Introduction
Lung cancer is the leading cause of cancer-related
deaths worldwide. Approximately 85% of all lung cancers are
non-small cell lung cancer (NSCLC). Despite advances in early
detection and standard treatments, NSCLC is often diagnosed at an
advanced stage and has a poor prognosis (1). Metastases are the cause of 90% of
human cancer-related deaths (2).
Epithelial-to-mesenchymal transition (EMT) is a
cellular process during which epithelial cells lose their polarized
organization and cell-cell junctions, undergo changes in cell shape
and in cytoskeletal organization and acquire mesenchymal
characteristics, such as fibroblast-like cell morphology and
increased cell migration and invasion (3). Transcriptional factors which are
essential for EMT, such as Snail, Slug and ZEB1 are induced by
growth factors in most cases. The endogenous presence or the forced
expression of these transcriptional factors in cancer cells have
been linked with the loss of E-cadherin and the gain of vimentin
(4). EMT endows cells with
migratory and invasive properties, induces stem cell properties,
prevents apoptosis and senescence and contributes to
immunosuppression (5).
Statins, which inhibit the
3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoAR) enzyme,
are the most effective drugs used in the reduction of intracellular
synthesis of cholesterol (6). The
interactions between statins and HMG-CoA reductase prevent the
conversion of HMG-CoA to L-mevalonate resulting in the inhibition
of downstream cholesterol biosynthesis, and numerous isoprenoid
metabolites such as geranylgeranyl pyrophosphate (GGPP) and
farnesyl pyrophosphate (FPP) (7).
The isoprenoids are lipid moieties that are added to various
proteins, including G-proteins and the G-protein subunits RAS, Rho,
Rac and Cdc42, during post-translational modification (prenylation)
and anchor these proteins to the cell membrane (8). Prenylation also occurs in many
cellular and systemic regulatory pathways that are partly
responsible for the pleiotropic effects of statins (9). Other pleiotropic effects may be
independent of prenylation or inhibition of cholesterol production
such as cell cycle arrest (10).
Numerous studies have confirmed that statins reduce the risk of
multiple types of cancers (11–14).
Statin use in cancer patients is also associated with reduced
cancer-related mortality (15).
The sphingosine kinase 1 (SphK1) can convert
sphingosine to sphingosine 1-phosphate (16), and can be upregulated by TGF-β in
fibroblasts and myofibroblasts (17). TGF-β can upregluate SphK1 in A549
cells (18). All of these facts
indicate that SphK1 has roles in the TGF-β lower signaling
pathway.
EMT, a complicated process programmed by many genes,
is an important mechanism for cancer metastasis. In the present
research, we evaluated the effect and mechanism of atorvastatin on
the EMT process in NSCLC cells by establishing an EMT model in
vitro induced by TGF-β1, and we investigated the effects of
atorvastatin on the lower signaling pathway of TGF-β1 as well as
its effect on SphK1.
Materials and methods
Cell culture and reagents
Human lung adenocarcinoma A549 and NCI-H1975 cells
were obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA). A549 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) (Gibco,
Grand Island, NY, USA) at 37°C in a humidified 5% CO2
atmosphere. NCI-H1975 cells were maintained in RPMI-1640 medium
with 10% FBS. The cells were trypsinized and seeded in 12-well
tissue culture plates, and were treated with atorvastatin (Sigma,
St. Louis, MO, USA) or TGF-β1 (PeproTech Inc., Rocky Hill, NJ, USA)
for different time periods in serum-free medium. The rabbit
anti-human antibodies, such as E-cadherin, phospho-Smad2
(S465/467), Smad2, phospho-Smad3 (Ser423/425), Smad3, phospho-AKT
(S473), AKT, phospho-ERK (T202/Y204), ERK, Snail and Slug were
purchased from Cell Signaling Technology (Danvers, MA, USA). The
monoclonal mouse anti-human antibodies, such as vimentin, SphK1 and
ZEB1 were purchased from Abcam (Cambridge, UK). The polyclonal
rabbit anti-human β-actin antibody was purchased from
Sigma-Aldrich. The monoclonal mouse anti-human
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody,
horseradish peroxidase-conjugated anti-mouse and anti-rabbit
secondary antibodies were all purchased from KangChen Bio-tech
(Shanghai, China). BCA protein assay kit was purchased from
Beyotime Biotechnology (Shanghai, China). The pBABE-SphK1 plasmid
was a kind gift synthesized by the Shanghai Institute of Materia
Medica (Chinese Academy of Sciences).
Transwell assay
The migration assay was carried out using
8-µm pore size Transwell filters (6.5-mm diameter; Corning
Inc., Corning, NY, USA). Following pretreatment with atorvastatin
for 24 h, the A549 cells were detached and single-cell suspensions
were placed at 1×105 cells/well into the upper chamber
in 0.1 ml of serum-free medium. In the lower chamber, DMEM
supplemented with 10% FBS was placed as a chemoattractant.
Atorvastatin and TGF-β1 were added at constant concentration to
both the upper and the lower chambers. After 18 h of incubation in
5% CO2 at 37°C, the filters were fixed with 90% ethanol
for 30 min and stained with crystal violet. Cells from the upper
surface of the chamber were removed with gentle swabbing and the
invasive cells on the lower surface of the filters were examined by
bright field microscopy. The average number from five randomly
chosen fields was counted.
Immunofluorescence analysis
Stock solutions of FITC-phalloidin were made in DMSO
at 0.2 mg/ml. Final staining solutions in phosphate-buffered saline
(PBS) were at the concentration of 5 µg/ml. Monolayers of
A549 cells were serum-starved and treated with atorvastatin on
glass cover-slips for 24 h, and then treated with atorvastatin and
TGF-β1 for 24 h. The A549 cells were washed with PBS and fixed for
10 min in 4% polyformaldehyde, and then extensively washed in PBS
three times. After being permeabilized with 0.1% Triton X-100 in
PBS and washed again in PBS, the A549 cells were stained with 5
µg/ml FITC-phalloidin. Cover slides were mounted onto slides
with 1% 4′6-diamidine-2′-phenylindole dihydrochloride (DAPI). Cells
were examined under an Olympus Fluoview FV1000 confocal microscope
(Olympus, Tokyo, Japan).
Western blot analysis
The cells were harvested, rinsed with cold PBS and
lysed in buffer supplemented with protease inhibitors for 1 h as
previously described (19). The
protein samples were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and electrophoretically
transferred to nitrocellulose. The nitrocellulose membrane was
blocked with 5% milk in Tris-buffered saline and Tween-20 (TBST)
for 1 h at room temperature and incubated with primary antibodies
at 4°C overnight. Then, the membrane was washed with TBST for 15
min each time for three times and incubated with secondary
antibodies at room temperature for 1 h. To ensure that equal
amounts of sample protein were applied for electrophoresis and
immunoblotting, GAPDH or β-actin was used as a protein loading
control. All experiments were performed at least three independent
times.
Transient transfection of the SphK1
plasmid
A549 cells were plated in 12-well plates in DMEM
supplemented with 10% FBS overnight to reach ~80–90% confluency.
Then, the cells were transfected with the pBABE-SphK1 plasmid and
pBABE-puro using Lipofectamine 2000 reagent (Invitrogen, Carlsbad,
CA, USA) according to the manufacturer's instructions and replaced
with culture medium containing 1 µM atorvastatin at 5 h
after transfection. Twenty-four hours later, the cells were treated
with atorvastatin and TGF-β1 for another 24 h. Then, the cells were
harvested for protein quantitative analysis using the BCA protein
assay kit and western blot analysis.
RNA extraction and real-time PCR for
SphK1 mRNA expression
A549 cells were maintained in 12-well plates with
serum-free medium and exposed to atorvastatin for 24 h before being
treated with TGF-β1 for 48 h. Total RNA was extracted using Takara
MiniBEST universal RNA Extraction kit (Takara Bio, Dalian, China)
according to the manufacturer's instructions. cDNA was synthesized
with PrimeScript reverse transcriptase using oligo(dT) primer and 2
µg of total RNA (Takara PrimeScript™ RT Master Mix) (both
from Takara Bio). Real-time PCR assays were conducted using SYBR
Premix Ex Taq™ according to the manufacturer's instructions (Takara
Bio). Amplification was performed in 20 µl reactions (0.4
µl primer, 10 µl SYBR Premix Ex Taq™) containing 2
µl of cDNA in 40 cycles at 95°C (3 sec) and 60°C (30 sec)
following pre-denaturation at 95°C (30 sec). Data normalization was
performed using GAPDH as a reference gene. Primer sequences of
SphK1 were: F, 5′-CTGGCAGCTTCCTTGAACCAT-3′ and R,
5′-TGTGCAGAGACAGCAGGTTCA-3′; the control primer sequences of GAPDH
were: F, 5′-GGGAGCCAAAAGGGTCATCATCTC-3′ and R,
5′-CCATGCCAGTGAGCTTCCCGTTC-3′. The relative quantification of SphK1
mRNA was obtained by calculating ΔΔCt.
Statistical analysis
Data are presented as mean ± standard deviation
(SD). SPSS 10.0 was used for statistic analysis and differences
between groups were analyzed by the Student's t-test or ANOVA.
Differences between groups were considered significant at
P<0.05.
Results
Atorvastatin partially inhibits the EMT
process in A549 cells induced by TGF-β1
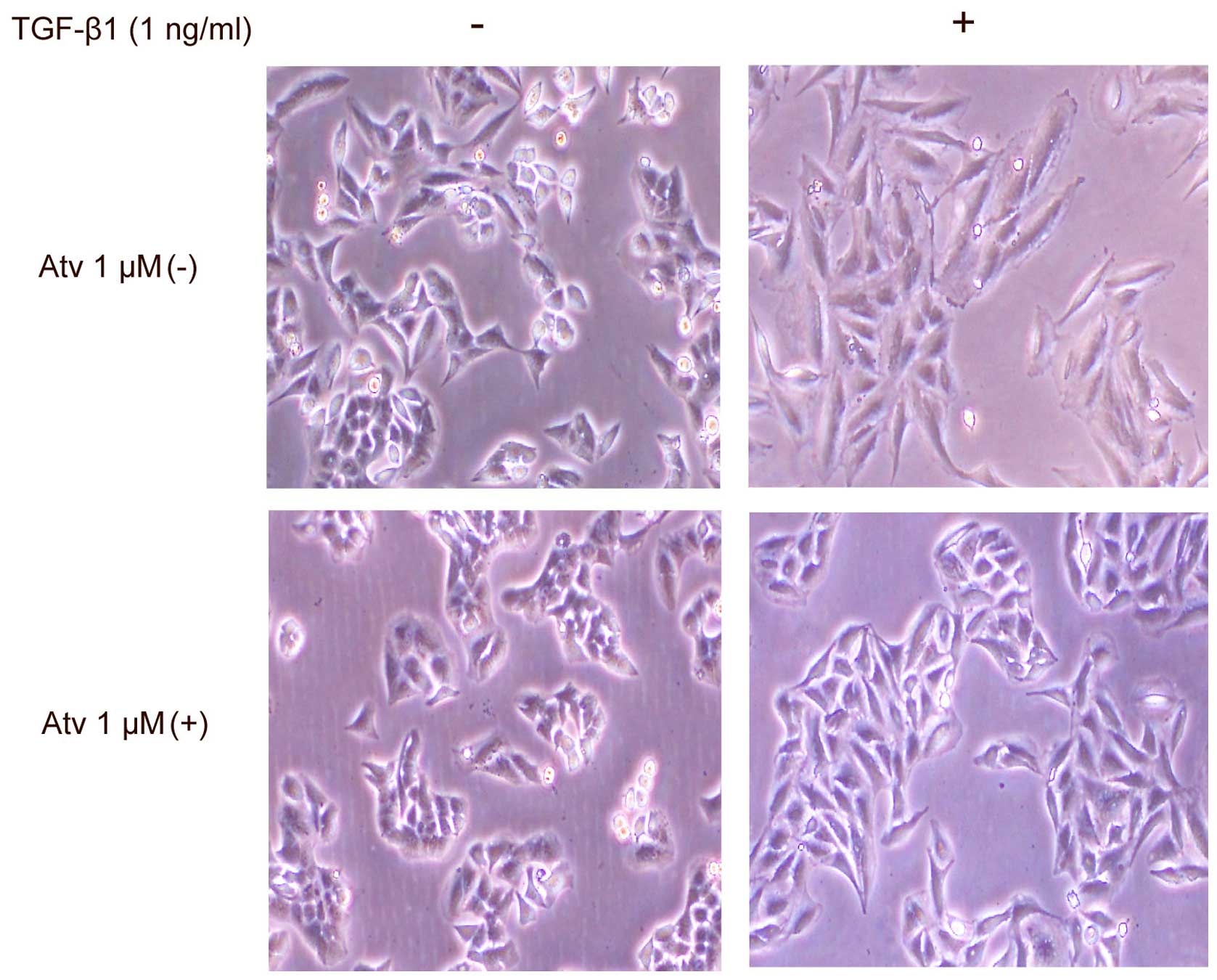
A549 cells were serum-starved and treated with
atorvastatin for 24 h, and then exposed to atorvastatin and TGF-β1
for another 48 h. Cells displayed a complete transformation to
fibroblast-like characteristics after 48 h with TGF-β1 stimulation.
However, when pretreated with atorvastatin for 24 h, cells had a
partial transition to a fibroblast-like morphology stimulated by
TGF-β1 for 48 h. In addition, there was no recognizable change in
cell shape after stimulation with atorvastatin alone for 48 h
(Fig. 1). The result indicated that
atorvastatin inhibited the TGF-β1-stimulated morphological
transition of the A549 cells to fibroblast-like cells.
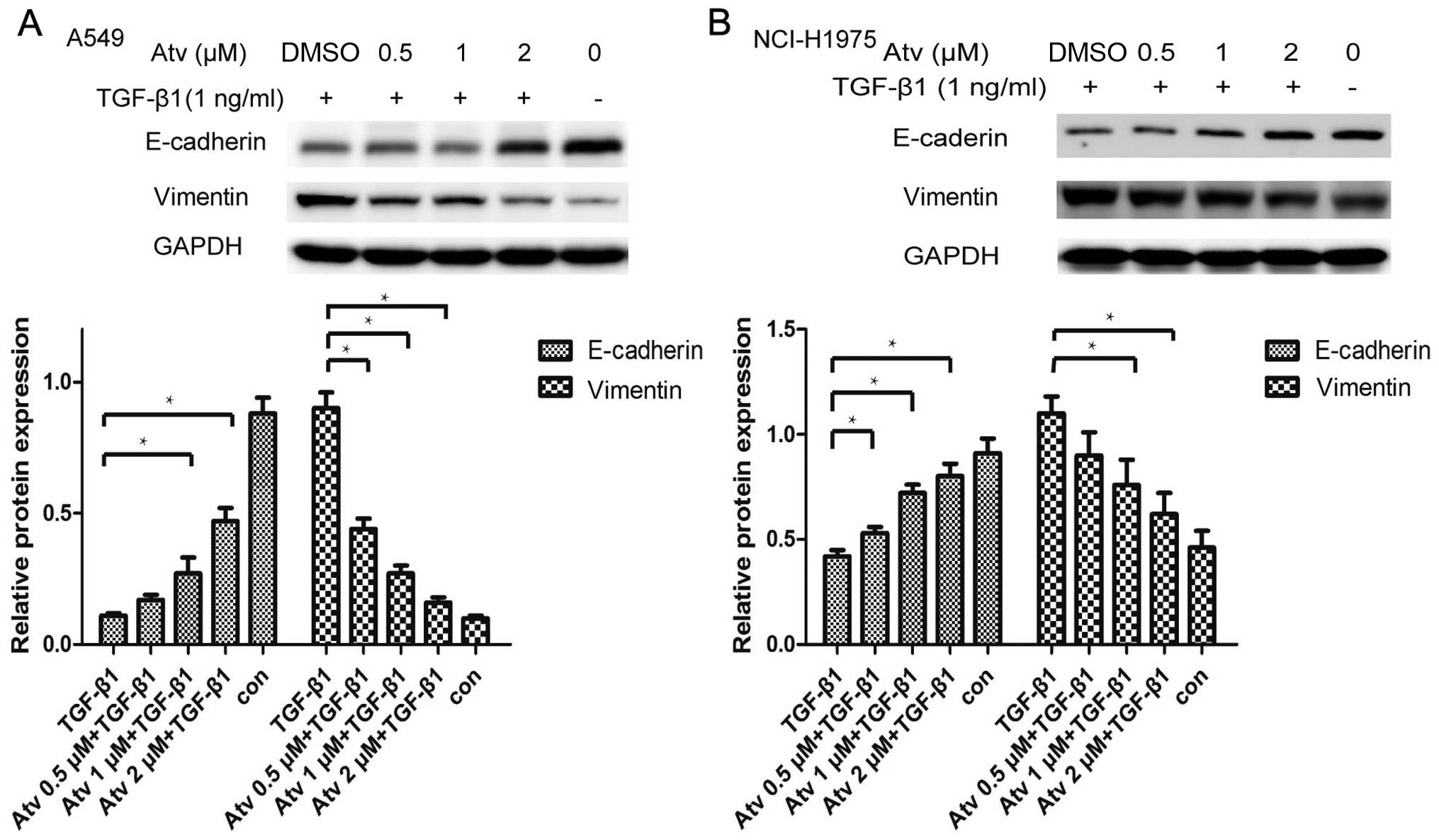
The expression of E-cadherin and vimentin was not
affected by treatment of various concentrations of atorvastatin.
Atorvastatin pretreatment effectively weakened TGF-β1-stimulated
downregulation of E-cadherin as well as upregulation of vimentin.
The alterations of EMT marker proteins such as E-cadherin and
vimentin were further confirmed in the NCI-H1975 cells (Fig. 2). The result further confirmed that
atorvastatin can partially inhibit the EMT process in A549
cells.
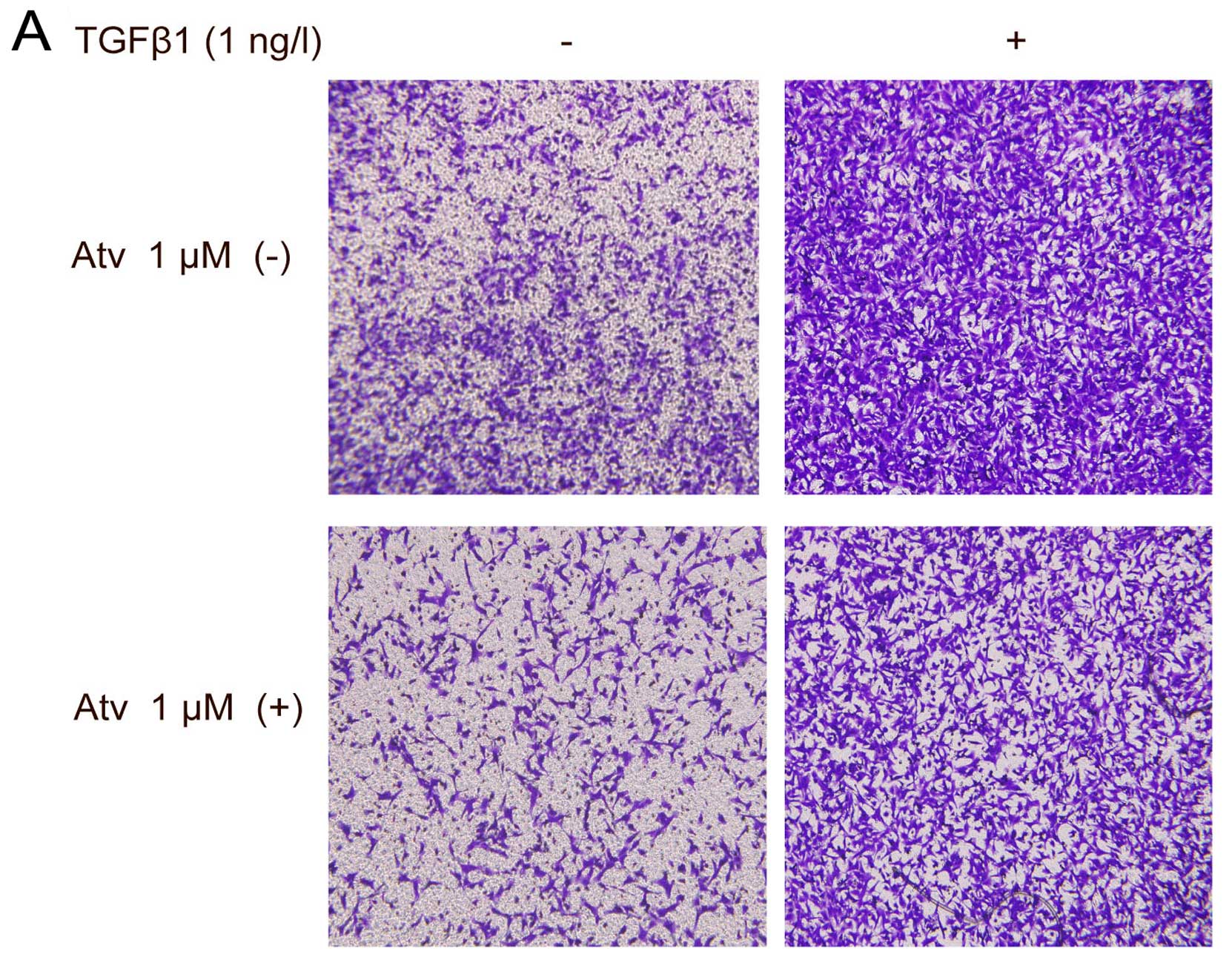
Atorvastatin inhibits cell migration and
actin filament remodeling induced by TGF-β1
It is known that EMT renders tumor cells with
increased migratory potential (4).
We evaluated the effect of atorvastatin on the elevated migration
capabilities induced by TGF-β1 in the A549 cells using Transwell
assay. A549 cells were seeded on the upper chamber of the Transwell
wells in the presence of TGF-β1. A significant decrease in the
number of A549 cells that migrated to the lower filters was
observed when cells were pretreated with 1 µM atorvastatin
for 24 h (Fig. 3).
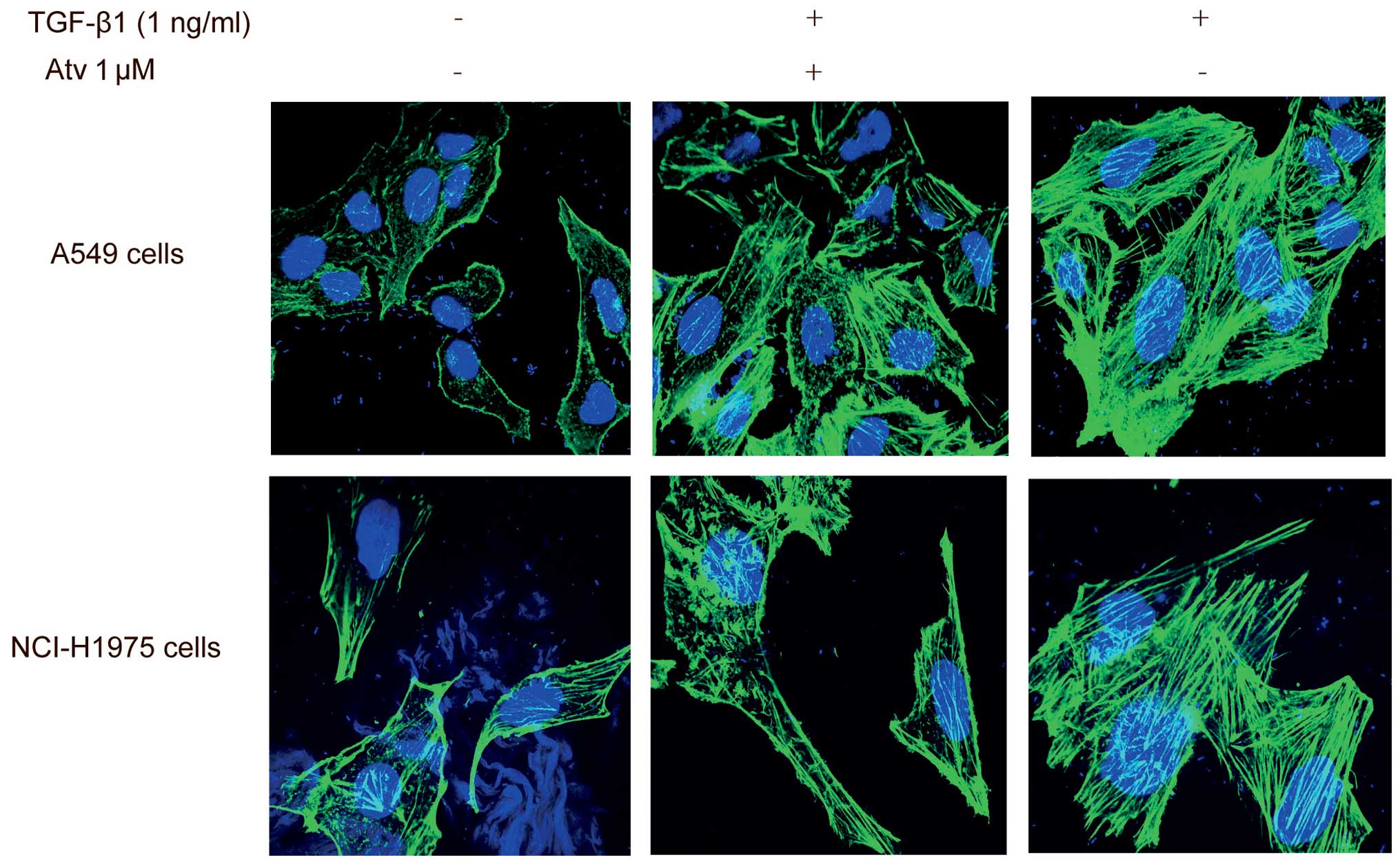
Remodeling of actin filaments is necessary for
TGF-β1-stimulated EMT. Actin filaments in epithelial cells, which
are organized in cortical thin bundles, are bundled into thick
contractile stress fibers after the EMT process (20). Following exposure to TGF-β1 for 24
h, atorvastatin pretreatment made the actin fibers of A549 cells
less and thinner. This finding was confirmed in the NCI-H1975 cells
and the results indicate that atorvastatin can impede the actin
filament remodeling process (Fig.
4).
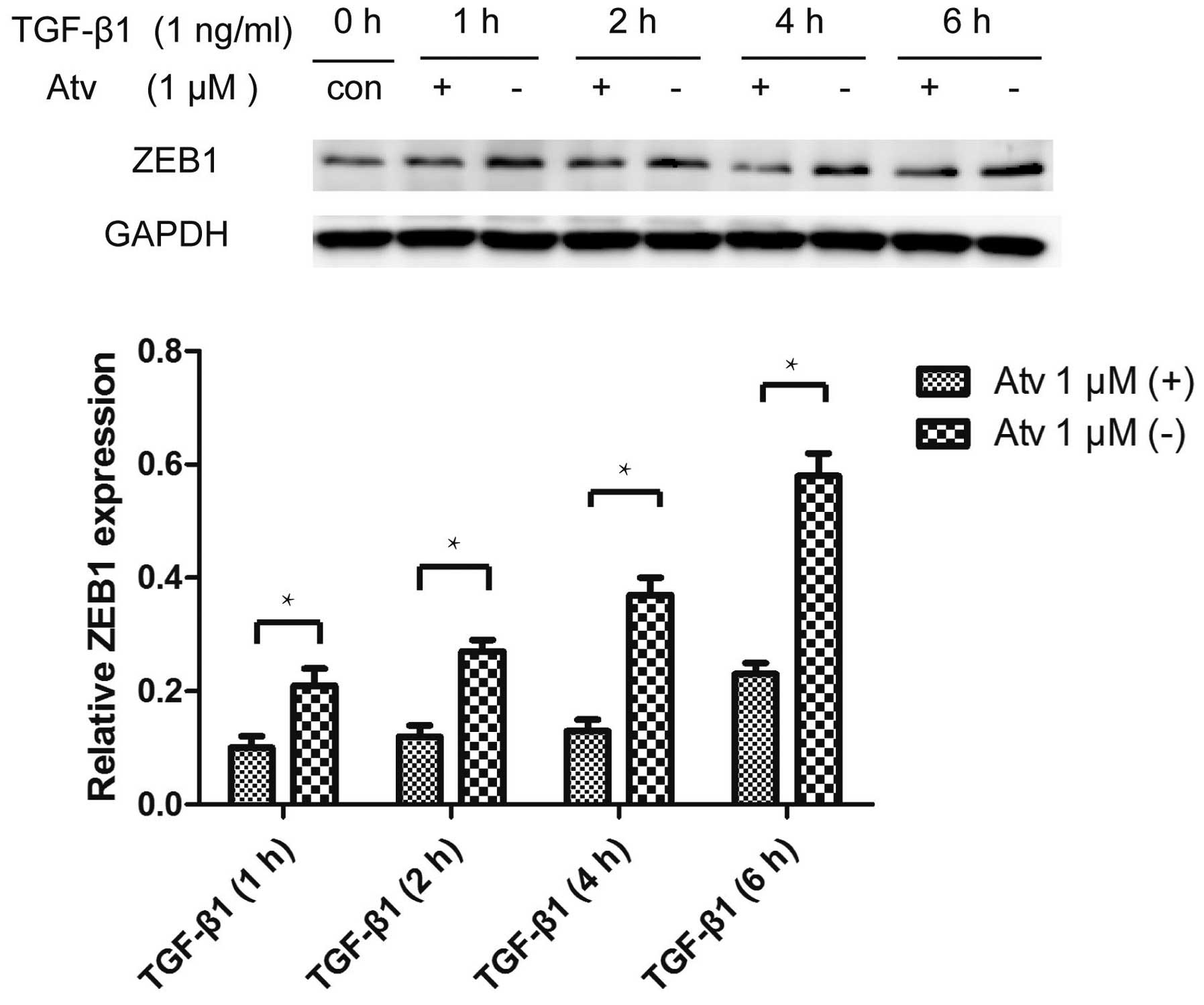
Atorvastatin inhibits the
TGF-β1-stimulated upregulation of ZEB1
Numerous transcription factors which are essential
for EMT, such as Snail, Slug and ZEB1, can repress genes important
for maintaining epithelial polarity and organization (3). Our experiment found that atorvastatin
inhibited the upregulation of ZEB1 induced by TGF-β1. However, we
did not find that Snail and Slug were inhibited by atorvastatin
pretreatment (Fig. 5).
The activation of Smad2/3 by TGF-β1 represents the
canonical TGF-β1 signaling system, and non-canonical signaling
systems including MAPK-ERK1/2, PI3K-AKT and small GTP-binding
proteins (21). In our research,
atorvastatin did not inhibit the TGF-β1-stimulated activation of
Smad2/3, AKT and ERK1/2.
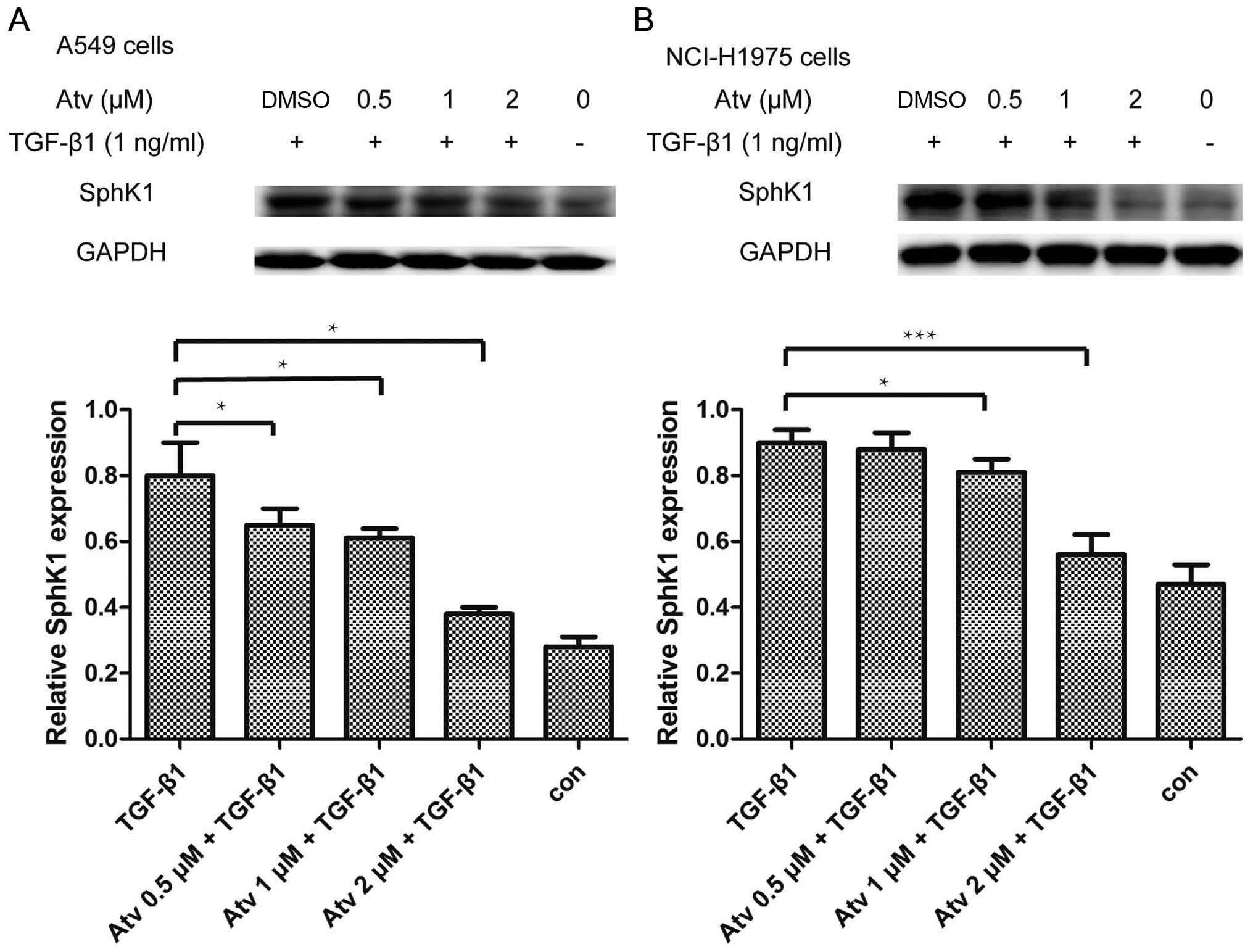
Atorvastatin attenuates the upregulation
of SphK1 induced by TGF-β1
It has been confirmed that TGF-β1 upregulates SphK1
protein expression in A549 cells (18). In our experiment, we found that the
upregulation of protein SphK1 induced by TGF-β1 was also inhibited
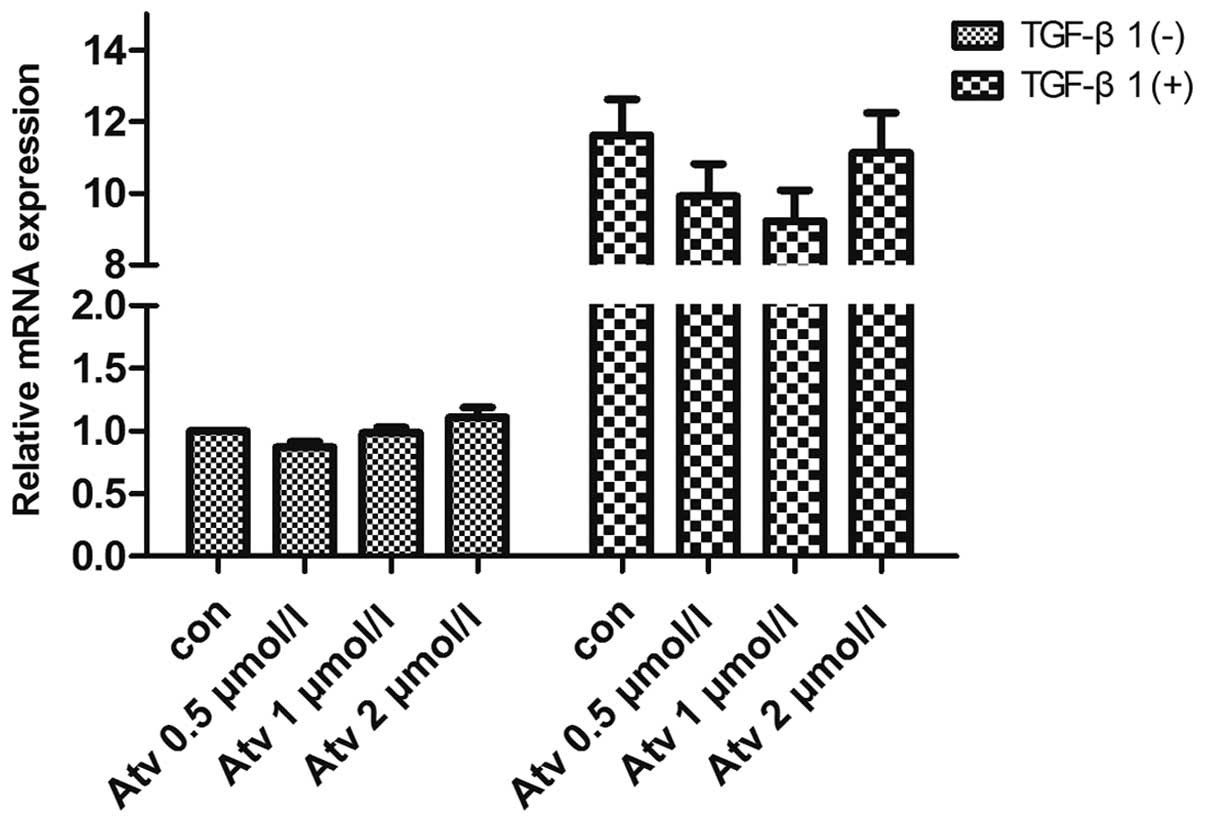
by atorvastatin pretreatment at 1 µM (Fig. 6). However, at the mRNA level,
real-time PCR found that atorvastatin did not inhibit the
upregulation of SphK1 mRNA, which indicates that atorvastatin
inhibits the translation of SphK1 at the post-transcriptional level
when exposed to TGF-β1 (Fig.
7).
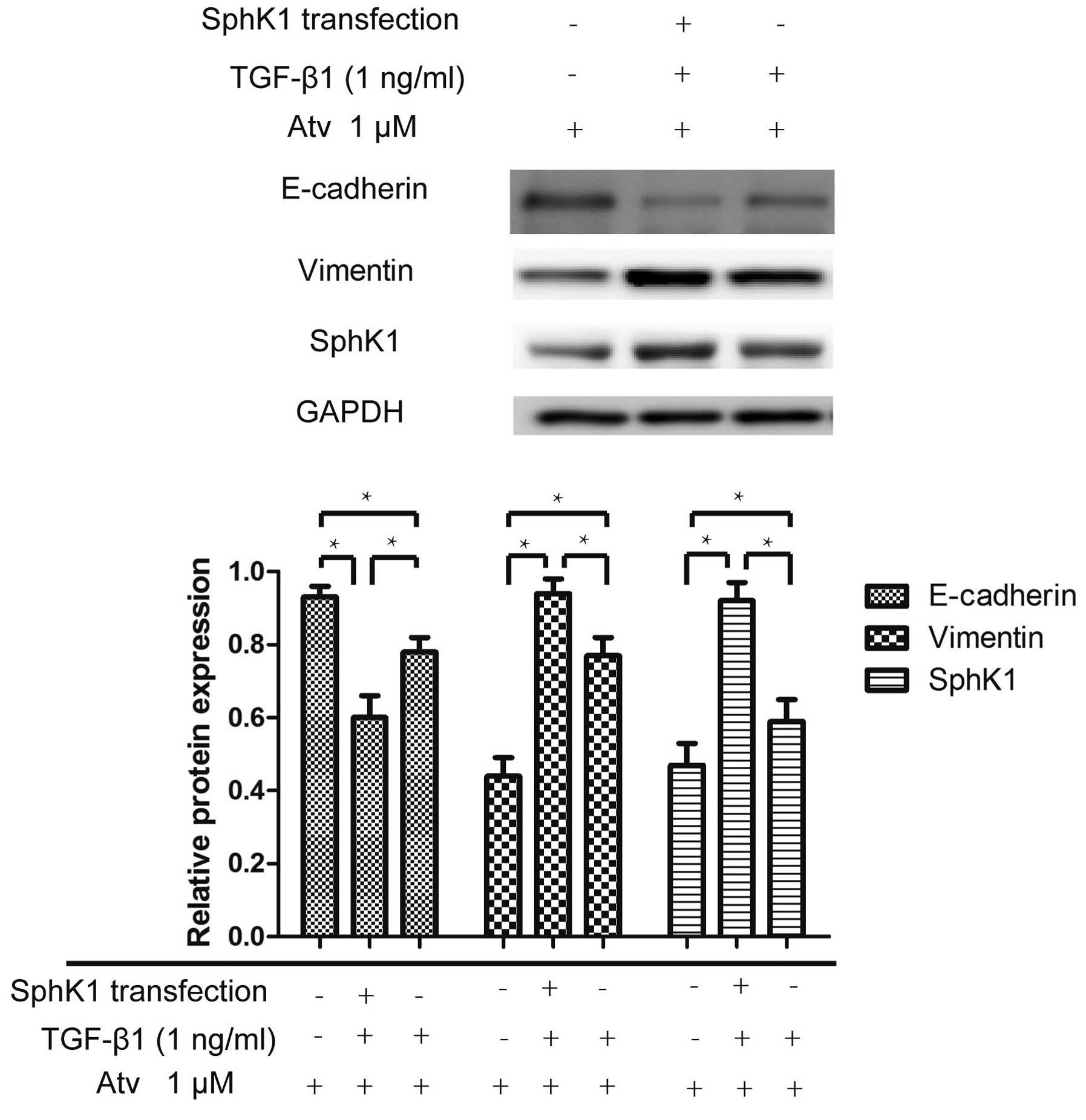
Transient transfection of the SphK1
plasmid strengthens the EMT process induced by TGF-β1 in the
presence of atorvastatin
A549 cells were transiently transfected with the
p-BABE SphK1 plasmid using Lipofectamine 2000. The EMT process
induced by TGF-β1 in the A549 cells, which were transfected with
the SphK1 plasmid was strengthened. Expression of the EMT marker
E-cadherin was lower in the cells transfected with the SphK1
plasmid. The result indicated that SphK1 transfection weakens the
inhibitory effect of atorvastatin on the EMT process (Fig. 8).
Discussion
Epithelial-to-mesenchymal transition (EMT) is an
important mechanism for cancer metastasis. Recent research has
confirmed that EMT is a potential biomarker of therapeutic
resistance and is a potential drug target in breast cancer that
warrants further investigation (22). Statins, HMG-CoA reductase
inhibitors, are used extensively in the treatment of
hyperlipidemia. Evidence shows that statins have anticancer
effects. These secondary actions are known as pleiotropic effects.
In the present study, we designed a series of experiments to
evaluate the effect of atorvastatin on the EMT process induced by
TGF-β1 in NSCLC cells. MTT assay found that atorvastatin at 15
µM significantly reduced the cell viability by 80%. In
addition, atorvastatin at 1–5 µM did not affect the cell
viability (23). In the present
study, the cells were treated with atorvastatin at 0.5–2 µM,
which had no effect on cell proliferation.
Differentiated epithelial cancer cells convert into
migratory mesenchymal cancer cells via EMT, which may lead to
cancer invasion, systemic cancer cell dissemination and metastasis.
The metastatic potential is acquired by the loss of epithelial
markers and the acquisition of mesenchymal markers. Our research
found that atorvastatin treatment alone did not change the
expression of the EMT markers, E-cadherin and vimentin, as well as
cell morphology. However, after pretreatment with atorvastatin for
24 h, the EMT process in A549 cells induced by TGF-β1 was partially
inhibited. The results indicated that atorvastatin can suppress the
downregulation of E-cadherin and upregulation of vimentin induced
by TGF-β1, impede the formation of actin stress fibers and hinder
cancer cell migration. The conversion of the cell shape into a
fibroblast-like cell morphology was inhibited by atorvastatin
pretreatment. These experiments demonstrated that atorvastatin can
inhibit the EMT process induced by TGF-β1 in vitro.
Transcription factors such as Snail, Slug and ZEB1
can induce EMT in cancer cells (24,25).
We further investigated whether these transcription factors are
inhibited by atorvastatin during the EMT process. The results
showed that atorvastatin impeded the upregulation of ZEB1 induced
by TGF-β1 and did not affect the expression of Snail and Slug.
These findings imply that atorvastatin may block the
TGF-β1 lower signal transduction in one manner or another. However,
in our experiments, atorvastatin did not inhibit the activation of
Smad2/3, ERK1/2 and AKT. Sphingosine kinase type 1 (SphK1), which
catalyzes the phosphorylation of sphingosine to
sphingosine-1-phosphate (S1P), has been shown to regulate various
processes important for cancer progression (26). TGF-β1 upregulates SphK1 in A549
cells and TGF-β1-induced EMT in the A549 cells was inhibited by an
antagonist of SphK1, providing evidence for crosstalk between SphK1
and TGF-β1 (18). We found that
atorvastatin pretreatment suppressed the upregulation of SphK1
protein induced by TGF-β1, while the mRNA expression of SphK1 was
not inhibited by atorvastatin as determined by the RT-PCR
experiment. The results indicated that atorvastatin inhibited the
transcription of SphK1 induced by TGF-β1 stimulation. As S1P,
catalyzed by SphK1 with sphingosine, can activate Smad2/3 in A549
cells (27), we considered that
atorvastatin indirectly inhibits the activation of Smad2/3 by
inhibiting the upregulation of SphK1 and downregulating the
expression of transcription factor ZEB1.
When A549 cells were transiently transfected with
the SphK1 plasmid, cells pretreated with atorvastatin had a lower
expression of E-cadherin and a higher expression of vimentin
compared with cells transfected with the SphK1-negative plasmid
induced by TGF-β1, indicating that the ability of atorvastatin to
inhibit the EMT process was attenuated in cells highly expressing
SphK1. This result further illustrated that atorvastatin inhibited
the EMT process through the SphK1 pathway.
In summary, the present study suggests that
atorvastatin partially inhibits the EMT process in A549 cells
induced by TGF-β1 by attenuating the upregulation of SphK1. As EMT
is one of the main mechanisms for the promotion of cancer invasion,
and is a survival mechanism of cancer cells, statins may have
beneficial effects for cancer patients in diverse ways.
References
|
1
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mehlen P and Puisieux A: Metastasis: A
question of life or death. Nat Rev Cancer. 6:449–458. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tiwari N, Gheldof A, Tatari M and
Christofori G: EMT as the ultimate survival mechanism of cancer
cells. Semin Cancer Biol. 22:194–207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thiery JP, Acloque H, Huang RYJ and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liao JK: Clinical implications for statin
pleiotropy. Curr Opin Lipidol. 16:624–629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gazzerro P, Proto MC, Gangemi G, Malfitano
AM, Ciaglia E, Pisanti S, Santoro A, Laezza C and Bifulco M:
Pharmacological actions of statins: A critical appraisal in the
management of cancer. Pharmacol Rev. 64:102–146. 2012. View Article : Google Scholar
|
|
8
|
Demierre MF, Higgins PD, Gruber SB, Hawk E
and Lippman SM: Statins and cancer prevention. Nat Rev Cancer.
5:930–942. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Menter DG, Ramsauer VP, Harirforoosh S,
Chakraborty K, Yang P, Hsi L, Newman RA and Krishnan K:
Differential effects of pravastatin and simvastatin on the growth
of tumor cells from different organ sites. PLoS One. 6:e288132011.
View Article : Google Scholar
|
|
10
|
Rao S, Porter DC, Chen X, Herliczek T,
Lowe M and Keyomarsi K: Lovastatin-mediated G1 arrest is
through inhibition of the proteasome, independent of hydroxymethyl
glutaryl-CoA reductase. Proc Natl Acad Sci USA. 96:7797–7802. 1999.
View Article : Google Scholar
|
|
11
|
Liu Y, Tang W, Wang J, Xie L, Li T, He Y,
Deng Y, Peng Q, Li S and Qin X: Association between statin use and
colorectal cancer risk: A meta-analysis of 42 studies. Cancer
Causes Control. 25:237–249. 2014. View Article : Google Scholar
|
|
12
|
Khurana V, Bejjanki HR, Caldito G and
Owens MW: Statins reduce the risk of lung cancer in humans: A large
case-control study of US veterans. Chest. 131:1282–1288. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi M, Zheng H, Nie B, Gong W and Cui X:
Statin use and risk of liver cancer: An update meta-analysis. BMJ
Open. 4:e0053992014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lustman A, Nakar S, Cohen AD and Vinker S:
Statin use and incident prostate cancer risk: Does the statin brand
matter? A population-based cohort study. Prostate Cancer Prostatic
Dis. 17:6–9. 2014. View Article : Google Scholar
|
|
15
|
Nielsen SF, Nordestgaard BG and Bojesen
SE: Statin use and reduced cancer-related mortality. N Engl J Med.
367:1792–1802. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baker DL, Pham TC and Sparks MA: Structure
and catalytic function of sphingosine kinases: Analysis by
site-directed mutagenesis and enzyme kinetics. Biochim Biophys
Acta. 1831:139–146. 2013. View Article : Google Scholar
|
|
17
|
Kono Y, Nishiuma T, Nishimura Y, Kotani Y,
Okada T, Nakamura S and Yokoyama M: Sphingosine kinase 1 regulates
differentiation of human and mouse lung fibroblasts mediated by
TGF-beta1. Am J Respir Cell Mol Biol. 37:395–404. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Milara J, Navarro R, Juan G, Peiró T,
Serrano A, Ramón M, Morcillo E and Cortijo J:
Sphingosine-1-phosphate is increased in patients with idiopathic
pulmonary fibrosis and mediates epithelial to mesenchymal
transition. Thorax. 67:147–156. 2012. View Article : Google Scholar
|
|
19
|
Ai J, Tang Q, Wu Y, Xu Y, Feng T, Zhou R,
Chen Y, Gao X, Zhu Q, Yue X, et al: The role of polymeric
immunoglobulin receptor in inflammation-induced tumor metastasis of
human hepatocellular carcinoma. J Natl Cancer Inst. 103:1696–1712.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haynes J, Srivastava J, Madson N, Wittmann
T and Barber DL: Dynamic actin remodeling during
epithelial-mesenchymal transition depends on increased moesin
expression. Mol Biol Cell. 22:4750–4764. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wendt MK, Allington TM and Schiemann WP:
Mechanisms of the epithelial-mesenchymal transition by TGF-beta.
Future Oncol. 5:1145–1168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu M, Bardia A, Wittner BS, Stott SL, Smas
ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al:
Circulating breast tumor cells exhibit dynamic changes in
epithelial and mesenchymal composition. Science. 339:580–584. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miraglia E, Högberg J and Stenius U:
Statins exhibit anticancer effects through modifications of the
pAkt signaling pathway. Int J Oncol. 40:867–875. 2012.
|
|
24
|
Argast GM, Krueger JS, Thomson S,
Sujka-Kwok I, Carey K, Silva S, O'Connor M, Mercado P, Mulford IJ,
Young GD, et al: Inducible expression of TGFβ, Snail and Zeb1
recapitulates EMT in vitro and in vivo in a NSCLC model. Clin Exp
Metastasis. 28:593–614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alves CC, Carneiro F, Hoefler H and Becker
KF: Role of the epithelial-mesenchymal transition regulator Slug in
primary human cancers. Front Biosci. 14:3035–3050. 2009. View Article : Google Scholar
|
|
26
|
Shida D, Takabe K, Kapitonov D, Milstien S
and Spiegel S: Targeting SphK1 as a new strategy against cancer.
Curr Drug Targets. 9:662–673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xin C, Ren S, Kleuser B, Shabahang S,
Eberhardt W, Radeke H, Schäfer-Korting M, Pfeilschifter J and
Huwiler A: Sphingosine 1-phosphate cross-activates the Smad
signaling cascade and mimics transforming growth
factor-beta-induced cell responses. J Biol Chem. 279:35255–35262.
2004. View Article : Google Scholar : PubMed/NCBI
|