Introduction
Breast cancer is the most common type of malignant
tumor and the most deadly cancer among women worldwide (1). In the Asia-Pacific region, the
incidence of breast cancer has increased in recent years and is
rising year by year (2). Although
sophisticated methods including surgery, chemotherapy, radiotherapy
and biotherapy are being used in breast cancer treatment, the
mortality rate of breast cancer patients is still high and this
trend is anticipated to continue (3). Among numerous causes, metastasis is
the major cause of cancer-related mortality in breast cancer
patients (4). Thus, the molecular
mechanism of metastasis is a key focus of breast cancer
research.
Epithelial-mesenchymal transition (EMT) which plays
an important role in cancer metastasis, is an embryonic
transdifferentiation process converting adjacent epithelial cells
with polarity to mesenchymal cells. (5,6).
During the EMT process, cells lose epithelial markers, such as
E-cadherin and β-catenin, and gain increased levels of mesenchymal
markers, such as N-cadherin and vimentin. Once EMT occurs, the
intercellular adhesion complexes in epithelial cells are disrupted
and at the same time, the apico-basal polarity is lost. The basal
cytoskeleton disorganizes and the basement membrane is broken down
by proteases. EMT induces cells to achieve migratory and invasive
properties which allow them to migrate through the extracellular
matrix (7). TGFβ/Smads,
Wnt/β-catenin and Notch pathways induce EMT by targeting
transcription factors including Snail, Slug, Zeb1/2 and Twist
(8). In addition, receptor tyrosine
kinases (RTKs), including fibroblast growth factor (FGF) and
epidermal growth factor (EGF), can also promote the migration and
invasion of cancer cells and induce EMT in malignancies.
FOXO1 is one of the members of the FOXO subfamily of
forkhead transcription factors (9)
and participates in various cellular events such as cell cycle and
apoptosis control (10). As a
tumor-suppressor, FOXO1 expression is low in endome-trioid
endometrial cancer cells (11) and
non-small cell lung cancer (12)
and inhibits prostate cancer cell migration and invasion (13). Recently, FOXO1 was proven to play
key roles in drug resistance in tongue (14), non-small cell lung (15), ovarian (16) and prostate cancer (17).
FYN, a 59-kDa protein, is one of the members of the
Src family of kinases (SFKs) which are types of non-receptor
tyrosine kinases (NRTKs) overexpressed in various types of cancers
(18). FYN contains four domains
including SH1, SH2, SH3 and SH4 (19), which is similar with other members
of the SFKs such as Src, Lyn, Lck and Yes (20). The SH1 domain is the C-terminal
domain with kinase activity, SH2 is a structurally conserved region
taking part in protein interactions, which can bind to
phosphotyrosine residues with the pYEEI sequence, while another
highly conserved domain SH3 can recognize PXXP-like sequences. The
N-terminal domain SH4 is associated with the cell membrane through
palmitoylation or myristoylation (21). FYN plays important roles in
Alzheimer's disease by regulating Aβ production, mediating
Aβ-induced synaptic deficits and neurotoxicity and inducing
tyrosine phosphorylation of tau. In addition, FYN can regulate T
cell development and T cell receptor signal transduction. In
addition, FYN is overexpressed in prostate cancer (22), and participates in cell growth,
migration and apoptosis as a downstream target of the PI3K/AKT
pathway (23). Recently, FYN was
found to play a key role in the Wnt receptor Frizzled2 which drives
EMT (24), and to predict early
recurrence in patients treated with endocrine therapy as an
important molecule in tamoxifen resistance (25). However, the role and mechanism of
FYN in breast cancer progression are still unclear.
In the present study, we showed that FYN was
overexpressed in breast cancer and promoted breast cancer cell
proliferation, migration and invasion. Furthermore, FYN was
transcriptionally inhibited by FOXO1 and mediated FGF2-induced EMT
through both the PI3K/AKT and ERK/MAPK pathways.
Materials and methods
Cell culture
MCF10A, MDA-MB-231, MDA-MB-435, MCF-7 and T47D cell
lines were purchased from the Cell Bank of the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China).
MDA-MB-231 and MDA-MB-435 cell lines were cultured in RPMI-1640
medium with 10% fetal bovine serum (FBS) (both from Gibco, Grand
Island, NY, USA) while MCF-7 and T47D cell lines were supplemented
with Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT,
USA) with 10% FBS. MCF10A cells were maintained in DMEM/F12 medium
(HyClone) supplemented with 5% horse serum (Gibco), 10 mg/ml
insulin, 0.5 mg/ml hydrocortisone (both from Sigma-Aldrich, St.
Louis, MO, USA), 20 ng/ml EGF (R&D Systems, Minneapolis, MN,
USA), and 100 ng/ml cholera toxin (Sigma-Aldrich). All of the cell
lines were supplemented with 1% penicillin/streptomycin (Gibco), in
a 5% CO2 and humidified atmosphere at 37°C.
Antibodies, reagents and
transfection
The anti-ERK1/2, anti-phospho-ERK1/2, anti-AKT,
anti-phospho-AKT, anti-FAK, anti-phospho-FAK and anti-Snail
antibodies were purchased from Cell Signaling Technology (Beverly,
MA, USA). The anti-E-cadherin, anti-cytokeratin 19,
anti-N-cadherin, anti-Slug, anti-FYN, anti-vimentin and anti-nestin
antibodies were purchased from Abcam (Cambridge, MA, USA). The
siRNAs targeting FYN or the control were obtained from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). FGF2 was purchased from
R&D Systems. PP1, BGJ398, AZD8931, LY294002 and PD98059 were
acquired from Selleck Chemicals (Houston, TX, USA).
Cells were seeded into 6-well plates. The cells were
then transfected with the siRNA or plasmid using FuGENE HD (Roche,
Mannheim, Germany) according to the manufacturer's
instructions.
Real-time quantitative PCR (RT-qPCR)
The total RNA was extracted with RNA Plus (Takara
Bio, Inc., Otsu, Shiga, Japan) according to the manufacturer's
recommendations. Then, the cDNA was synthesized from RNA by
PrimerScript RT Reagent kit (Takara) according to the
manufacturer's protocol. RT-qPCR was carried out using a SYBR-Green
PCR mix (Takara) on a Bio-Rad CFX96 Real-Time PCR system.
Quantification of the target gene expression was calculated by
normalizing the averaged Ct value of the target gene to the
averaged Ct value of the housekeeping gene β-actin (ΔCt), and
determined as 2−ΔCt.
Western blotting
Cells were lysed in RIPA lysis buffer containing
protease inhibitor cocktail. Protein lysates were resolved by 10%
SDS/PAGE and the separated proteins were transferred onto a
polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA,
USA). Non-specific binding was blocked by 5% skimmed milk in
Tris-buffered saline and Tween-20 (TBST) for 1 h at room
temperature. The membranes were incubated with the primary antibody
at an appropriate dilution overnight at 4°C and then incubated with
a suitable HRP-conjugated anti-rabbit or anti-mouse secondary
antibody at an appropriate dilution at room temperature for 1 h.
The blots were visualized with enhanced chemiluminescence (ECL)
reagent (Millipore).
Cell proliferation assay
The MTT assay was used to evaluate the ability of
cell proliferation. In brief, 5x103 cells were seeded
into 96-well plates/well. After incubation for the indicated time,
the cells were incubated with 10 µl MTT (0.5 mg/ml;
Sigma-Aldrich) at 37°C for 4 h. The medium was then removed, and
precipitated formazan was dissolved in 150 µl dimethyl
sulfoxide (DMSO). The absorbance at 570 nm was detected using a
microplate autoreader.
Transwell assay
The Transwell assay (BD Biosciences, San Diego, CA,
USA) was used to evaluate the ability of cell migration and
invasion. Briefly, relevant cells were seeded into the top chambers
with or without Matrigel (BD). Medium without serum was added to
the top chambers, while complete medium was added to the bottom
wells. The cells were fixed with 4% neutral formalin and stained
with crystal violet after incubation. The number of migrated cells
was counted under a microscope in five predetermined fields.
Immunofluorescence
Cells were seeded onto glass coverslips in 12-well
plates. After adherence, the cells were washed three times with
phosphate-buffered saline (PBS), then fixed with 4% neutral
formalin and permeabilized with 0.1% Triton X-100. Non-specific
binding was blocked by 3% BSA in PBS for 1 h at room temperature
and then the membranes were incubated with primary antibody at an
appropriate dilution overnight at 4°C. After being washed with PBS,
the membranes were incubated with a suitable FITC-conjugated
anti-rabbit or anti-mouse secondary antibody (Cell Signaling
Technology) at an appropriate dilution at room temperature for 1 h,
then stained with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime,
Jiangsu, China) and glass coverslips were observed with a
fluorescence microscope.
Luciferase assay
Cells were transiently transfected with luciferase
reporter constructs, mixtures of expression plasmids encoding
FOXO1, along with the pRL-TK vector (Promega, Madison, WI, USA).
Luciferase activity was measured after 24 h by the Dual Luciferase
Reporter Assay (Promega).
ChIP assay
ChIP assay was performed using kits from Active
Motif (Carlsbad, CA, USA). Cells were fixed in formaldehyde. Cells
were lysed and nuclei were pelleted by centrifugation. Nuclei were
resuspended and sheared using a sonicator (Misonix, Inc.,
Farmingdale, NY, USA). Sheared chromatin was immunoprecipitated
with the anti-FOXO1 or control IgG antibody. The cross-links were
reversed overnight at 65°C and deproteinated with 20 g/ml
proteinase K. The DNA samples were semi-quantified by PCR using the
following primer sets containing the FOXO1 binding region on the
FYN promoter: forward, 5′-CCTTGTTTACTTC-3′ and reverse,
5′-TGTTGTTTACNTT-3′.
Statistical analysis
Data are presented as the mean ± SEM, and were
analyzed by one-way analysis of variance (ANOVA). All experiments
were carried out at least three times independently to confirm the
conclusions. Statistical significance was set at P<0.05.
Results
FYN is overexpressed in breast cancer and
promotes cell proliferation, migration and invasion
Previous studies indicate that FYN is overexpressed
in prostate cancer (22), and
induces proliferation and migration in the HEK 293T cell line
(26). To investigate the role of
FYN in breast cancer, we determined the expression of FYN in breast
cancer cell lines and the normal breast epithelial MCF10A cells by
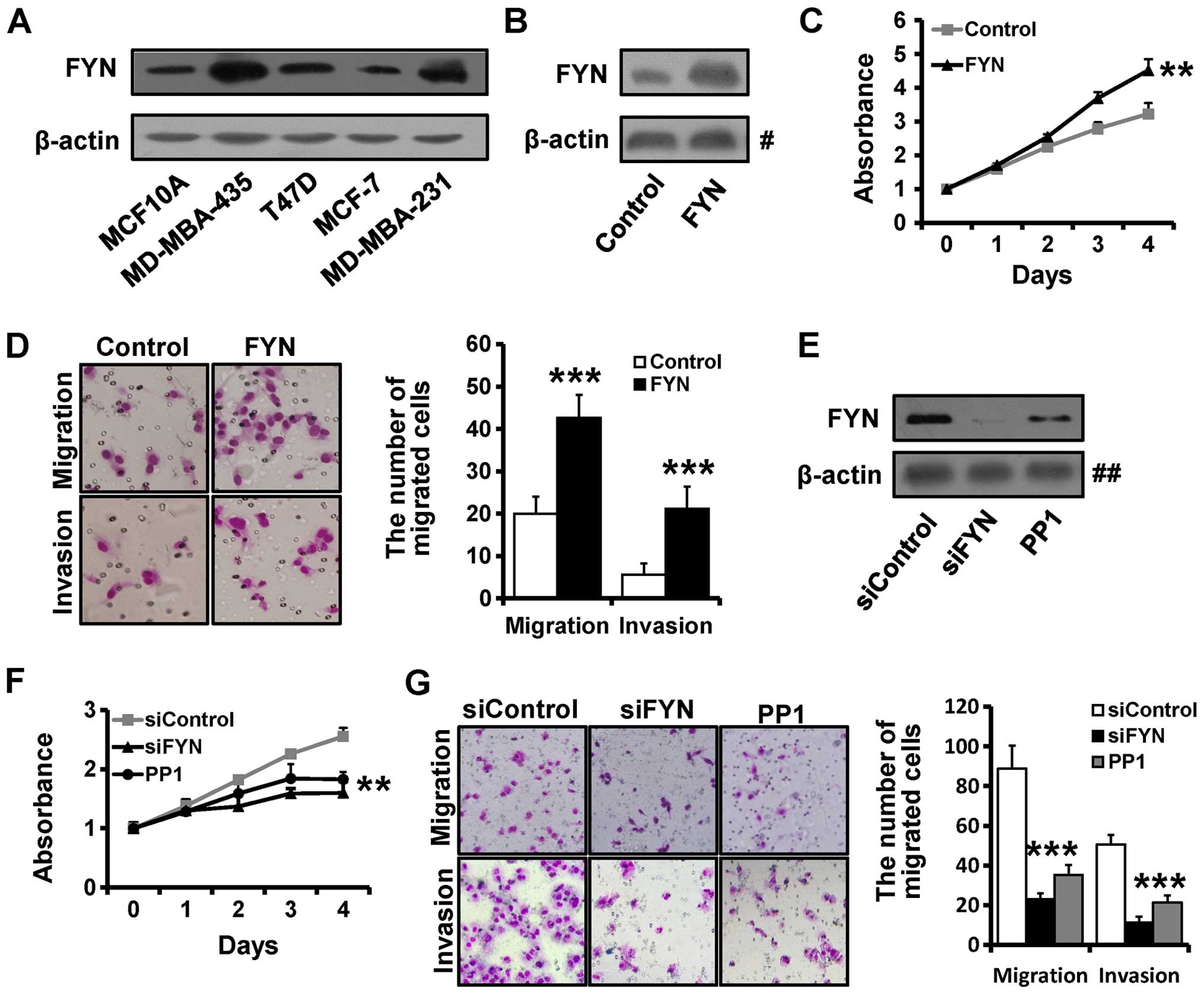
western blotting (Fig. 1A). The
results showed that FYN expression was higher in the MDA-MB-231 and
MDA-MB-435 cell lines and in contrast, it was lower in the MCF-7 an
MCF10A cell lines.
To further investigate the role of FYN in breast
cancer development and progression, we overexpressed FYN by
transfecting the FYN plasmid into the MCF10A cell line. We found
that FYN expression was significantly higher in the FYN-transfected
MCF10A cells by western blotting (Fig.
1B). We next examined the migration and invasion abilities of
the the FYN-overexpressed and the control MCF10A cells. MTT
analysis showed that MCF10A-FYN cells grew much faster than the
control cells (Fig. 1C). We also
observed that the number of migrated cells was much higher in the
FYN-transfected MCF10A cells than that in the control cells
(Fig. 1D).
Conversely, to determine the effect of decreased FYN
expression on breast cancer progression, we silenced FYN with
siRNAs targeting FYN or inhibited FYN with FYN inhibitor PP1 in the
MBA-MB-231 cell line. The FYN expression was significantly
decreased in the FYN siRNA-transfected MDA-MB-231 cells when
compared to the level in the control cells (Fig. 1E). The MTT assay showed that
depletion of FYN inhibited cell proliferation in the MDA-MB-231
cells (Fig. 1F). The Transwell
assay showed that depletion of FYN inhibited breast cancer cell
migration and invasion in the MDA-MB-231 cells (Fig. 1G), suggesting that FYN promotes
breast cancer cell proliferation, migration and invasion.
FYN induces EMT in the breast cancer
cells
As previously described, FYN promotes breast cancer
cell migration and invasion. We therefore speculated that FYN may
be involved in the process of EMT consequently promoting breast
cancer metastasis. To test this contention, we next investigated
the role of FYN in breast cancer cell EMT. EMT results in loss of
epithelial markers and concomitant acquisition of mesenchymal
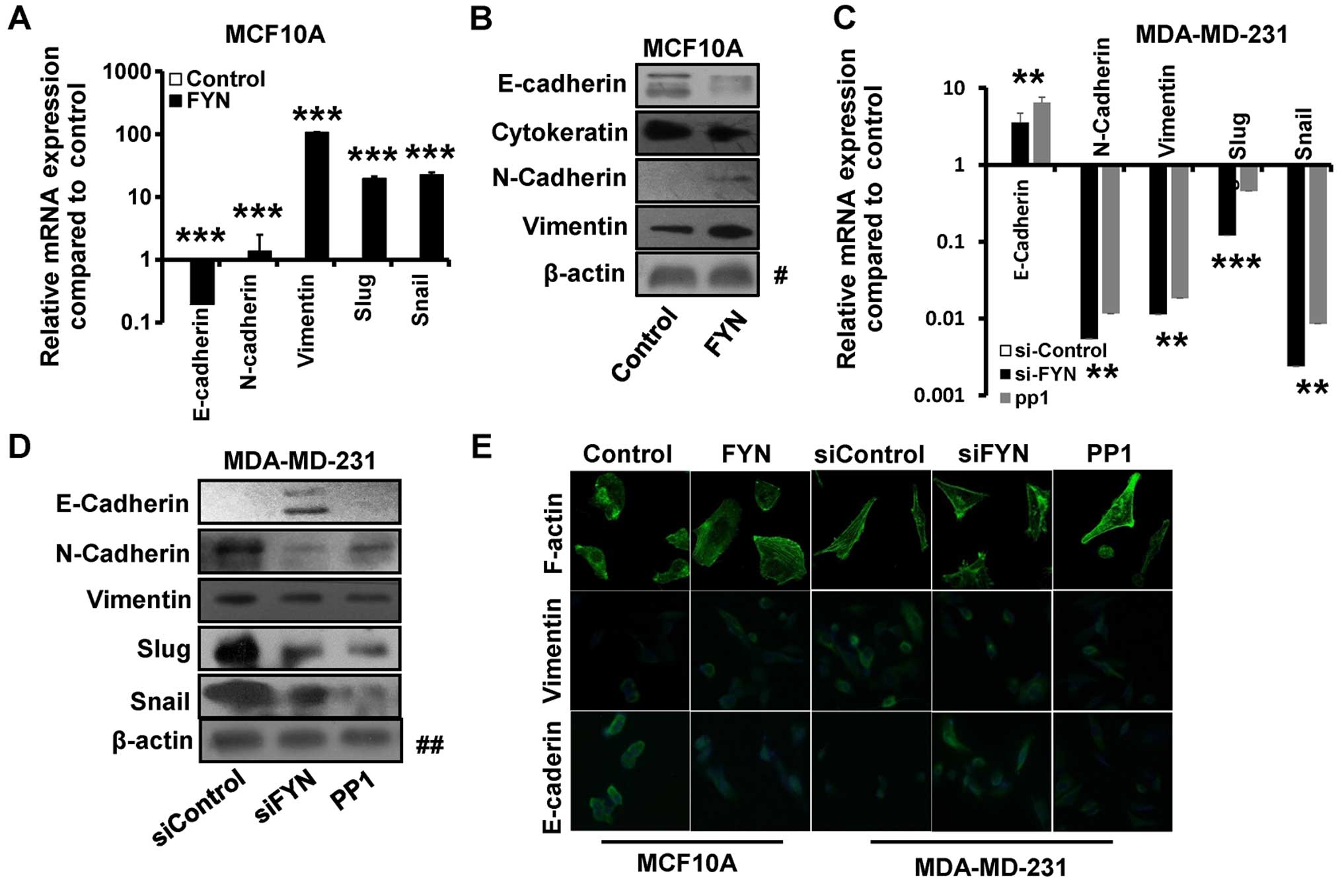
markers. Our results showed that the mRNA expression of E-cadherin
was decreased, whereas the expression of N-cadherin and vimentin
was significantly increased in the FYN-transfected MCF10A cells by
RT-qPCR (Fig. 2A). In addition, the
expression of EMT-related transcription factors Slug and Snail was
significantly elevated in the FYN-overexpressed MCF10A cells when
compared with that of the control cells (Fig. 2A). Furthermore, overexpression of
FYN in the MCF10A cells led to decreased expression of epithelial
markers E-cadherin and cytokeratin 19 and increased the expression
of mesenchymal markers N-cadherin and vimentin by western blotting
(Fig. 2B).
In contrast, the expression of mesenchymal markers
vimentin and N-cadherin was downregulated and the expression of
epithelial marker E-cadherin was upregulated in the FYN-depleted
MDA-MB-231 cells by RT-qPCR (Fig.
2C) and western blotting (Fig.
2D). In addition, the expression of EMT-related transcription
factors Slug and Snail was significantly reduced in the
FYN-depleted MDA-MB-231 cells compared with that of the control
cells by RT-qPCR (Fig. 2C) and
western blotting (Fig. 2D).
Immunofluorescence staining also revealed that the E-cadherin
expression was decreased in the FYN-overexpressing MCF10A cells and
increased in the FYN-depleted MDA-MB-231 cells, whereas the
vimentin expression was increased in the FYN-overexpressing MCF10A
cells and decreased in the FYN-depleted MDA-MB-231 cells (Fig. 2E). Moreover, EGF-mediated remodeling
of the cytoskeleton from cortical actin to stress fibers was noted,
as determined by phalloidin staining (Fig. 2E).
FYN mediates EGF and FGF2-induced
EMT
Previous studies have indicated that FYN is a
downstream target of the TGFTβ/Smad pathway and can be upregulated
by TGFβ-1 (27). In addition, the
TGFβ/Smad pathway, the receptor tyrosine kinase pathway can also
promote cell proliferation, migration and induce EMT. Thus, EGF and
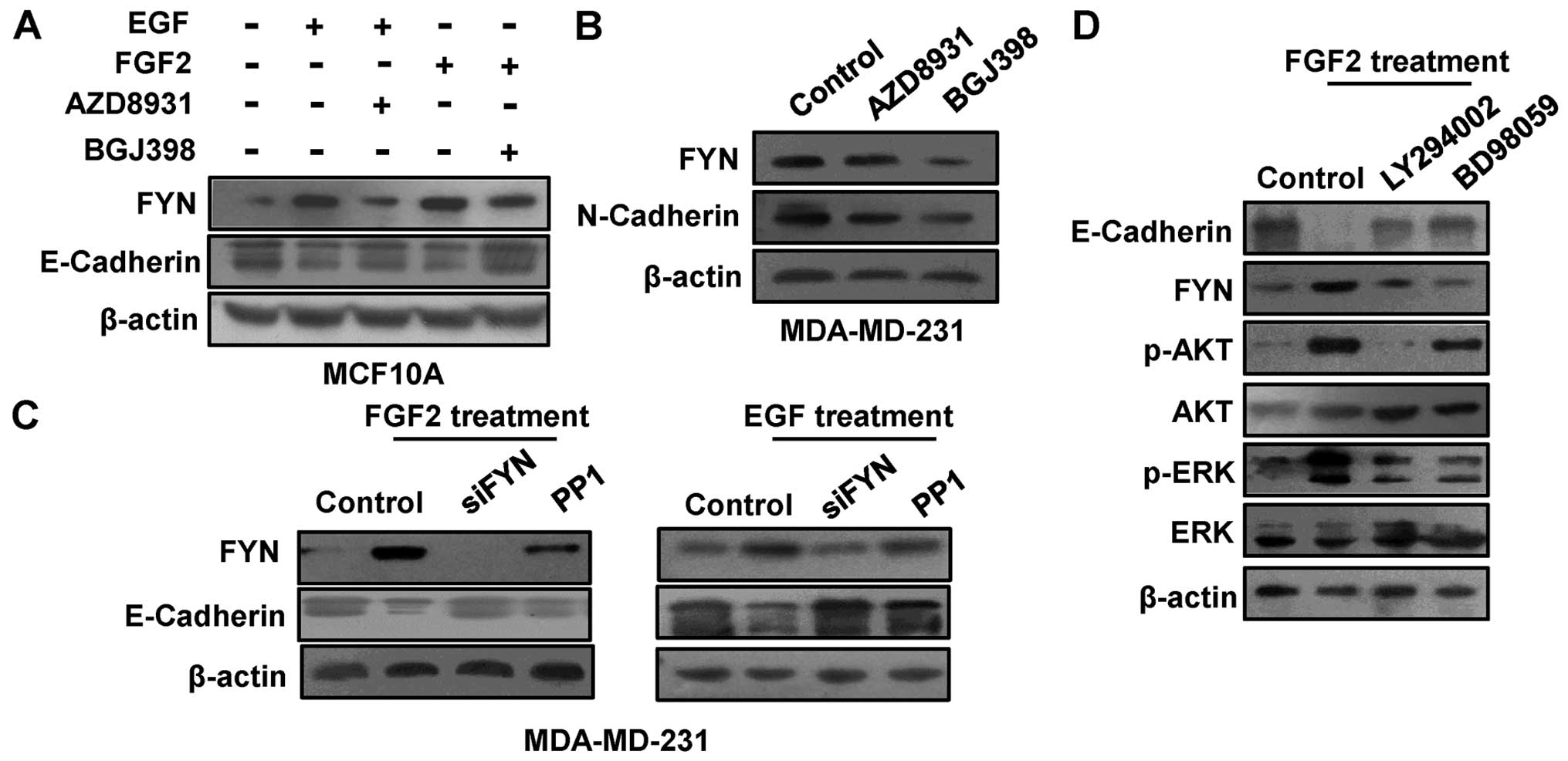
FGF2, and their inhibitors AZD8931 and BGJ398 were used to
determine whether FYN is a downstream target of the receptor
tyrosine kinase pathway. After treatment with EGF or FGF2 for 2
days, the expression of FYN was increased in the MCF10A cell line,
while the FYN expression was decreased after treatment with their
inhibitors (AZD8931 and BGJ398) (Fig.
3A). Furthermore, FYN expression was decreased in the
MDA-MB-231 cells after treatment with AZD8931 or BJ398 (Fig. 3B). Moreover, the FYN expression was
negatively related to the E-cadherin expression in the MCF10A cells
(Fig. 3A), whereas it was
positively related to N-cadherin expression in the MDA-MB-231 cells
(Fig. 3B). To further investigate
the role of FYN in FGF2 and EGF-induced EMT, we depleted the FYN
expression by FYN siRNA and inhibitor PP1 after EGF or FGF2
treatment. The results showed that FYN depletion reduced the EGF
and FGF2 induced-EMT (Fig. 3C). The
PI3K/AKT and ERK/MAPK pathways are involved in FGF2-induced EMT
(28). To further investigate the
role of FYN in the PI3K/AKT and MAPK/ERK pathways, we examined the
FYN expression after treatment with PI3K inhibitor LY294002 or MEK
1/2 inhibitor PD98059. The results showed that FYN expression was
decreased after treatment with LY294002 or PD98059 (Fig. 3D).
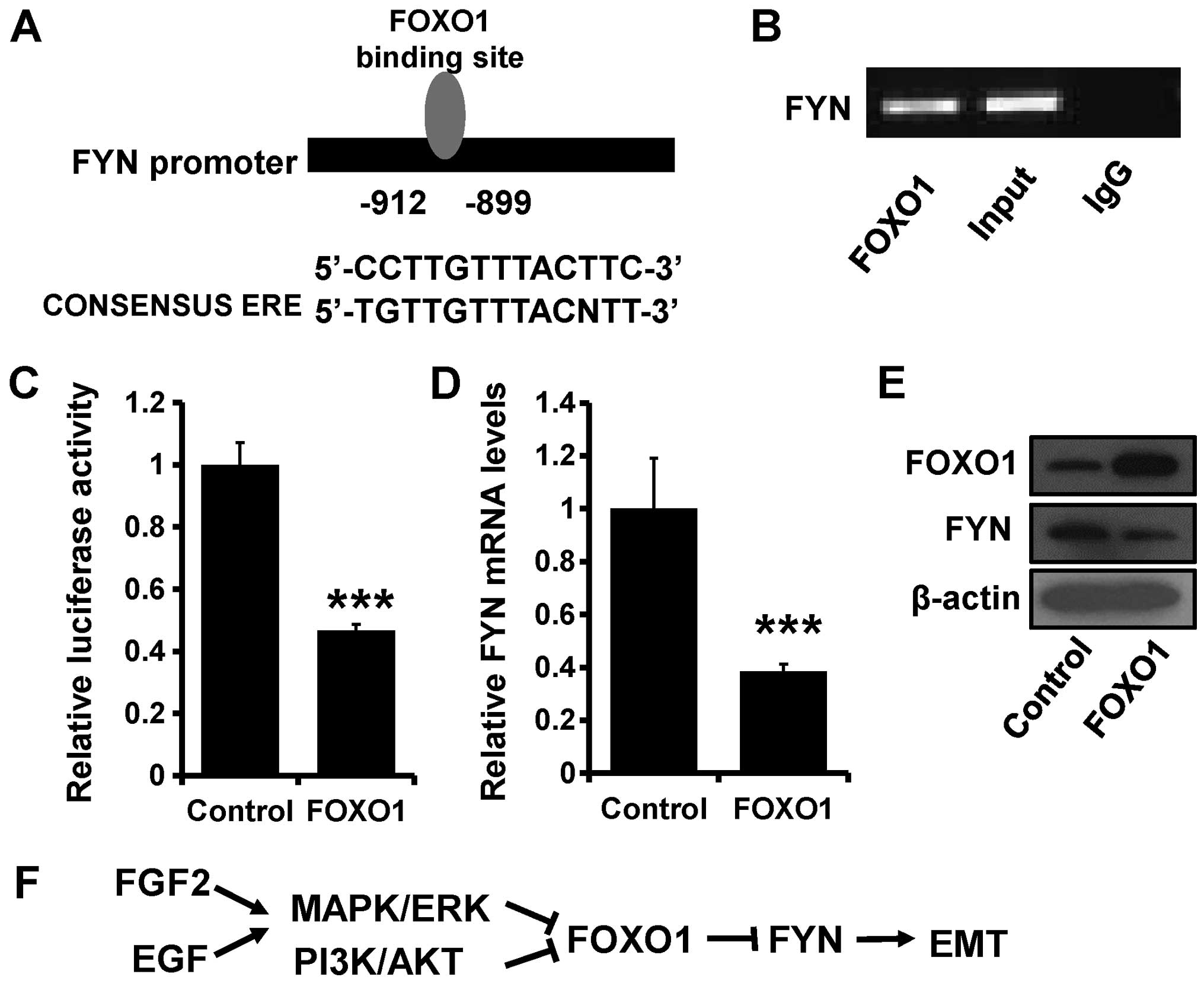
FOXO1 transcriptionally inhibits FYN
Transcription factor FOXO1 can suppress tumor
progression, while PI3K/AKT or ERK/MAPK pathway activation can
suppress FOXO1 expression (29). We
designed a FYN promoter containing the FOXO1 binding site (Fig. 4A) for luciferase assay to explore
the relationship between FYN and FOXO1. ChIP and luciferase assay
confirmed that the FOXO1 protein can combine with FYN DNA fragments
(Fig. 4B) and inhibit FYN
transcription (Fig. 4C).
Furthermore, FOXO1 transfection decreased the expression of FYN at
both the mRNA (Fig. 4D) and protein
levels (Fig. 4E). Thus, we can
conclude that FGF2 and EGF can decrease FOXO1 expression through
the PI3K/AKT and ERK/MAPK pathways, which suppress the
transcriptional inhibition of FYN and induce EMT (Fig. 4F).
Discussion
Previous studies have found that FYN is
overexpressed in a variety of solid tumors and hematologic
malignancies, such as prostate cancer, squamous cell carcinoma of
the head and neck, melanoma and chronic myeloid leukemia (22,30,31).
Consistent with these studies, we found that FYN was upregulated in
breast cancer cells when compared to the levels in normal breast
cells. Moreover, as a member of the SRC family, FYN overexpression
in normal cells was capable of changing cell morphology. For
instance, overexpression of FYN in normal fibroblast NIH3T3 cells
induced anchorage-independent growth and morphological
transformation, even in the fully tumorigenic phenotype (32). In the present study, we observed
that FYN was highly expressed in the high-invasive breast cancer
cell lines (MDA-MB-231 and MDA-MB-435) than in the low-invasive
breast cancer cell lines (MCF-7 and T47D) and in the normal breast
mammary cell line MCF10A. In addition, it has been reported that
FYN plays an important role in integrin-mediated cell adhesion and
migration (33). Depletion of FYN
reduced cell migration and invasion in 293T cells (26). Consistent with previous studies, we
observed that depletion of FYN in the highly invasive MDA-MB-231
cells reduced cell migration and invasion. Furthermore,
over-expression of FYN increased cell motility in the low-invasive
MCF10A cell line, suggesting that FYN promotes breast cancer cell
migration and invasion.
E-cadherin plays a crucial role in epithelial
cell-cell adhesion and loss of E-cadherin can be regarded as a
hallmark of EMT (34). Interferon-γ
was found to reduce the expression of EMT epithelial marker
E-cadherin in a FYN-dependent manner, which can be inhibited by Src
kinase selective inhibitor PP1 (35). TGF-β1 was proven to repress
E-cadherin expression in A549 cells through the Fyn-p38-Snail
signaling pathway (36). In the
present study, we also observed that E-cadherin was repressed in
FYN-overexpressing MCF10A cells and was elevated in the
FYN-depleted MDA-MB-231 cells. Snail and Slug, as EMT-related
transcription factors and EMT inducers, were able to repress
E-cadherin expression (37). We
found that FYN upregulated Snail and Slug expression in the breast
cancer cells. In addition to E-cadherin, Slug also induced the
expression of an important EMT mesenchymal marker vimentin
(38). Our results indicated that
overexpression of FYN induces EMT processes in human breast cancer
cells, including upregulation of mesenchymal markers (N-cadherin
and vimentin) and downregulation of epithelial markers (E-cadherin
and cytokeratin 19). These studies indicated that FYN promotes
breast cancer cell proliferation, migration and invasion through
induction of EMT.
Previous study have indicated that the PI3K/AKT and
ERK/MAPK pathways are regulated by FGF2 (28) and Ras induces the expression of FYN
through the PI3K/AKT signaling pathway (23). In the present study, we found that
FYN was the downstream target of the receptor tyrosine kinase
pathway. After FGF2 stimulation, the expression of FYN and the
phosphorylation level of AKT and ERK were increased, while the
expression of E-cadherin was decreased. Moreover, the expression of
FYN and E-cadherin was decreased after treatment with the PI3K or
MEK1/2 inhibitor, suggesting that FGF2 regulates FYN expression
through both the PI3K/AKT and ERK/MAPK pathways. In addition, FYN
was found to be able to induce the activity of PI3K/AKT (26) and ERK/MAPK (39), which indicates the existence of a
crosstalk between PI3K/AKT, ERK/MAPK and FYN.
Tumor suppressor FOXO1 is in the downstream of the
PI3K/AKT and MAPK/ERK pathways and it can be activated by PI3K/AKT
and MAPK/ERK inhibition (29). We
found that FOXO1 decreased the expression of FYN by inhibiting FYN
transcription. FOXO1 was previously found to inhibit Runx2
transcriptional activity and prostate cancer cell migration and
invasion (13). In the present
study, FOXO1 regulated FGF2-inducing EMT in breast cancer cells by
transcriptional inhibition of FYN.
In addition to cell proliferation, migration and
invasion, FYN is also involved in apoptosis and drug resistance,
however, the mechanisms of these functions of FYN are still
unclear, and more studies must be performed to solve these issues.
As research progresses, FYN may be regarded as a novel biomarker
for cancer prognosis and diagnosis.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (nos. 81372843, 81472472 and
81502518), the National Science and Technology Support Program (no.
2015BAI12B15), and the Tianjin Municipal Natural Science Foundation
(no. 13JCYBJC21800).
References
|
1
|
Ha R, Chow D and Wynn R: Global trend in
breast cancer imaging research 1992–2012: Bibliometric study. AJR
Am J Roentgenol. 202:696–697. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Youlden DR, Cramb SM, Yip CH and Baade PD:
Incidence and mortality of female breast cancer in the Asia-Pacific
region. Cancer Biol Med. 11:101–115. 2014.PubMed/NCBI
|
|
3
|
Shi XJ, Au WW, Wu KS, Chen LX and Lin K:
Mortality characteristics and prediction of female breast cancer in
China from 1991 to 2011. Asian Pac J Cancer Prev. 15:2785–2791.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang J, Liang Q, Lei Y, Yao M, Li L, Gao
X, Feng J, Zhang Y, Gao H, Liu DX, et al: SOX4 induces
epithelial-mesenchymal transition and contributes to breast cancer
progression. Cancer Res. 72:4597–4608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ansieau S: EMT in breast cancer stem cell
generation. Cancer Lett. 338:63–68. 2013. View Article : Google Scholar
|
|
6
|
Sarkar FH, Li Y, Wang Z and Kong D:
Pancreatic cancer stem cells and EMT in drug resistance and
metastasis. Minerva Chir. 64:489–500. 2009.PubMed/NCBI
|
|
7
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
The importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Anderson MJ, Viars CS, Czekay S, Cavenee
WK and Arden KC: Cloning and characterization of three human
forkhead genes that comprise an FKHR-like gene subfamily. Genomics.
47:187–199. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gross DN, van den Heuvel AP and Birnbaum
MJ: The role of FoxO in the regulation of metabolism. Oncogene.
27:2320–2336. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goto T, Takano M, Albergaria A, Briese J,
Pomeranz KM, Cloke B, Fusi L, Feroze-Zaidi F, Maywald N, Sajin M,
et al: Mechanism and functional consequences of loss of FOXO1
expression in endometrioid endometrial cancer cells. Oncogene.
27:9–19. 2008. View Article : Google Scholar
|
|
12
|
Maekawa T, Maniwa Y, Doi T, Nishio W,
Yoshimura M, Ohbayashi C, Hayashi Y and Okita Y: Expression and
localization of FOXO1 in non-small cell lung cancer. Oncol Rep.
22:57–64. 2009.PubMed/NCBI
|
|
13
|
Zhang H, Pan Y, Zheng L, Choe C, Lindgren
B, Jensen ED, Westendorf JJ, Cheng L and Huang H: FOXO1 inhibits
Runx2 transcriptional activity and prostate cancer cell migration
and invasion. Cancer Res. 71:3257–3267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng G, Jia X, Peng C, Deng Y, Yin J,
Zhang Z, Li N, Deng M, Liu X, Liu H, et al: The
miR-491-3p/mTORC2/FOXO1 regulatory loop modulates chemo-sensitivity
in human tongue cancer. Oncotarget. 6:6931–6943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu ZH, Shun WW, Hang JB, Gao BL and Hu JA:
Posttranslational modifications of FOXO1 regulate epidermal growth
factor receptor tyrosine kinase inhibitor resistance for non-small
cell lung cancer cells. Tumour Biol. 36:5485–5495. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Yang H, Li W, Xu H, Yang X and Gan
L: Thioredoxin 1 upregulates FOXO1 transcriptional activity in drug
resistance in ovarian cancer cells. Biochim Biophys Acta.
1852:395–405. 2015. View Article : Google Scholar
|
|
17
|
Duan X, Kong Z, Liu Y, Zeng Z, Li S, Wu W,
Ji W, Yang B, Zhao Z and Zeng G: β-Arrestin2 contributes to cell
viability and proliferation via the down-regulation of FOXO1 in
castration-resistant prostate cancer. J Cell Physiol.
230:2371–2381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang YM, Kung HJ and Evans CP:
Nonreceptor tyrosine kinases in prostate cancer. Neoplasia.
9:90–100. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sato I, Obata Y, Kasahara K, Nakayama Y,
Fukumoto Y, Yamasaki T, Yokoyama KK, Saito T and Yamaguchi N:
Differential trafficking of Src, Lyn, Yes and Fyn is specified by
the state of palmitoylation in the SH4 domain. J Cell Sci.
122:965–975. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Szalmás A, Gyöngyösi E, Ferenczi A, László
B, Karosi T, Csomor P, Gergely L, Veress G and Kónya J: Activation
of Src, Fyn and Yes non-receptor tyrosine kinases in keratinocytes
expressing human papillomavirus (HPV) type 16 E7 oncoprotein. Virol
J. 10:792013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu X, Hu X, Song L, An L, Duan M, Chen S
and Zhao S: SH2 domain is crucial for function of Fyn in neuronal
migration and cortical lamination. BMB Rep. 48:97–102. 2015.
View Article : Google Scholar :
|
|
22
|
Posadas EM, Al-Ahmadie H, Robinson VL,
Jagadeeswaran R, Otto K, Kasza KE, Tretiakov M, Siddiqui J, Pienta
KJ, Stadler WM, et al: FYN is overexpressed in human prostate
cancer. BJU Int. 103:171–177. 2009. View Article : Google Scholar :
|
|
23
|
Yadav V and Denning MF: Fyn is induced by
Ras/PI3K/Akt signaling and is required for enhanced
invasion/migration. Mol Carcinog. 50:346–352. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gujral TS, Chan M, Peshkin L, Sorger PK,
Kirschner MW and MacBeath G: A noncanonical Frizzled2 pathway
regulates epithelial-mesenchymal transition and metastasis. Cell.
159:844–856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elias D, Vever H, Laenkholm AV, Gjerstorff
MF, Yde CW, Lykkesfeldt AE3 and Ditzel HJ: Gene expression
profiling identifies FYN as an important molecule in tamoxifen
resistance and a predictor of early recurrence in patients treated
with endocrine therapy. Oncogene. 2014.PubMed/NCBI
|
|
26
|
Ninio-Many L, Grossman H, Shomron N,
Chuderland D and Shalgi R: microRNA-125a-3p reduces cell
proliferation and migration by targeting Fyn. J Cell Sci.
126:2867–2876. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu L, Lin Q, Liao H, Feng J, Dong X and Ye
J: TGF-β1 induces podocyte injury through Smad3-ERK-NF-κB pathway
and Fyn-dependent TRPC6 phosphorylation. Cell Physiol Biochem.
26:869–878. 2010. View Article : Google Scholar
|
|
28
|
Hardy KM, Yatskievych TA, Konieczka J,
Bobbs AS and Antin PB: FGF signalling through RAS/MAPK and PI3K
pathways regulates cell movement and gene expression in the chicken
primitive streak without affecting E-cadherin expression. BMC Dev
Biol. 11:202011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roy SK, Srivastava RK and Shankar S:
Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of
FOXO transcription factor, leading to cell cycle arrest and
apoptosis in pancreatic cancer. J Mol Signal. 5:102010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saito YD, Jensen AR, Salgia R and Posadas
EM: Fyn: A novel molecular target in cancer. Cancer. 116:1629–1637.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Singh MM, Howard A, Irwin ME, Gao Y, Lu X,
Multani A and Chandra J: Expression and activity of Fyn mediate
proliferation and blastic features of chronic myelogenous leukemia.
PLoS One. 7:e516112012. View Article : Google Scholar
|
|
32
|
Kawakami T, Kawakami Y, Aaronson SA and
Robbins KC: Acquisition of transforming properties by FYN, a normal
SRC-related human gene. Proc Natl Acad Sci USA. 85:3870–3874. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yeo MG, Oh HJ, Cho HS, Chun JS,
Marcantonio EE and Song WK: Phosphorylation of Ser 21 in Fyn
regulates its kinase activity, focal adhesion targeting, and is
required for cell migration. J Cell Physiol. 226:236–247. 2011.
View Article : Google Scholar
|
|
34
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: Acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smyth D, Leung G, Fernando M and McKay DM:
Reduced surface expression of epithelial E-cadherin evoked by
interferon-gamma is Fyn kinase-dependent. PLoS One. 7:e384412012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim AN, Jeon WK, Lim KH, Lee HY, Kim WJ
and Kim BC: Fyn mediates transforming growth factor-beta1-induced
down-regulation of E-cadherin in human A549 lung cancer cells.
Biochem Biophys Res Commun. 407:181–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wakahashi S, Sudo T, Oka N, Ueno S,
Yamaguchi S, Fujiwara K, Ohbayashi C and Nishimura R: VAV1
represses E-cadherin expression through the transactivation of
Snail and Slug: A potential mechanism for aberrant epithelial to
mesenchymal transition in human epithelial ovarian cancer. Transl
Res. 162:181–190. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vuoriluoto K, Haugen H, Kiviluoto S,
Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB and Ivaska J:
Vimentin regulates EMT induction by Slug and oncogenic H-Ras and
migration by governing Axl expression in breast cancer. Oncogene.
30:1436–1448. 2011. View Article : Google Scholar
|
|
39
|
Lee SJ, Jung YH, Oh SY, Yong MS, Ryu JM
and Han HJ: Netrin-1 induces MMP-12-dependent E-cadherin
degradation via the distinct activation of PKCalpha and FAK/Fyn in
promoting mesenchymal stem cells motility. Stem Cells Dev.
23:1870–1882. 2014. View Article : Google Scholar : PubMed/NCBI
|