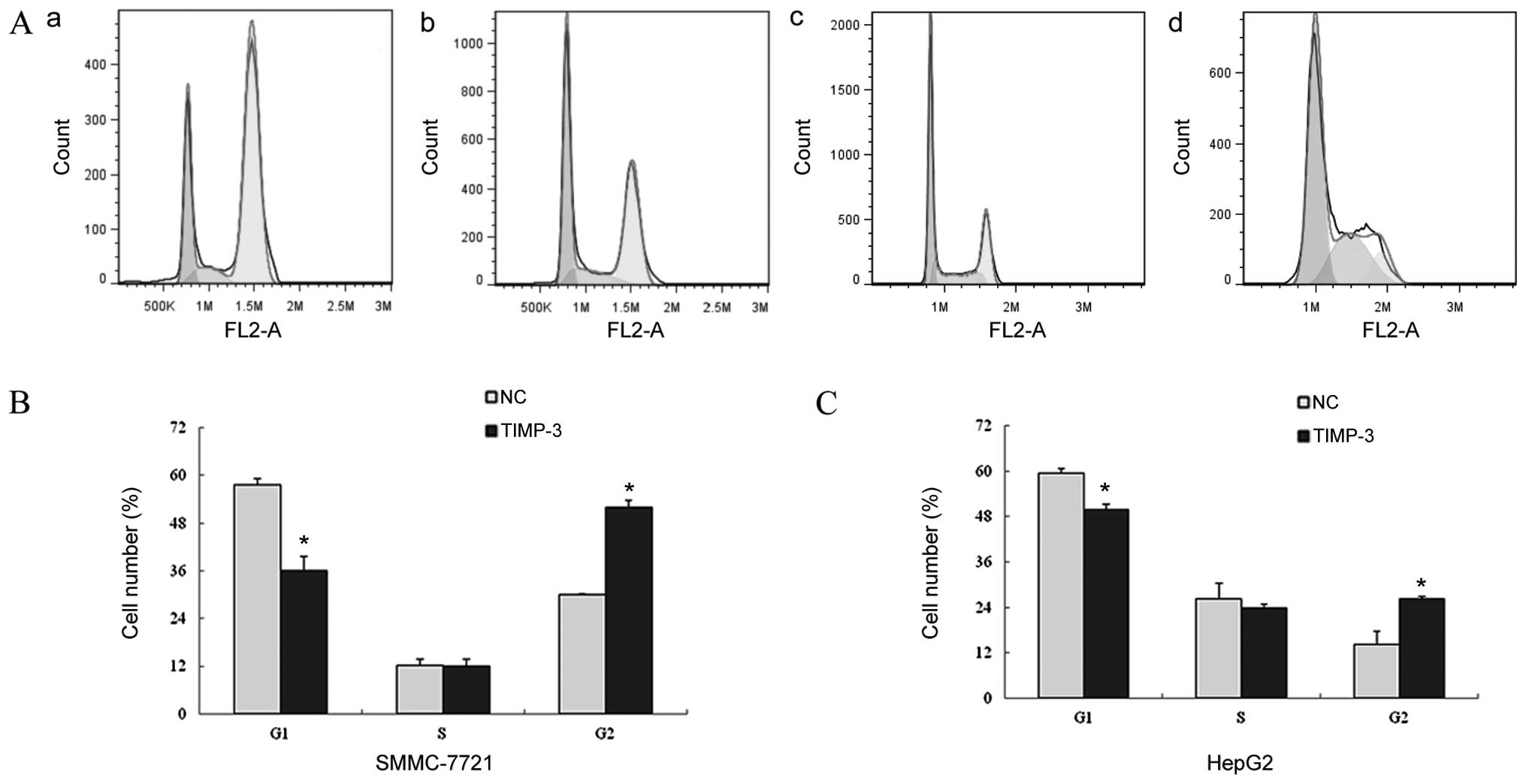
P<0.05). The flow cytometry results depicted in Fig. 6A, band c revealed an induction of
cell cycle arrest in TIMP-3 transfected cells, as evidenced by a
decreased mean number of G0/G1 cells compared
to that of the NC group (SMMC-7721: 36.08±3.53% vs. 57.72±1.53%,
P<0.05; HepG2: 49.82±1.46% vs. 59.47±1.27%, P<0.05) and an
increased mean number of G2/M cells compared to that of
the NC group (SMMC-7721: 51.91±1.71% vs. 30.11±0.20%, P<0.05;
HepG2: 26.27±0.54% vs. 14.15±3.61%, P<0.05).
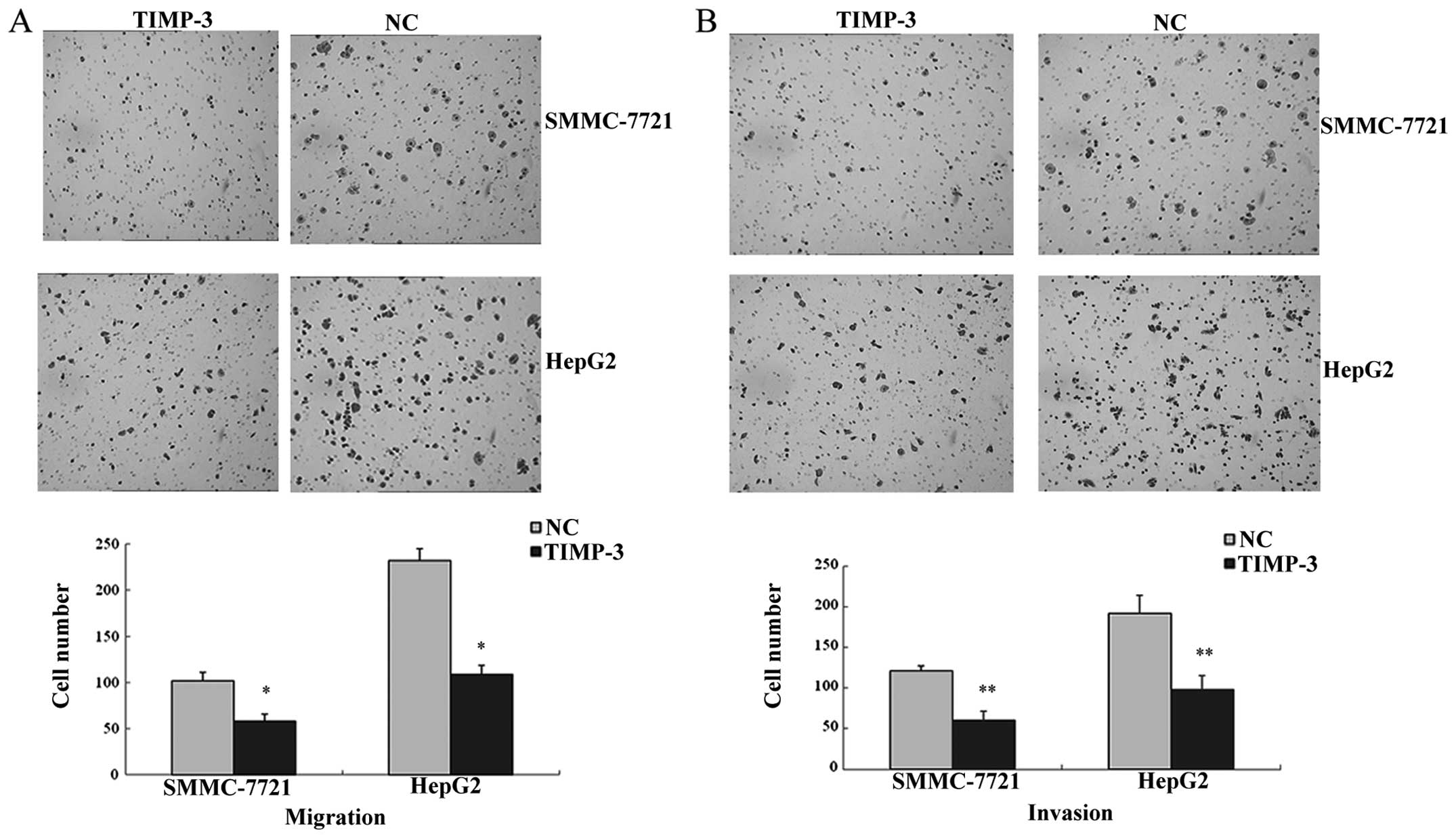
Effect of TIMP-3 on SMMC-7721 and HepG2
invasion and migration
The number of cells penetrating through the Matrigel
membrane reflects the cell invasion ability. As shown in Fig. 7A, the mean number of
TIMP-3-transfected invading cells was lower compared to that of the
NC group (SMMC-7721: 60±11 vs. 121±17, P<0.05; HepG2: 98±6 vs.
192±22, P<0.05). Results of the migration assay (Fig. 7B) revealed a lower mean number of
TIMP-3 transfected migrating cells compared to that of the NC group
(SMMC-7721: 53±8 vs. 102±10, P<0.01; HepG2: 109±9 vs. 232±13,
P<0.01).
Discussion
In this study, we characterized the methylation
status of the TIMP-3 promoter in HCC cells and have examined the
effects of TIMP-3 promoter methylation and TIMP-3 expression
on several parameters of HCC cell function. The results from our
study allow us to elucidate the biological role of TIMP-3 in HCC
and the molecular basis for the TIMP-3 effects on HCC cells. We
found decreased TIMP-3 expression in a significant number of tissue
samples from patients with HCC. Methylation status of the
TIMP-3 promoter was positive in three HCC cell lines (C3A,
HepG2, and Hep-3B), and the expression of TIMP-3 mRNA significantly
increased after demethylation with 5-Aza-CdR. These results
indicate that TIMP-3 expression in HCC is downregulated, and the
mechanism responsible for this downregulation, at least in part, is
methylation of the TIMP-3 promoter. These findings are
similar to those of other studies that revealed methylation of
TIMP-3 in brain, lung, kidney, colorectal, and other tumors
(7,30,31).
To determine whether TIMP-3 has antitumor activity
in HCC, we upregulated TIMP-3 expression in HCC cells via gene
transfection. After incubation with selective media, nearly all the
remaining cells expressed tGFP, as evidenced by the observed green
fluorescence. This indicates that the TIMP-3 plasmid was
transfected with relatively high efficiency. These transfected HCC
cells exhibited robust expression of TIMP-3 protein, which was
associated with decreased cell proliferation. Upregulation of
TIMP-3 expression results in significantly increased expression of
cyclin-dependent kinase inhibitor, p27 (30). It has been shown that p27, by
binding to and inhibiting cyclin-cyclin-dependent kinase complexes
directly involved in cell cycle control (31,32),
is a key inhibitor of cell proliferation. The loss of p27
expression promotes hepatocarcinogenesis through STAT3 signaling
activation (33).
Along with reduced cell proliferation, we also
observed a significant increase in apoptosis in TIMP-3 transfected
HCC cells. There are two major apoptotic pathways that cells can
follow: the extrinsic Fas death receptor-mediated pathway and the
intrinsic, mitochondrial-associated caspase-dependent pathway.
Binding of the Fas receptor to its ligand (Fas-L) can trigger a
signaling cascade via the intrinsic caspase-dependent pathway that
results in apoptosis (34,35), and it has been shown that TIMP-3 can
induce this intrinsic apoptotic pathway that is initiated via the
extrinsic Fas-associated mechanism (17). Another study demonstrated that
increased expression of TIMP-3, via silencing the APRIL gene, leads
to decreased expression of the antiapoptotic proteins, Bcl-2 and
Bcl-xL, and increased expression of the proapoptotic protein, Bax
(36). Additional evidence suggests
that TIMP-3-induced upregulation of p27 can result in increased
expression of apoptosis-related proteins (33).
We used flow cytometry analysis to evaluate the
effects of TIMP-3 on cell cycling. The decreased number of
G0/G1 and increased number G2/M
TIMP-3-transfected HCC cells suggest that TIMP-3 can induce
G2/M phase arrest, with a concomitant reduction in
cellular proliferation. G2/M arrest by TIMP-3 is linked
to p53, which controls the entry of cells into the mitosis phase at
the G2 checkpoint (37).
Cells prepare for mitosis by increasing the level of cyclin B1 and
activating Cdc2, the cyclin-dependent kinase required to enter
mitosis (36). The mechanism by
which p53 blocks entry into the mitosis phase partly involves
inhibition of Cdc2 by several transcription factors: p21,
14-3-3σ, and Gadd45. p21 can bind to and inhibit the
cyclin B1/Cdc2 complex, thereby inactivating Cdc2 (38). When the cyclin B1-Cdc2 complexes are
inactivated, the cells are arrested in the G2/M phase
(39).
Invasion and migration ability are typical features
of HCC and other malignant cell types. Compared to non-transfected
cells, we observed significantly lower numbers of transfected HCC
cells that were able to penetrate through the Matrigel and cross
the membrane to the lower chamber. These results indicate that
increased expression of TIMP-3 suppresses the HCC cell ability to
metastasize and invade surrounding tissues. As observed for other
kinds of malignant tumors, our study provides evidence that TIMP-3
has antitumor activity against HCC and that TIMP-3 may exert this
antitumor activity via multiple mechanisms.
Having observed the TIMP-3 protein expression and
suppressive function clinically and in vitro, we plan to
explore the molecular mechanism underlying the inhibitory effect of
TIMP-3 on HCC in our next project. TIMP-3 is considered a tumor
suppressor gene that may be capable of upregulating other
antitumorigenic genes. Thus, the precise downstream pathways by
which it mediates such effects are worthy of future studies.
In conclusion, TIMP-3 can act as a
tumor-suppressor gene in HCC. TIMP-3 is expressed at low levels in
HCC, but its expression can be activated by treatment with
demethylation agents, such as 5-Aza-CdR. TIMP-3 exerts its
anticancer effects on HCC via suppressing cell proliferation,
inducing apoptosis and G2/M phase arrest, and inhibiting
invasion and migration.
Acknowledgments
The study was supported by the National Natural
Science Foundation of China (no. 81270037), the Research Fund for
the Doctoral Program of Higher Education of China (no.
20114423110005) and Medical Scientific Research Foundation of
Guangdong Province (no. A2014550).
References
|
1
|
French SW, Lee J, Zhong J, Morgan TR,
Buslon V, Lungo W and French BA: Alcoholic liver disease -
Hepatocellular carcinoma transformation. J Gastrointest Oncol.
3:174–181. 2012.PubMed/NCBI
|
|
2
|
Mann CD, Neal CP, Garcea G, Manson MM,
Dennison AR and Berry DP: Prognostic molecular markers in
hepatocellular carcinoma: A systematic review. Eur J Cancer.
43:979–992. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Apte SS, Mattei MG and Olsen BR: Cloning
of the cDNA encoding human tissue inhibitor of metalloproteinases-3
(TIMP-3) and mapping of the TIMP3 gene to chromosome 22. Genomics.
19:86–90. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu WH, Yu S, Meng Q, Brew K and Woessner
JF Jr: TIMP-3 binds to sulfated glycosaminoglycans of the
extracellular matrix. J Biol Chem. 275:31226–31232. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anand-Apte B, Bao L, Smith R, Iwata K,
Olsen BR, Zetter B and Apte SS: A review of tissue inhibitor of
metalloproteinases-3 (TIMP-3) and experimental analysis of its
effect on primary tumor growth. Biochem Cell Biol. 74:853–862.
1996. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kallio JP, Hopkins-Donaldson S, Baker AH
and Kähäri VM: TIMP-3 promotes apoptosis in nonadherent small cell
lung carcinoma cells lacking functional death receptor pathway. Int
J Cancer. 128:991–996. 2011. View Article : Google Scholar
|
|
7
|
Bachman KE, Herman JG, Corn PG, Merlo A,
Costello JF, Cavenee WK, Baylin SB and Graff JR:
Methylation-associated silencing of the tissue inhibitor of
metalloproteinase-3 gene suggest a suppressor role in kidney,
brain, and other human cancers. Cancer Res. 59:798–802.
1999.PubMed/NCBI
|
|
8
|
Shen B, Chu ES, Zhao G, Man K, Wu CW,
Cheng JT, Li G, Nie Y, Lo CM, Teoh N, et al: PPARgamma inhibits
hepatocellular carcinoma metastases in vitro and in mice. Br J
Cancer. 106:1486–1494. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Almog N, Ma L, Schwager C, Brinkmann BG,
Beheshti A, Vajkoczy P, Folkman J, Hlatky L and Abdollahi A:
Consensus microRNAs governing the switch of dormant tumors to the
fast-growing angiogenic phenotype. PLoS One. 7:e440012012.
View Article : Google Scholar
|
|
10
|
Nagao Y, Hisaoka M, Matsuyama A, Kanemitsu
S, Hamada T, Fukuyama T, Nakano R, Uchiyama A, Kawamoto M,
Yamaguchi K, et al: Association of microRNA-21 expression with its
targets, PDCD4 and TIMP3, in pancreatic ductal adenocarcinoma. Mod
Pathol. 25:112–121. 2012. View Article : Google Scholar
|
|
11
|
Yu D, Zhou H, Xun Q, Xu X, Ling J and Hu
Y: microRNA-103 regulates the growth and invasion of endometrial
cancer cells through the downregulation of tissue inhibitor of
metalloproteinase 3. Oncol Lett. 3:1221–1226. 2012.PubMed/NCBI
|
|
12
|
Guo JX, Tao QS, Lou PR, Chen XC, Chen J
and Yuan GB: miR-181b as a potential molecular target for
anticancer therapy of gastric neoplasms. Asian Pac J Cancer Prev.
13:2263–2267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Panda H, Chuang TD, Luo X and Chegini N:
Endometrial miR-181a and miR-98 expression is altered during
transition from normal into cancerous state and target PGR, PGRMC1,
CYP19A1, DDX3X, and TIMP3. J Clin Endocrinol Metab. 97:E1316–E1326.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang C, Zhang J, Hao J, Shi Z, Wang Y,
Han L, Yu S, You Y, Jiang T, Wang J, et al: High level of
miR-221/222 confers increased cell invasion and poor prognosis in
glioma. J Transl Med. 10:1192012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lü GL, Wen JM, Xu JM, Zhang M, Xu RB and
Tian BL: Relationship between TIMP-3 expression and promoter
methylation of TIMP-3 gene in hepatocellular carcinoma. Zhonghua
Bing. Li Xue Za Zhi. 32:230–233. 2003.In Chinese.
|
|
16
|
Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ and
Kang GH: Aberrant CpG island hypermethylation along multistep
hepatocarcinogenesis. Am J Pathol. 163:1371–1378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bond M, Murphy G, Bennett MR, Newby AC and
Baker AH: Tissue inhibitor of metalloproteinase-3 induces a
Fas-associated death domain-dependent type II apoptotic pathway. J
Biol Chem. 277:13787–13795. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Masson D, Rioux-Leclercq N, Fergelot P,
Jouan F, Mottier S, Théoleyre S, Bach-Ngohou K, Patard JJ and Denis
MG: Loss of expression of TIMP3 in clear cell renal cell carcinoma.
Eur J Cancer. 46:1430–1437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoque MO, Begum S, Brait M, Jeronimo C,
Zahurak M, Ostrow KL, Rosenbaum E, Trock B, Westra WH, Schoenberg
M, et al: Tissue inhibitor of metalloproteinases-3 promoter
methylation is an independent prognostic factor for bladder cancer.
J Urol. 179:743–747. 2008. View Article : Google Scholar
|
|
20
|
Gu P, Xing X, Tänzer M, Röcken C, Weichert
W, Ivanauskas A, Pross M, Peitz U, Malfertheiner P, Schmid RM, et
al: Frequent loss of TIMP-3 expression in progression of esophageal
and gastric adenocarcinomas. Neoplasia. 10:563–572. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stetler-Stevenson WG: The role of matrix
metalloproteinases in tumor invasion, metastasis, and angiogenesis.
Surg Oncol Clin N Am. 10:383–392. x2001.PubMed/NCBI
|
|
22
|
Amălinei C, Căruntu ID, Giuşcă SE and
Bălan RA: Matrix metalloproteinases involvement in pathologic
conditions. Rom J Morphol Embryol. 51:215–228. 2010.
|
|
23
|
Jiao Y, Feng X, Zhan Y, Wang R, Zheng S,
Liu W and Zeng X: Matrix metalloproteinase-2 promotes αvβ3
integrin-mediated adhesion and migration of human melanoma cells by
cleaving fibronectin. PLoS One. 7:e415912012. View Article : Google Scholar
|
|
24
|
Chetty C, Vanamala SK, Gondi CS, Dinh DH,
Gujrati M and Rao JS: MMP-9 induces CD44 cleavage and CD44 mediated
cell migration in glioblastoma xenograft cells. Cell Signal.
24:549–559. 2012. View Article : Google Scholar :
|
|
25
|
Yu Q and Stamenkovic I: Cell
surface-localized matrix metalloproteinase-9 proteolytically
activates TGF-beta and promotes tumor invasion and angiogenesis.
Genes Dev. 14:163–176. 2000.PubMed/NCBI
|
|
26
|
Yu BF, Wu J, Zhang Y, Sung HW, Xie J and
Li RK: Ultrasound-targeted HSVtk and Timp3 gene delivery for
synergistically enhanced antitumor effects in hepatoma. Cancer Gene
Ther. 20:290–297. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hama R, Watanabe Y, Shinada K, Yamada Y,
Ogata Y, Yoshida Y, Tamura T, Hiraishi T, Oikawa R, Sakurai J, et
al: Characterization of DNA hypermethylation in two cases of
peritoneal mesothelioma. Tumour Biol. 33:2031–2040. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang H, Wang YS, Han G and Shi Y: TIMP-3
gene transfection suppresses invasive and metastatic capacity of
human hepatocarcinoma cell line HCC-7721. Hepatobiliary Pancreat
Dis Int. 6:487–491. 2007.PubMed/NCBI
|
|
29
|
Tian H, Huang ML, Liu KY, Jia ZB, Sun L,
Jiang SL, Liu W, McDonald Kinkaid HY, Wu J and Li RK: Inhibiting
matrix metalloproteinase by cell-based timp-3 gene transfer
effectively treats acute and chronic ischemic cardiomyopathy. Cell
Transplant. 21:1039–1053. 2012. View Article : Google Scholar
|
|
30
|
Wu DW, Tsai LH, Chen PM, Lee MC, Wang L,
Chen CY, Cheng YW and Lee H: Loss of TIMP-3 promotes tumor invasion
via elevated IL-6 production and predicts poor survival and relapse
in HPV-infected non-small cell lung cancer. Am J Pathol.
181:1796–1806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ju HX, An B, Okamoto Y, Shinjo K,
Kanemitsu Y, Komori K, Hirai T, Shimizu Y, Sano T, Sawaki A, et al:
Distinct profiles of epigenetic evolution between colorectal
cancers with and without metastasis. Am J Pathol. 178:1835–1846.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lacy ER, Filippov I, Lewis WS, Otieno S,
Xiao L, Weiss S, Hengst L and Kriwacki RW: p27 binds cyclin-CDK
complexes through a sequential mechanism involving binding-induced
protein folding. Nat Struct Mol Biol. 11:358–364. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo J, Ma Q, Zhou X, Fan P, Shan T and
Miao D: Inactivation of p27kip1 promotes chemical
hepatocarcinogenesis through enhancing inflammatory cytokine
secretion and STAT3 signaling activation. J Cell Physiol.
228:1967–1976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ding WX, Ni HM, DiFrancesca D, Stolz DB
and Yin XM: Bid-dependent generation of oxygen radicals promotes
death receptor activation-induced apoptosis in murine hepatocytes.
Hepatology. 40:403–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hsu CH, Peng KL, Kang ML, Chen YR, Yang
YC, Tsai CH, Chu CS, Jeng YM, Chen YT, Lin FM, et al: TET1
suppresses cancer invasion by activating the tissue inhibitors of
metalloproteinases. Cell Rep. 2:568–579. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang J, Ding W, Sun B, Jing R, Huang H,
Shi G and Wang H: Targeting of colorectal cancer growth,
metastasis, and anti-apoptosis in BALB/c nude mice via APRIL siRNA.
Mol Cell Biochem. 363:1–10. 2012. View Article : Google Scholar
|
|
37
|
Bunz F, Dutriaux A, Lengauer C, Waldman T,
Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B:
Requirement for p53 and p21 to sustain G arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Smits VA, Klompmaker R, Arnaud L, Rijksen
G, Nigg EA and Medema RH: Polo-like kinase-1 is a target of the DNA
damage checkpoint. Nat Cell Biol. 2:672–676. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schwartz GK and Shah MA: Targeting the
cell cycle: A new approach to cancer therapy. J Clin Oncol.
23:9408–9421. 2005. View Article : Google Scholar : PubMed/NCBI
|