Introduction
Despite intensive initial treatments that include
surgery, radioiodine therapy, thyroid-stimulating hormone (TSH)
suppression with levothyroxine, poorly differentiated thyroid
cancer including anaplastic thyroid cancer are aggressive and
refractory to conventional therapies (1). As the knowledge of molecular
pathogenesis of thyroid cancer increases, targeted therapies are
being innovated for these patients (2). Unfortunately, side-effects and
cellular resistance of single agent multikinase or
BRAFV600E inhibitor often leads to termination of the
targeted therapy. To overcome resistance and to reduce
side-effects, combined administration containing multikinase or
BRAFV600E inhibitor should be investigated. In contrast
to development of new compounds, screening FDA-approved drugs may
identify anticancer drugs and facilitate initiation of early
clinical trials. Propranolol hydrochloride, although originally
used for the treatment of hypertension, cardiovascular disorders
and hemangiomas, has been shown to have anticancer property for
many cancers including pancreatic, breast and gastric cancer,
leukemia, neuroblastoma, and head and neck squamous cell carcinoma
(3–9). In addition, retrospective studies have
demonstrated that cancer patients taking β-blockers may have
improved outcomes and decreased incidence of secondary malignances
(9–11). Based on the basic research and
clinical evidence, we hypothesized that the β-blockers propranolol
may have potential efficacy in inhibiting the proliferation and/or
inducing the apoptosis of anaplastic thyroid cancer.
In the present study we observed that
β-adrenoreceptors (ADRBs), particularly β2-AR (ADRB2), are
expressed in both well-differentiated and poorly-differentiated
thyroid cancer cell lines. Propranolol reduced the viability of
8505C cells through inhibition of proliferation and induction of
apoptosis. Propranolol induced apoptosis was associated with
suppressed expression of anti-apoptotic Bcl-2 while inhibited
proliferation was the result of decreased expression of cyclin D1.
In addition, the levels of glucose metabolism proteins, hexokinase
2 (HK2) and glucose transporter 1 (GLUT1), also decreased in the
propranolol-treated group. Furthermore, propranolol inhibited the
growth of ATC xenografts in vivo at a dose of 10 mg/kg/day.
Consistently 18F-FDG PET/CT imaging revealed tumor
shrinkage in the propranolol-treated group. Immunohistochemistry of
the tumor specimens validated the downregulation of Bcl-2 and
cyclin D1 observed in vitro. Notably, we found that
propranolol pretreatment sensitized K1 cells to the cytotoxicity of
vemurafenib.
Taken together, our present findings suggest that
propranolol is an effective agent in inhibiting the growth of
thyroid cancers, and that the combination therapy consisting
propranolol and BRAFV600 inhibitor may provide clinical
benefits with minimal side-effects to BRAFV600E mutant
advanced thyroid cancer patients.
Materials and methods
Cell culture
Human PTC cell line K1, BCPAP and ATC cell line
8505C were purchased from the Institute of Biochemistry and Cell
Biology (SIBS, CAS, Shanghai, China). BHP27 cell line was a kind
gift from Professor Li-Bo Chen in our hospital. Both the cell lines
used in the present study have been confirmed for identity (Fumed
Biotech Bio-Medicine; SIBS, CAS). All cell lines used in these
experiments were maintained in RPMI-1640 medium (cat. #11875-093)
supplemented with 10% fetal bovine serum (FBS) (cat. #16000-044)
(both from Gibco, Carlsbad, CA, USA) in a 5% CO2-95% air
atmosphere at 37°C.
Cell Counting Kit-8 (CCK-8), colony
formation and apoptosis assay
In order to determine the half maximal inhibitory
concentration (IC50) of propranolol, atenolol and
ICI118551, both from Sigma (St. Louis, MO, USA); ICI118551 from
MedChem Express (Monmouth Junction, NJ, USA), and the effect of
isoprenaline hydrochloride (Sigma) on growth, a CCK-8 assay
(Yeasen, Shanghai, China) was performed. The cells were seeded into
96-well plates at a density of 1×104 cells/well for 24
h, and then incubated for 24 h with increasing concentrations of
the various compounds under study (10 and 40 µM for
isoprenaline; 50, 100, 150, 200, 250, 300, 350 and 400 µM
for propranolol, atenolol and ICI118551). Control cells were
allowed to grow in the absence of any inhibitors for the same
period of time. The samples were assayed in sextuplet and at least
in three independent experiments, and the mean value for each
experiment was calculated. Results are presented as mean (± SEM)
and are expressed as percentage of the control group. For
clonogenic survival studies, K1 cells were pretreated with ABT-737
(1 µM) or propranolol (200 µM) for 24 h before
treatment with vemurafenib (PLX-4032) (5 µM) for 72 h, and
then the media were changed and colonies were stained with crystal
violet 10 days after treatment and imaged. For nuclear
fragmentation assay, 8505C cells were treated with propranolol (0,
200 and 500 µM) for 24 h and the cells were stained with
Hoechst 33342 and imaged by fluorescent microscopy.
Flow cytometric analysis
Before flow cytometric apoptosis analysis, 8505C
cells were pretreated for 24 h with increasing concentrations of
propranolol (0, 100, 200, 300, 400 and 500 µM). At each
concentration half a million cells were collected, washed 2 times
with cold phosphate-buffered saline (PBS). Fixed cells were stained
with Annexin V-FITC/PI (Yeasen). For cell cycle analysis, cells
(106) were treated with propranolol (100 and 200
µmol/l) for 0, 24 and 48 h. After the treatment, the cells
were trypsinized and centrifuged at 800 rpm for 5 min.
Subsequently, cells were collected and washed 2 times with PBS.
Cell pellets were resuspended in 300 µl ice-cold PBS and
fixed overnight by adding 700 µl ethanol. After washing with
PBS, cell pellets were resuspended and incubated in 300 µl
PBS (containing 20 µl Rnase A) at 37°C for 30 min. In
addition, 400 µl propidium iodide (PI) solution was added
and incubated at 4°C for 30–60 min in the dark. Samples were
analyzed on a flow cytometer (Beckman Coulter, Brea, CA, USA).
Western blot analysis
8505C cells were plated at a density of
20×104 cells/well in 6-well plates. At confluence,
propranolol and ICI118551 at doses of 0, 25, 50 and 100 µM
were incubated with 8505C cells for 24 h. Cells were harvested in
RIPA lysis buffer containing proteinase and phosphatase inhibitors.
Protein was quantified using a protein assay kit (Bicinchoninic
Acid; Yeasen). Equal amounts of cell lysates were separated by 12%
SDS-PAGE, and electrophoretically transferred to a polyvinylidene
fluoride (PVDF) membrane. The membrane was blocked with
Tris-buffered saline (TBS) containing 5% skimmed milk powder for 1
h, and then probed with specific antibodies [anti-ADRB1 from Aviva
Systems Biology Corporation (San Diego, CA, USA); anti-ADRB2,
anti-Akt, anti-Bcl-2, anti-MCL1, anti-Bcl-xL, anti-Bax, anti-CCND1,
anti-HK2 and anti-GAPDH from ProteinTech (Chicago, IL, USA);
anti-mTOR, anti-phospho-mTOR and anti-phospho-Akt from Signalway
Antibody (SAB; Signalway Antibody, College Park, MD, USA);
anti-GLUT1 from novus Biologicals (Littleton, CO, USA);
anti-β-actin from Sigma] overnight at 4°C and followed by
horseradish peroxidase (HRP)-labeled goat anti-mouse IgG or
HRP-labeled goat anti-rabbit IgG (both from Abcam, Cambridge, MA,
USA) for 1 h. The membranes were developed using the enhanced plus
chemiluminescence assay (Thermo Fisher Scientific, Waltham, MA,
USA) according to the manufacturer's instructions. Images were
analyzed using Image-Pro Plus 6.0.
Xenograft studies and PET/CT imaging
All protocols involving mice were evaluated and
approved by our Institutional Animal Care and Use Committee and
performed under veterinary supervision. Nude mice (5-week-old) were
injected with 2×106 8505C cells in RPMI-1640 medium
subcutaneously in the left flank. When tumors reached 50
mm3 mice were injected subcutaneously with propranolol
(Sigma) dissolved in PBS (n=15) or PBS alone (n=10) at a dose of 10
mg/kg/day for up to 15 days. Tumor growth was monitored every two
days and tumor volume was calculated (volume = length x
width2/2). Following in vivo pharmacologic
intervention, 130–200 µCi 18F-FDG in 200
µl of saline were injected into the tail vein of each mouse.
Anesthesia was performed with isoflurane anesthesia system. The
PET/CT data acquisition procedure was performed on Siemens Inveon
PET-CT when the mice were fully anesthetized. Body temperature was
maintained using a heating pad equipped with the micro PET/CT
system. All PET/CT images were processed and analyzed using
Intrasense software. After PET/CT imaging, mice bearing ATC were
sacrificed and resected tumors were weighted, followed by fixation
of the specimens.
Histopathology and
immunohistochemistry
The resected tumor specimens of ATC xenografts were
fixed in 10% neutral buffered formalin and embedded in paraffin.
Sections were cut on a microtome and mounted on glass slides.
Sections were dewaxed and hydrated in graded alcoholic solutions
and then distilled water. Routine hematoxylin and eosin (H&E)
staining was carried out. Immunohistochemical staining for Bcl-2,
CCND1 and Ki-67 were performed using the SABC kit according to the
manufacturer's instructions.
Statistical analysis
Statistical analyses were performed using the
Statistical Package for the Social Sciences, version 20.0 (SPSS,
Inc., Chicago, IL, USA) and GraphPad prism version 5.0 (GraphPad
Software, Inc., La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant result.
Results
β-adrenergic receptors are expressed in
8505C and K1 cell lines
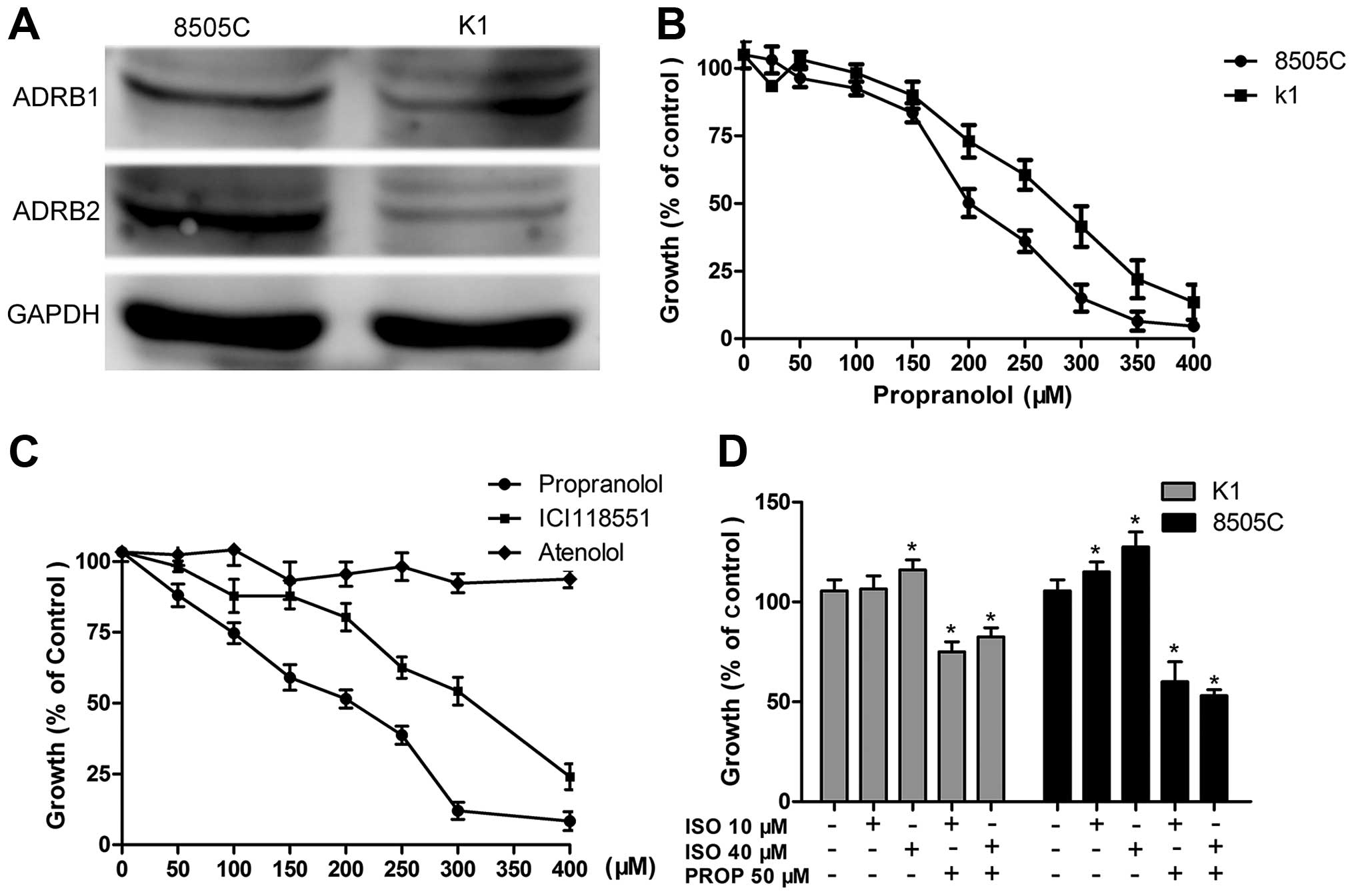
As is shown in Fig.
1A, western blotting of 8505C and K1 lysates demonstrated that
ADRB 1/2 could be detected in both 8505C and K1 cell lines. ADRB2
is relatively higher expressed in 8505C cells than that expressed
in K1 cells. It seems that the expressions of ADRB1 between the two
cell lines are comparable. Positive expression of ADRB1/2 suggests
that propranolol may influence the viability of thyroid cancer
cells through targeting ADRB1/2.
Propranolol inhibits the growth of 8505C
and K1 cells in vitro
To determine the effect of propranolol on thyroid
cancer cells, CCK-8 assay was used to determine the half-maximal
inhibitory concentration (IC50). Growth curves of 8505C
and K1 cells treated with increasing doses of propranolol for 24 h
are shown in Fig. 1B. For 8505C and
K1, the IC50 was 200 and 280 µM, respectively,
which were higher than that reported in neuroblastoma (5) and similar to those reported in other
cancer cells (3).
Growth inhibition of propranolol is
specific to β2-AR
Next, we explored whether β1-AR or β2-AR is involved
in the inhibitory effect of propranolol, 8505C cells were treated
with non-selective β-blocker propranolol, β1-specific antagonist
atenolol and β2-specific antagonist ICI118551 hydrochloride. With
the concentrations increased, propranolol and ICI118551 showed
equivalent inhibitory function on the growth of 8505C while
atenolol had no effect, suggesting that propranolol induces cell
death via blocking β2-AR rather than β1-AR (Fig. 1C). In contrast to the growth
suppression in response to β-blockers, we observed that
isoproterenol induced a dose-dependent cell growth in both 8505C
and K1 cells and this effect can be reversed by pretreatment using
propranolol (50 µM) (Fig.
1D).
Propranolol induces apoptosis of 8505C
cells in vitro
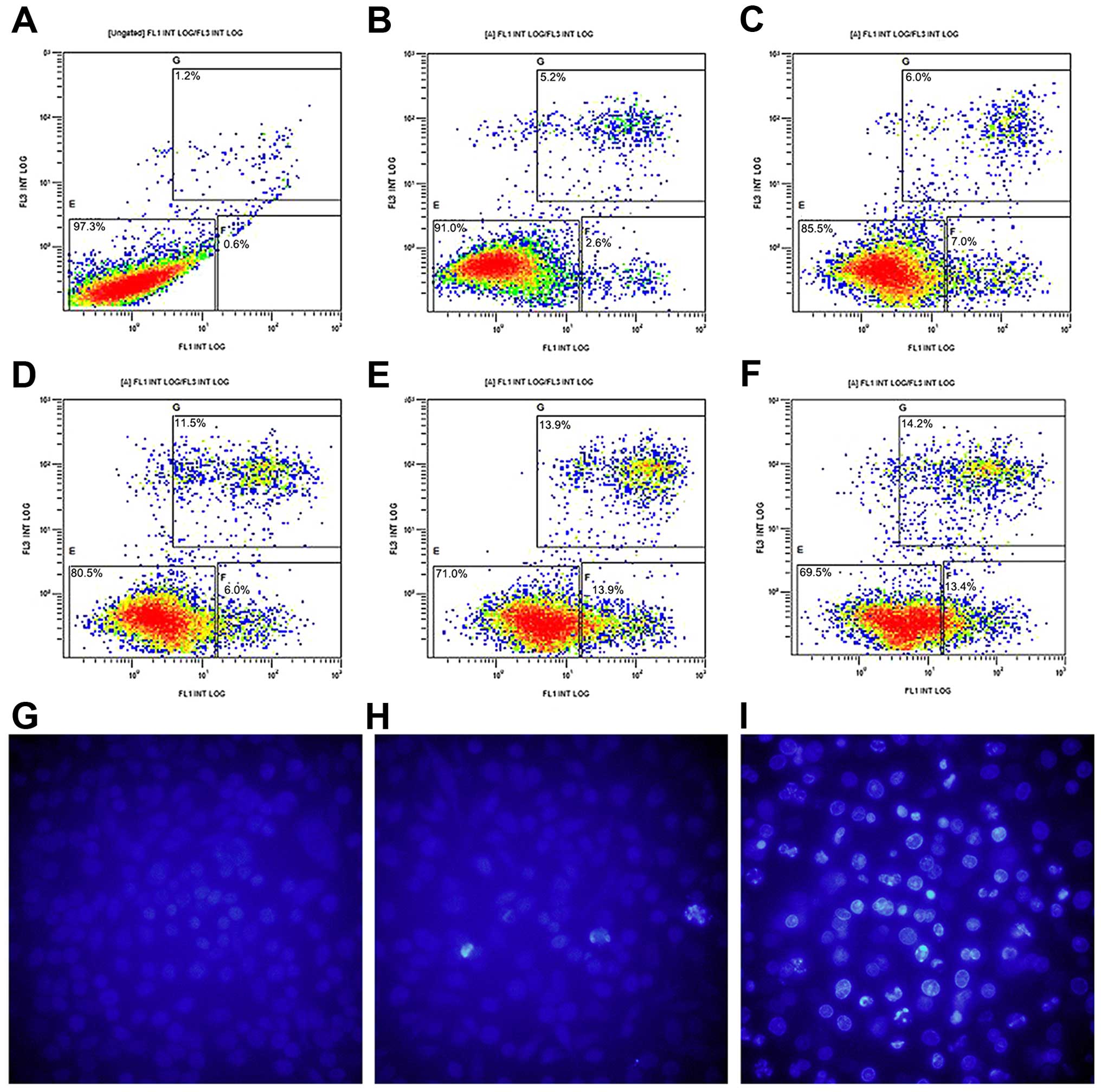
In order to study the mechanism of
propranolol-induced inhibition on the viability of 8505C cells, we
detected cell apoptosis in the normal control and
propranolol-treated 8505C cells as depicted in Fig. 2. Fig.
2A–F shows that following the propranolol treatment (0–500
µM) for 24 h, FCM revealed a dose-dependent increase in
Annexin V-FITC/PI-positive apoptotic cells (apoptosis rate from 0
to 500 µM: 1.8, 7.8, 13, 17.5, 27.8 and 27.6%). In addition,
propranolol induced apoptosis was further characterized by nuclear
fragmentation, a hallmark feature of apoptosis (Fig. 2G–I). These data suggested that
propranolol was able to induce apoptosis of 8505C cells in a
dose-dependent manner in vitro.
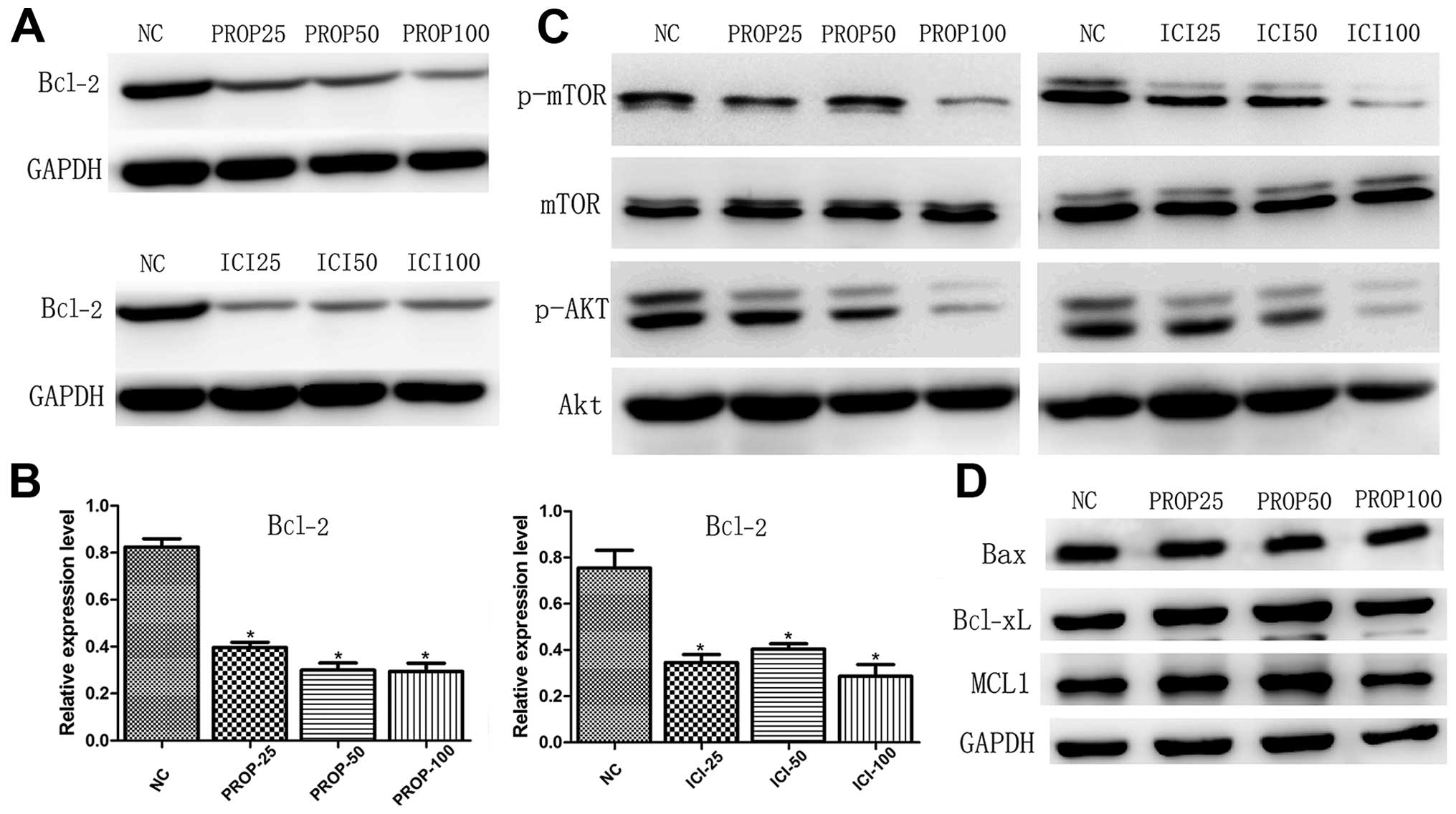
Propranolol treatment decreases the
expressions of Bcl-2, p-Akt and p-mTOR
Bcl-2 family members, including Bcl-2, Mcl-1,
Bcl-xL, Bim, Bax and Bak, have been reported to be involved in the
mitochondrial manner of apoptosis, of which anti-apoptotic Bcl-2 is
overexpressed in certain solid tumors and inhibition of Bcl-2 may
enhance apoptosis and primary responses to targeted therapy
(12–14). Following treatment with increasing
concentrations of propranolol and ICI118551 for 24 h, Bcl-2
decreased after treatment with propranolol and ICI118551, and these
two drugs yielded similar inhibitory effect in 8505C cells.
Notably, expression levels of other Bcl-2 family members did not
change following propranolol treatment (Fig. 3D). Given the central role of
PI3K/Akt/mTOR pathway in tumorigenesis, the fact that AKT directly
phosphorylates pro-apoptotic BAD and restores anti-apoptotic Bcl-xL
and Bcl-2 (15–17), we next investigated the expression
profiles of Akt and mTOR. Western blotting of 8505C lysates
validated that propranolol and ICI118551 significantly suppressed
the expression levels of p-Akt and p-mTOR in 8505C cells at a
relatively higher concentration (Fig.
3C).
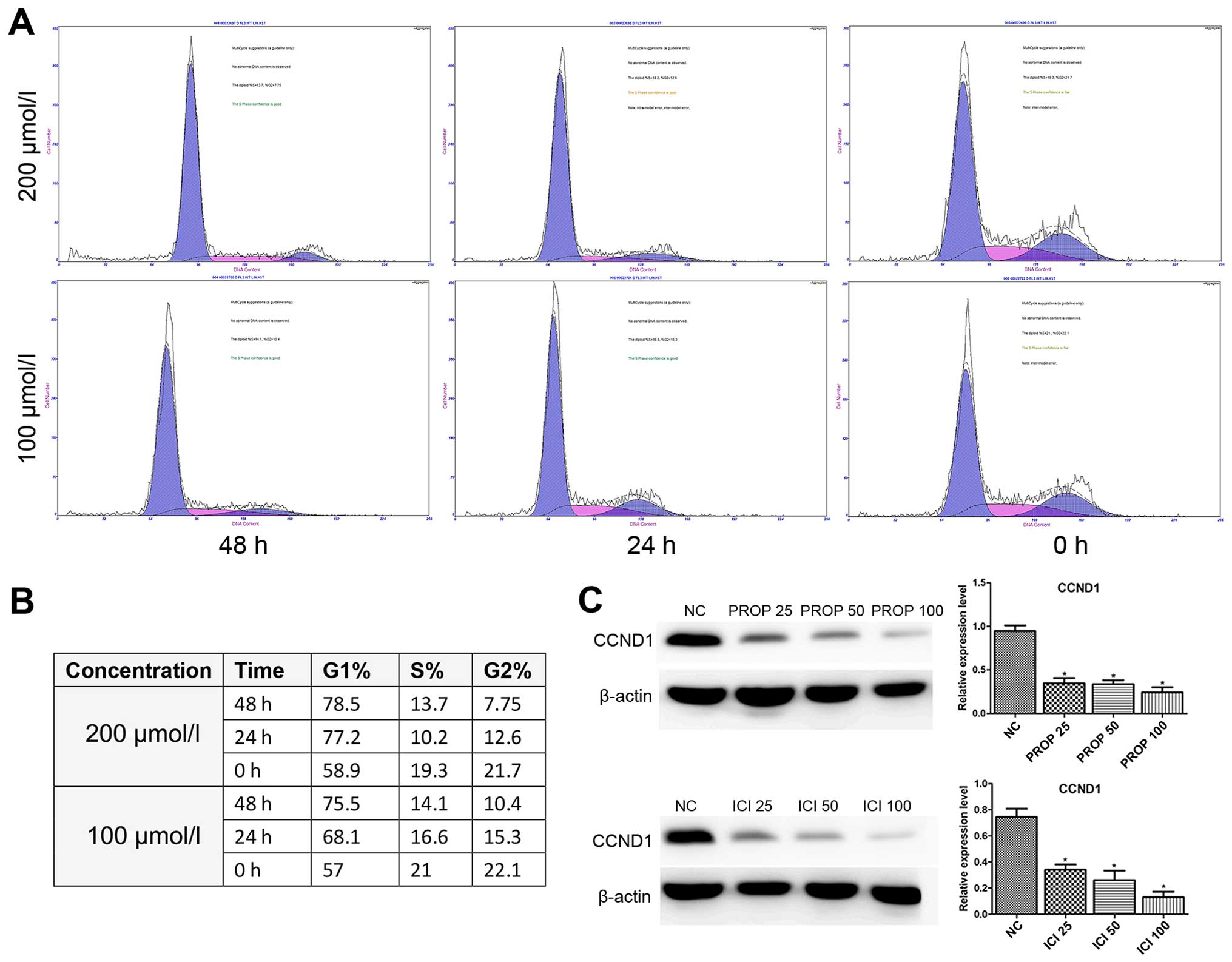
Propranolol induces 8505C cell cycle
arrest through downregulating cyclin D1
Furthermore, we performed cell cycle analysis in the
propranolol-treated 8505C cells as shown in Fig. 3A and B. Exposure of 8505C cells to
propranolol resulted in the enrichment of G0/G1 phase accompanied
by a decrease in the S phase in a dose- and time-dependent manner.
The cell cycle regulation protein cyclin D1, which is responsible
for cell cycle progression (18),
was also investigated in the present study. The expression level of
cyclin D1 was dose-dependent and significantly decreased in both
propranolol and ICI118551 treated groups when compared to the
normal controls (Fig. 4C).
Similarly Zhang et al also reported decreased expression of
Bcl-2 and cyclin D1 induced by propranolol in pancreatic cancer
cells (8).
Propranolol intervention downregulates
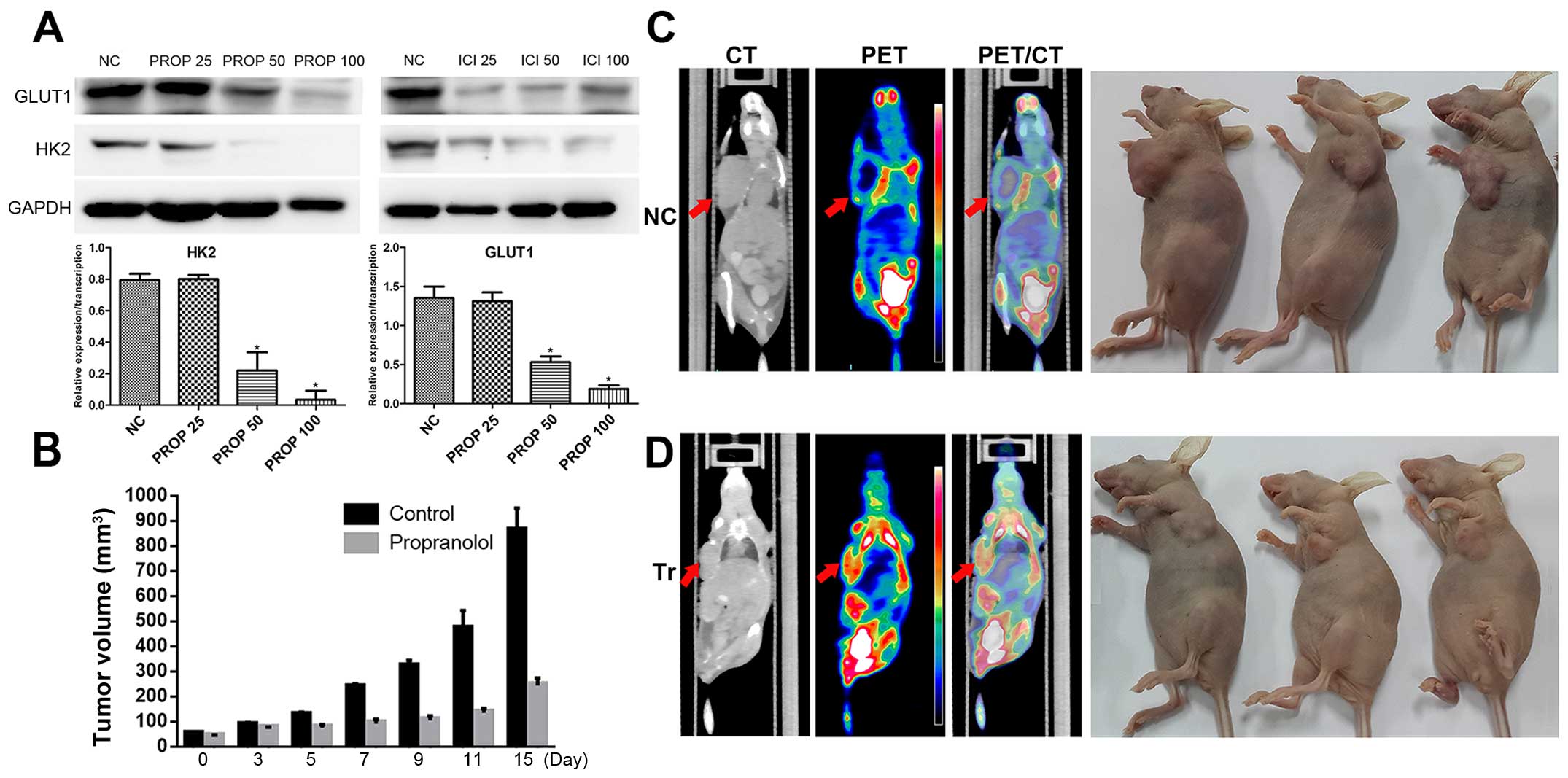
the expression of HK2 and GLUT1
During dedifferentiation process from differentiated
thyroid cancer to anaplastic thyroid cancer, an inverse
relationship between radioiodine and fluorodeoxyglucose uptake was
observed (19). Among the major
proteins regulating the transportation and metabolism of glucose,
GLUT-1 and HK-2 are closely related to the rate of
18F-FDG uptake in cancers. Therefore, we investigated
the relationship between the blockage of ADRB and the expression of
GLUT-1 and HK-2 in 8505C cells in vitro. The results of the
in vitro propranolol and ICI118551 intervention revealed
that the expression of both GLUT-1 and HK-2 significantly decreased
after treatment using β-blockers (Fig.
5A).
In vivo propranolol intervention and
18F-FDG PET/CT imaging
Finally we investigated the effect of propranolol on
the growth of 8505C xenografts in vivo. Under the
circumstances that ATC is high metabolic and the tumors in the
treatment group shrinked (Fig. 5B),
first we assessed the nude mouse tumors using 18F-FDG
PET/CT scan. Coronal computed tomography validated that tumor of
the control group was bigger than that of the propranolol treated
group (Fig. 5C and D).
SUVmax of the control group is statistically higher than
that of the propranolol intervention group (SUVmax 8.9
vs. SUVmax 2.1). It was noteworthy that the mean body
weights of the two groups were not statistically significant (data
not shown), suggesting limited toxicity of propranolol in the
short-term. Although no low density areas appeared on the CT scans
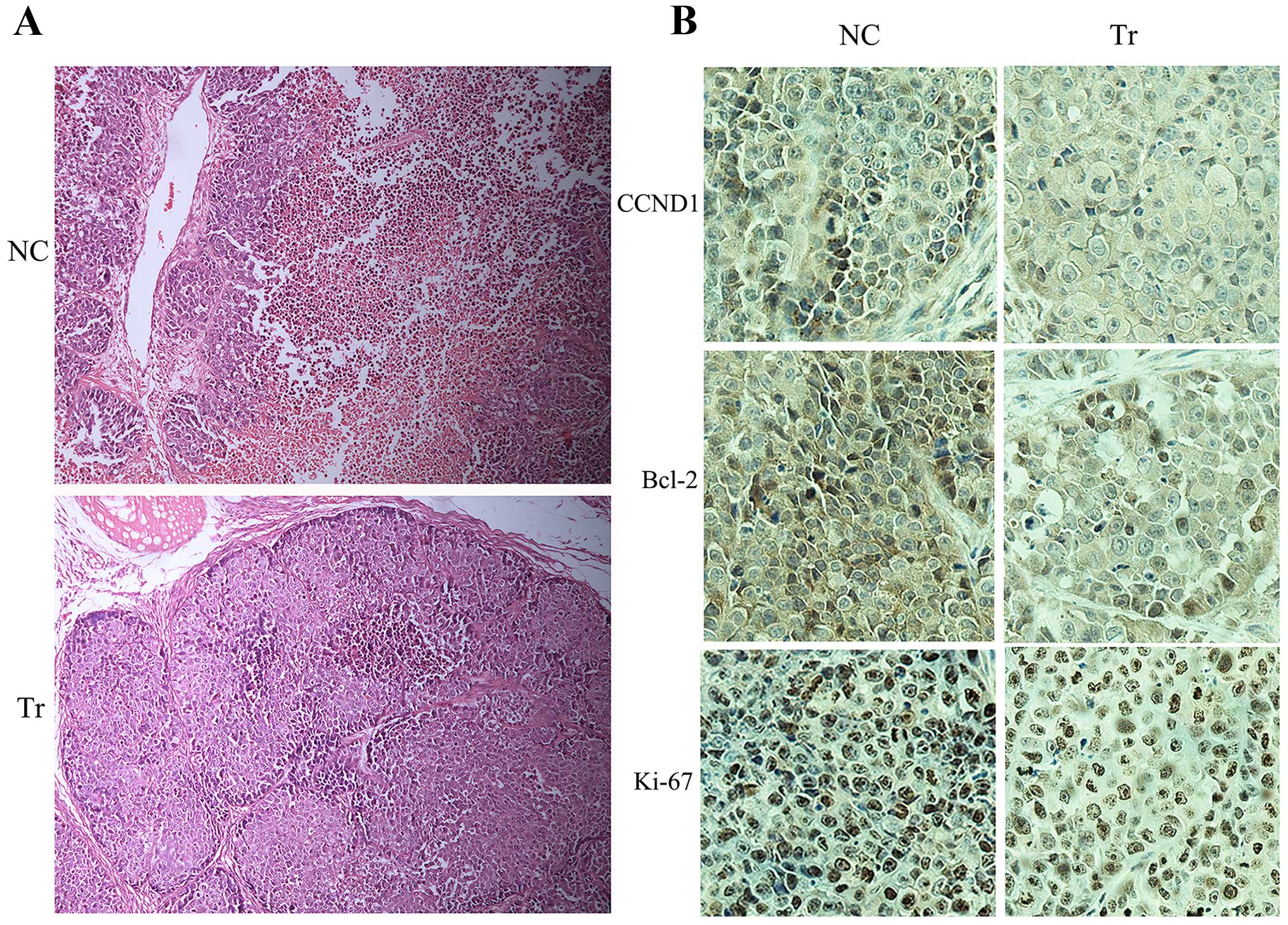
of the control mice, H&E staining of the tumor specimens of the
control group showed necrosis in the middle area of the slice
(Fig. 6A), partially reflecting the
aggressive property of ATC in the control group mainly caused by
rapid tumor progression. Consistent with western blot results,
immunohistochemistry analysis of the resected tumor tissue showed
decreased cyclin D1 and Bcl-2 in the propranolol treated group when
compared with normal controls (Fig.
6B). Delayed tumor proliferation in the experimental group was
further supported by immunohistochemical staining of tumor cells
with the proliferation marker Ki-67 (Fig. 6B).
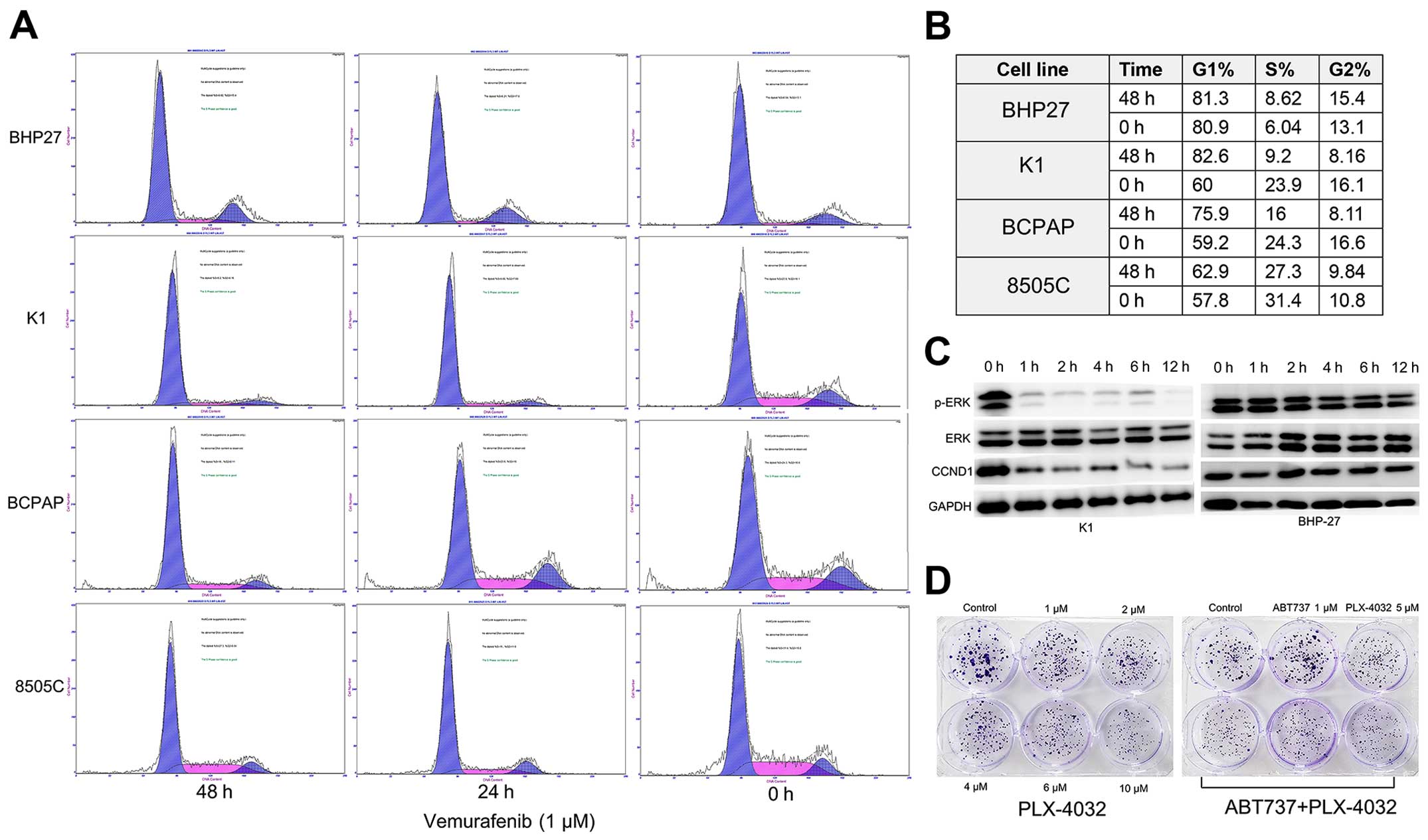
Propranolol sensitizes thyroid cancer
cells to vemurafenib
The present study confirmed the previous observation
that BRAFV600E inhibitor vemurafenib induces cell cycle
arrest of BRAF mutant cell lines K1 and BCPAP, but had little
impact on the 8505C and BRAF-WT BHP27 cells (Fig. 7A and B). In addition, we found that
G0/G1 cell cycle arrest was correlated with reduced phosphorylation
of ERK1/2 and reduced expression of cyclin D1 in K1 cells
time-dependently (Fig. 7C).
However, as was seen from the colony formation assay, vemurafenib
(PLX-4032) alone only inhibited the proliferation of K1 cells, but
was insufficient to induce apoptosis. Neither ABT-737 (a Bcl-2
inhibitor) nor the combination of ABT-737 and vemurafenib resulted
in profound synergism or extensive tumor cell death (Fig. 7D; data not shown), although ABT-737
has been proven to be potent when used along with MEK inhibitor or
vemurafenib in melanoma (13,20).
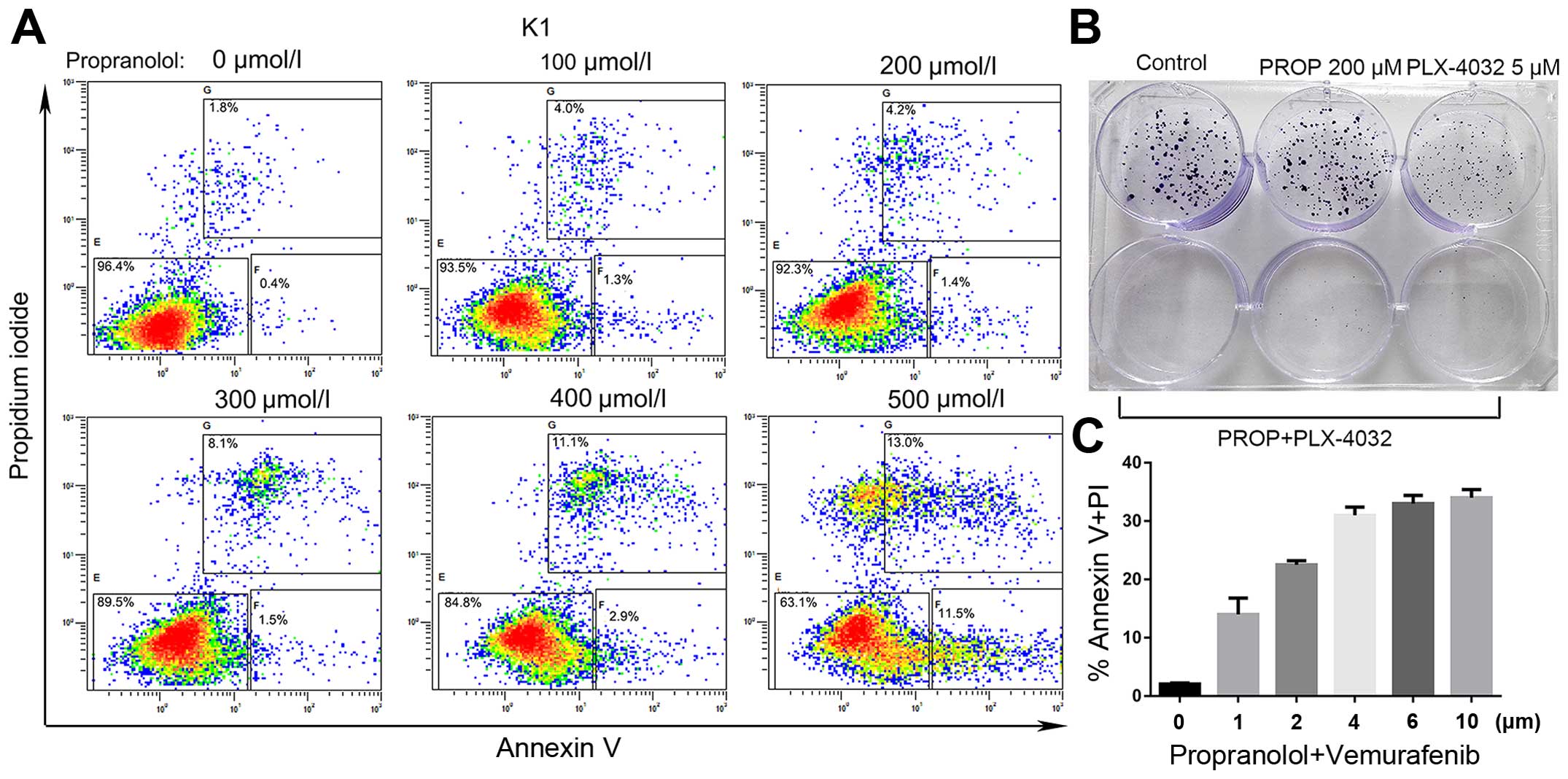
Notably propranolol alone induced apoptosis of K1 cells at a
relatively higher concentration (Fig.
8A). Most importantly, propranolol pretreatment (200 µM)
sensitized K1 cells to the cytotoxicity of vemurafenib
characterized by the loss of clonogenic survival and enhanced
apoptosis (Fig. 8B and C). These
preliminary results demonstrated that propranolol may enhance the
cytotoxicity and minimize the side-effects of the targeted
molecular therapy.
Discussion
While stress-induced activation of β-adrenergic
signaling stimulates tumor cell proliferation, migration, invasion
and suppresses apoptosis (21,22),
treatment with β-antagonist propranolol reversed these
stress-induced effects (23,24).
Population based studies have also demonstrated that breast cancer
patients with propranolol intake history were less likely
associated with a T4 or N2/N3/M1 tumor stage at initial diagnosis
and breast cancer-specific mortality was significantly lower for
propranolol users, and that propranolol usage was also associated
with improved relapse-free survival for triple-negative breast
cancer patients (10,25).
In the present study, we found that β-adrenergic
receptors are expressed in both 8505C and K1 cell lines and that
blockage of β2-AR, but not β1-AR, inhibited the growth of 8505C
cells in vitro and in vivo. We have also explored the
many potential underlying mechanisms, and discovered that
inhibition of β2-AR using either propranolol or ICI118551 was
inversely correlated with the expressions of p-Akt, p-mTOR, Bcl-2,
cyclin D1, HK2 and GLUT1. Furthermore, we clarified the impact of
propranolol on ATC xenografts and validated the shrinkage of tumors
using 18F-FDG PET/CT imaging. Immunohistochemistry of
the tumor specimens affirmed the downregulation of Bcl-2 and cyclin
D1 revealed by western blotting. Finally we highlighted the
potential possibility of the combination therapy consisting of
propranolol and BRAF specific inhibitor.
Previous studies have demonstrated that propranolol
had an negative effect on the 18F-FDG uptake of brown
adipose tissue and expression of HK2 was mediated by propranolol in
breast cancer model (26–28), similarly we found that propranolol
suppressed both the expressions of HK2 and GLUT1 in ATC cell line
in vitro despite the intriguing PET imaging of the ATC
xenografts. The altered expression of HK2 was probably regulated at
the post-transcriptional level (28). HK2 has been reported to be
associated with lung and breast cancer development and its deletion
was therapeutic in mice bearing lung tumors (29,30),
therefore we suppose that downregulation of HK2 may account for the
propranolols anti-ATC properties to some extent. In addition,
vascular endothelial growth factor (VEGF) plays an important role
in thyroid carcinogenesis and its expression level correlates with
advanced disease (31). Propranolol
has been reported to inhibit VEGF and capillary vessel formation
in vivo (32,33), suggesting synergistic effects of
propranolol through various mechanisms. In addition, this kind of
effect may also rationalize the relative high concentration to
decrease the levels of target proteins in vitro (100
µM) to that needed to suppress tumor growth in vivo
(10 mg/kg/day).
Although the excellent prognosis of most thyroid
cancer cases, there are few treatment options for
radioiodine-resistant, metastatic differentiated thyroid cancer and
anaplastic thyroid cancer (34).
Targeted therapy has shown promise in clinical trials but cellular
resistance occurs, and sometimes termination of the targeted
therapy is unavoidable due to adverse effects (AE). Combination
therapy containing BRAFV600E inhibitor or multikinase
inhibitor is a promising option to prevent resistance and to reduce
AEs. Decreasing anti-apoptotic BCL-2 family members and lowering
the cellular threshold for apoptosis is highlighted by recent
studies (35,36). Serasinghe et al found that
although inhibition of BRAFV600E by PLX-4032 sensitized
melanoma cells to the mitochondrial manner of apoptosis but only a
fraction of cells eventually underwent apoptosis. Addition of
ABT-737 (a Bcl-2 and Bcl-xL inhibitor) to PLX-4032 promoted
apoptosis and reduced development of resistance to targeted therapy
(13). Cragg et al
demonstrated that addition of ABT-737 to MEK inhibitor converted
cytostatic effect of MEK inhibition to a cytotoxic effect and
induced long-term tumor regression in mice bearing melanoma,
successfully overcoming apoptotic resistance caused by
overexpression of Bcl-2 (20).
However, some preclinical studies have investigated effects of β-AR
signaling in the regulation of tumor cell apoptosis and anoikis.
Sastry et al determined that epinephrine via the β2-AR
reduces the sensitivity of prostate and breast cancer cells to
apoptosis (22). Sood et al
showed that the β-AR agonists epinephrine and norepinephrine not
only enhance the invasive potential but also protected ovarian
tumor cells from apoptosis and that this effect was inhibited by
the β1/β2-non-selective antagonist propranolol (37). Two further studies have shown that
inhibition of β2-AR signaling by propranolol or combined usage of a
β2-adrenergic receptor specific antagonist and gemcitabine induces
apoptosis in pancreatic cancer cells via downregulating Bcl-2
(7,38). In response to propranolol blockade
we also detected decreased levels of Bcl-2 and the phosphorylated
Akt in 8505C cells, along with previous studies we tend to believe
that propranolol mediate and induce apoptosis through lowering the
expression of the anti-apoptotic protein Bcl-2. Considering its
well-tolerated property, its function in inhibiting growth,
inducing apoptosis and lowering Bcl-2 level in thyroid cancer, we
supposed that propranolol may play a role in combination with
targeted agent in inhibiting refractory or progressive thyroid
cancer.
In conclusion, our results indicated that
propranolol, in addition to its primary action on cardiovascular
diseases such as hypertension and arrhythmias, has potential
anti-thyroid cancer properties. Studies investigating the combined
administration of propranolol and targeted molecular agent in
suppressing thyroid cancer should be conducted in the future.
Acknowledgments
The present study was sponsored by the National
Natural Science Foundation of China (nos. 81271611 and
81201115).
References
|
1
|
Haugen BR and Sherman SI: Evolving
approaches to patients with advanced differentiated thyroid cancer.
Endocr Rev. 34:439–455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wells SA Jr and Santoro M: Update: The
status of clinical trials with kinase inhibitors in thyroid cancer.
J Clin Endocrinol Metab. 99:1543–1555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hajighasemi F and Mirshafiey A: In vitro
sensitivity of leukemia cells to propranolol. J Clin Med Res.
1:144–149. 2009.PubMed/NCBI
|
|
4
|
Liao X, Che X, Zhao W, Zhang D, Bi T and
Wang G: The β-adrenoceptor antagonist, propranolol, induces human
gastric cancer cell apoptosis and cell cycle arrest via inhibiting
nuclear factor κB signaling. Oncol Rep. 24:1669–1676.
2010.PubMed/NCBI
|
|
5
|
Wolter JK, Wolter NE, Blanch A, Partridge
T, Cheng L, Morgenstern DA, Podkowa M, Kaplan DR and Irwin MS:
Anti-tumor activity of the beta-adrenergic receptor antagonist
propranolol in neuroblastoma. Oncotarget. 5:161–172.
2014.PubMed/NCBI
|
|
6
|
Wolter NE, Wolter JK, Enepekides DJ and
Irwin MS: Propranolol as a novel adjunctive treatment for head and
neck squamous cell carcinoma. J Otolaryngol Head Neck Surg.
41:334–344. 2012.PubMed/NCBI
|
|
7
|
Zhang D, Ma Q, Shen S and Hu H: Inhibition
of pancreatic cancer cell proliferation by propranolol occurs
through apoptosis induction: The study of beta-adrenoceptor
antagonist's anticancer effect in pancreatic cancer cell. Pancreas.
38:94–100. 2009. View Article : Google Scholar
|
|
8
|
Zhang D, Ma QY, Hu HT and Zhang M:
β2-adrenergic antagonists suppress pancreatic cancer cell invasion
by inhibiting CREB, NFκB and AP-1. Cancer Biol Ther. 10:19–29.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Powe DG, Voss MJ, Zänker KS, Habashy HO,
Green AR, Ellis IO and Entschladen F: Beta-blocker drug therapy
reduces secondary cancer formation in breast cancer and improves
cancer specific survival. Oncotarget. 1:628–638. 2010. View Article : Google Scholar
|
|
10
|
Barron TI, Connolly RM, Sharp L, Bennett K
and Visvanathan K: Beta blockers and breast cancer mortality: A
population-based study. J Clin Oncol. 29:2635–2644. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
De Giorgi V, Grazzini M, Gandini S,
Benemei S, Lotti T, Marchionni N and Geppetti P: Treatment with
β-blockers and reduced disease progression in patients with thick
melanoma. Arch Intern Med. 171:779–781. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Serasinghe MN, Missert DJ, Asciolla JJ,
Podgrabinska S, Wieder SY, Izadmehr S, Belbin G, Skobe M and Chipuk
JE: Anti-apoptotic BCL-2 proteins govern cellular outcome following
B-RAFV600E inhibition and can be targeted to reduce
resistance. Oncogene. 34:857–867. 2015. View Article : Google Scholar
|
|
14
|
Oltersdorf T, Elmore SW, Shoemaker AR,
Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges
J, Hajduk PJ, et al: An inhibitor of Bcl-2 family proteins induces
regression of solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feine U, Lietzenmayer R, Hanke JP, Wöhrle
H and Müller-Schauenburg W: 18FDG whole-body PET in differentiated
thyroid carcinoma. Flipflop in uptake patterns of 18FDG and 131I.
Nuklearmedizin. 34:127–134. 1995.In German. PubMed/NCBI
|
|
20
|
Cragg MS, Jansen ES, Cook M, Harris C,
Strasser A and Scott CL: Treatment of B-RAF mutant human tumor
cells with a MEK inhibitor requires Bim and is enhanced by a BH3
mimetic. J Clin Invest. 118:3651–3659. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Drell TL IV, Joseph J, Lang K, Niggemann
B, Zaenker KS and Entschladen F: Effects of neurotransmitters on
the chemokinesis and chemotaxis of MDA-MB-468 human breast
carcinoma cells. Breast Cancer Res Treat. 80:63–70. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sastry KSR, Karpova Y, Prokopovich S,
Smith AJ, Essau B, Gersappe A, Carson JP, Weber MJ, Register TC,
Chen YQ, et al: Epinephrine protects cancer cells from apoptosis
via activation of cAMP-dependent protein kinase and BAD
phosphorylation. J Biol Chem. 282:14094–14100. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sloan EK, Priceman SJ, Cox BF, Yu S,
Pimentel MA, Tangkanangnukul V, Arevalo JM, Morizono K, Karanikolas
BD, Wu L, et al: The sympathetic nervous system induces a
metastatic switch in primary breast cancer. Cancer Res.
70:7042–7052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Masur K, Niggemann B, Zanker KS and
Entschladen F: Norepinephrine-induced migration of SW 480 colon
carcinoma cells is inhibited by beta-blockers. Cancer Res.
61:2866–2869. 2001.PubMed/NCBI
|
|
25
|
Melhem-Bertrandt A, Chavez-Macgregor M,
Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD,
Hortobagyi GN and Gonzalez-Angulo AM: Beta-blocker use is
associated with improved relapse-free survival in patients with
triple-negative breast cancer. J Clin Oncol. 29:2645–2652. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Söderlund V, Larsson SA and Jacobsson H:
Reduction of FDG uptake in brown adipose tissue in clinical
patients by a single dose of propranolol. Eur J nucl Med Mol
Imaging. 34:1018–1022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Agrawal A, Nair N and Baghel NS: A novel
approach for reduction of brown fat uptake on FDG PET. Br J Radiol.
82:626–631. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang F, Ma W, Ma X, Shao Y, Yang W, Chen
X, Li L and Wang J: Propranolol inhibits glucose metabolism and
18F-FDG uptake of breast cancer through
posttranscriptional downregulation of hexokinase-2. J Nucl Med.
55:439–445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mathupala SP, Ko YH and Pedersen PL:
Hexokinase II: Cancer's double-edged sword acting as both
facilitator and gatekeeper of malignancy when bound to
mitochondria. Oncogene. 25:4777–4786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patra KC, Wang Q, Bhaskar PT, Miller L,
Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, et al:
Hexokinase 2 is required for tumor initiation and maintenance and
its systemic deletion is therapeutic in mouse models of cancer.
Cancer Cell. 24:213–228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu XM, Lo CY, Chan WF, Lam KY, Leung P and
Luk JM: Increased expression of vascular endothelial growth factor
C in papillary thyroid carcinoma correlates with cervical lymph
node metastases. Clin Cancer Res. 11:8063–8069. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Storch CH and Hoeger PH: Propranolol for
infantile haemangiomas: Insights into the molecular mechanisms of
action. Br J Dermatol. 163:269–274. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Filippi L, Dal Monte M, Casini G, Daniotti
M, Sereni F and Bagnoli P: Infantile hemangiomas, retinopathy of
prematurity and cancer: A common pathogenetic role of the
β-adrenergic system. Med Res Rev. 35:619–652. 2015. View Article : Google Scholar
|
|
34
|
McFarland DC and Misiukiewicz KJ:
Sorafenib in radioactive iodine-refractory well-differentiated
metastatic thyroid cancer. Onco Targets Ther. 7:1291–1299. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Champa D, Russo MA, Liao XH, Refetoff S,
Ghossein RA and Di Cristofano A: Obatoclax overcomes resistance to
cell death in aggressive thyroid carcinomas by countering Bcl2a1
and Mcl1 overexpression. Endocr Relat Cancer. 21:755–767. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Broecker-Preuss M, Viehof J, Jastrow H,
Becher-Boveleth N, Fuhrer D and Mann K: Cell death induction by the
BH3 mimetic GX15-070 in thyroid carcinoma cells. J Exp Clin Cancer
Res. 34:692015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sood AK, Bhatty R, Kamat AA, Landen CN,
Han L, Thaker PH, Li Y, Gershenson DM, Lutgendorf S and Cole SW:
Stress hormone-mediated invasion of ovarian cancer cells. Clin
Cancer Res. 12:369–375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shan T, Ma Q, Zhang D, Guo K, Liu H, Wang
F and Wu E: β2-adrenoceptor blocker synergizes with gemcitabine to
inhibit the proliferation of pancreatic cancer cells via apoptosis
induction. Eur J Pharmacol. 665:1–7. 2011. View Article : Google Scholar : PubMed/NCBI
|