Introduction
Recent studies have shown that approximately 80% of
the human genome is transcribed to RNA, with only 2% being
responsible for protein coding. According to their size, non-coding
RNAs (ncRNAs) are classified into small and long ncRNAs. Long
non-coding RNAs (lncRNAs) are a class of RNA molecules with >200
nucleotides that function as RNAs with little or no protein-coding
capacity (1). lncRNAs have been
implicated to play a functional role in carcinogenesis and cancer
growth (2). There is increasing
evidence that lncRNAs participate in a diversity of biological
processes. Expression of many lncRNAs is altered and is likely to
function in tumorigenesis. The functional diversity and mechanistic
role of lncRNAs are currently a field of intense investigation
(3).
Maternally expressed gene 3 (MEG3), an lncRNA, is an
imprinted gene located on chromosome 14q32.3 that functions as a
tumor suppressor (4,5). MEG3 is expressed in many normal human
tissues, with the highest expression in brain and pituitary gland.
However, its expression is lost or decreased in common human
tumors, such as glioma, colon cancer, hepatocellular cancer,
non-small cell lung cancer (NSCLC) and meningioma (2,6–9). We
and other investigators have demonstrated that promoter
hypermethylation or hypermethylation of the intergenic
differentially methylated region contributes to the loss of MEG3
expression in tumors (10).
The endoplasmic reticulum (ER) is the intracellular
organelle where protein synthesis, folding, and modifications
occur, as well as regulation of intracellular Ca2+
homeostasis (11). Various stimuli
can disturb ER homeostasis resulting in the accumulation of
unfolded or misfolded proteins leading to pathological
consequences, generally known as ER stress (12).
Slight stimulation activates an adaptive signaling
cascade to counteract the ER stress-associated damage, known as the
unfolded protein response (UPR) (13). During UPR, three major stress
sensors, comprised of the RNA-dependent protein kinase-like ER
kinase (PERK), activating transcription factor 6 (ATF6), and
inositol-requiring enzyme 1 (IRE1), reside on the ER membrane and
are activated by dissociating from the 78-kDa glucose-regulated
protein (GRP78). UPR can enhance cell survival (14). However, when the response is not
sufficient and ER stress persists, the UPR leads to apoptosis
through activation of C/EBP homologous protein (CHOP) (15), caspase-12 (16) and/or c-Jun NH2-terminal kinase (JNK)
(17).
Our previous study indicated that adenosine induces
ER stress and p53 activation, as well as the expression of MEG3 by
altering the methylation status of the MEG3 promoter (12), and MEG3 also induces cell apoptosis
through the p53-dependent pathway (10,18).
Moreover, p53 activation is thought to be related to ER stress
(19). However, the biological role
of MEG3 has not yet been completely understood; In particular,
little is known concerning the regulatory relationship between MEG3
and ER stress. Here, we explored a functional mechanism of MEG3
regulation via activating ER stress and p53 pathway to induce cell
apoptosis, and further defined its tumor-suppressor function.
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM) was
purchased from Invitrogen (Carlsbad, CA, USA). Primary antibodies
against GRP78, IRE1, PERK, ATF6, p53, and NF-κB were obtained from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Primary
antibodies against GAPDH and caspase-3 were purchased from Cell
Signaling Technology (CST; USA) and CHOP from MengZhouShi Ruiying
Biological Technology Co., Ltd. (China). Anti-rabbit and anti-mouse
secondary antibodies were purchased from LI-COR Biosciences (USA).
Alexa Fluor 555-labeled donkey anti-rabbit IgG was obtained from
Abcam (USA). The Annexin v/FITC Apoptosis Detection kit was
obtained from Beijing 4A Biotech Co., Ltd. (China). The primer
sequences for the MEG3 gene, used to verify transfection efficiency
were: forward, CTC AGG CAG GAT CTG GCA TA; and reverse, CCT GGA GTG
CTG TTG GAG AA and purchased from Invitrogen. Other materials, such
as Tris, SDS and glycine were obtained from Sangon (Shanghai,
China).
Cell culture and experimental groups
Human hepatoma HepG2 cells were obtained from the
American Type Culture Collection (Manassas, vA, USA) and cultured
in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS),
penicillin (100 U/ml), and streptomycin (100 mg/ml) under a
humidified atmosphere with 5% CO2 at 37°C. The cell
culture medium was changed every other day, and cells were passaged
at 80–90% confluency. For the immunofluorescence and
immunohistochemical assay, the cells were cultured on coverslips in
DMEM for 24 h before transfection.
For the recombinant lentiviral transfection
experiments, HepG2 cells were transfected with a lentiviral vector
containing MEG3 (Lv-MEG3) sequence diluted in DMEM with 2% FBS and
8 µg/ml Polybrene at a multiplicity of infection (MOI) of
100. The cell lines transfected with the empty lentiviral vector
(Lenti6.3-MIG) at MOI of 100 served as the control.
Construction of the recombinant
lentiviral vectors and HepG2 cell lines stably expressing MEG3
The MEG3 gene was designed and synthesized by means
of PCR according to the GenBank of human MEG3 (NR_002766) gene
sequence (10). The purified MEG3
gene fragment was inserted into a lentiviral vector
(pLenti6.3-MCS-IRES2-EGFP), and the insertion fragment was
identified by PCR, restriction endonuclease analysis and DNA
sequencing. The Lv-MEG3 sequence was then transferred into HEK
293FT cells to be packaged into mature lentivirus as
Lenti-hMEG3-IRES-EGFP (Lv-MEG3, MEG3).
To obtain cell lines stably expressing MEG3, the
lentiviral vector (Lv-MEG3, MEG3) was transfected into HepG2 cells.
RT-PCR was performed to examine the mRNA expression level of
MEG3.
RNA extraction and RT-PCR assay
HepG2 cells were transfected with Lv-MEG3 or
Lenti6.3-MIG for 48 h. Cells were harvested and total RNA was
extracted using TRIzol reagent according to the manufacturer's
protocol. For reverse transcriptase analysis, 1 µg of total
RNA was reverse transcribed using an EasyScript First-Strand cDNA
Synthesis SuperMix kit. Amplification of MEG3 and GAPDH was
performed using 1 µl cDNA as a template, 12.5 µl 2X
Taq PCR MasterMix (Tiangen Biotech Co., Ltd.), 9.5 µl
ddH2O and 1 µl of each primer (10 µM stock
concentration) in a total volume of 25 µl. The PCR reaction
was performed using the following primers: MEG3 forward,
5′-CTCAGGCAGGATCTGGCATA-3′ and reverse, 5′-CCTGGAGTGCTGTTGGAGAA-3′;
GAPDH forward, 5′-CACCATCTTCCAGGAGCGA-3′ and reverse,
5′-TCAGCAGAGGGGGCAGAGA-3′.
Samples were incubated at 94°C for 3 min followed by
38 cycles of denaturation at 94°C for 30 sec, annealing at 57°C for
30 sec and extension at 72°C for 1 min, and a final extension step
at 72°C for 5 min. The PCR products were separated on a 4% agarose
gel and visualized by ethidium bromide staining.
Determination of cell proliferation by
MTT assay
Viability of the cell post-transfection was
determined using the 3-(4,5-dimethyl
thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell
proliferation kit. Cells were inoculated into 96-well plates at
5×103 cells/well and allowed to attach for 24 h. The
HepG2 cells were transfected with Lv-MEG3 or Lenti6.3-MIG for 48 h.
The culture medium was then aspirated and the cells were washed
with PBS, followed by incubation with 0.5 mg/ml of MTT at 37°C for
4 h. After incubation, the supernatant was removed and the cells
were treated with DMSO to dissolve the formazan reaction product.
The concentration of formazan was determined by measuring the
absorbance at 540 nm using an enzyme-linked immunosorbent assay
reader (FilterMax F5; Molecular Devices, USA).
Tumor formation assay in a nude mouse
model
Five-week-old female athymic BALB/c mice were
purchased from Vital River (Beijing, China) and maintained under
specific pathogen-free conditions and manipulated according to
protocols approved by the Shanghai Medical Experimental Animal Care
Commission. HepG2 cells transfected with Lv-MEG3 or Lenti6.3-MIG
were harvested, washed with cold PBS and resuspended at a
concentration of 3×107 cells/ml. A volume of 0.15 ml
each of the suspending cells was subcutaneously injected into
either side of the forelimb flank of each nude mouse. Tumor growth
was examined every 3 days in mice from the MEG3 (n=6) or control
group (n=6), and tumor volumes were calculated using the equation:
v = 0.5 × D × d2 (v, volume; D, longitudinal diameter;
d, latitudinal diameter). Twenty-two days after injection, the mice
were euthanized and tumor weights were measured. RT-PCR analysis
was performed to detect MEG3 levels in the tumor tissues.
This study was carried out in strict accordance with
the recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health (20). The protocol was approved by the
Committee on the Ethics of Animal Experiments of Shantou University
Medical College. All surgery was performed under sodium
pentobarbital anesthesia, and all efforts were made to minimize
suffering in mice.
Flow cytometric detection of
apoptosis
HepG2 cells transfected with Lv-MEG3 or Lenti6.3-MIG
were harvested by trypsinization at 48 h, washed twice with cold
PBS, and resuspended in 1X binding buffer to a concentration of
1×106 cells/ml. The cells were then stained with 5
µl Annexin v/FITC and 10 µl propidium iodide (20
µg/ml) for 15 min at room temperature in the dark. Analyses
were performed with a BD Accuri™ C6 Flow Cytometer (BD Biosciences,
USA) with the FL1 and FL3 detector.
Western blot analysis
HepG2 cells were harvested at 48 h after MEG3
transfection, and washed with cold PBS three times. Total cellular
protein lysates were prepared with RIPA buffer containing
proteinase inhibitors. Nuclear and cytoplasmic protein fractions
were prepared using a Nuclear and Cytoplasmic Protein Extraction
kit (Beyotime Institute of Biotechnology). Protein concentration
was measured using a BCA Protein Assay kit (Thermo Fisher
Scientific, USA). Protein (50 µg) was separated on 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), and transferred to nitrocellulose membranes. The
membranes were blocked at room temperature for 60 min with 5%
non-fat milk in Tris-buffered saline (pH 7.6; TBS), and incubated
with primary antibodies against GAPDH (1:1,000), GRP78 (1:500),
PERK (1:500), IRE1 (1:500), ATF6 (1:500), CHOP (1:1,000), caspase-3
(1:1,000), p53 (1:3,000) and NF-κB (1:500) in TBS at 4°C overnight,
followed by TBST (with Tween-20) washes. Membranes were incubated
with fluorescent secondary antibodies (LI-COR) coupled to the
primary antibody at room temperature in the dark for 1 h, followed
by TBST washes, dried with neutral absorbent paper and scanned by
Odyssey detection system (LI-COR). Protein expression was analyzed
using Quantity One software (Bio-Rad, USA) and normalized to GAPDH
(for total cell fraction) or TBP (for nuclear fraction).
Immunofluorescence staining
HepG2 cells were seeded at 4×105
cells/well in 6-well flat bottomed plates and incubated in 10%
FBS-supplemented DMEM for 24 h. Cells were transfected and divided
into two groups: the MEG3-transfected group and the control group.
Cells were fixed in 4% paraformaldehyde for 15 min and
permeabilized in 0.3% Triton X-100 for 10 min at room temperature,
and then blocked with 3% BSA for 30 min. Subsequently, the cells
were incubated with the primary antibody against NF-κB (anti-p65,
1:100) or with PBS (blank group) at 4°C overnight. On the following
day, HepG2 cells were incubated with the Alexa Fluor 555-labeled
donkey anti-rabbit IgG (1:100) for 1 h at 37°C. The cell nuclei
were stained with DAPI for 10 min. Cells were viewed and captured
with a fluorescence microscope (Olympus BX51).
Immunohistochemical staining
For immunochemical analysis, the cells were fixed in
4% paraformaldehyde for 10 min, permeabilized in 0.3% Triton X-100
for 10 min at room temperature, respectively, and then blocked in
3% BSA for 30 min. Subsequently, the cells were incubated with the
primary antibody against NF-κB (anti-p65, 1:50) or with PBS
(negative control) at 4°C overnight. On the second day, the cells
were then incubated with biotinylated anti-mouse (1:400; Jackson
ImmunoResearch) at room temperature for 1 h, and then with an
avidin-horseradish peroxidase complex (vectastain ABC kit; Promega,
Madison, WI, USA) at room temperature for 30 min, and visualized
with amine nickel sulfate-enhanced 3,3′-diaminobenzidine (DAB).
Hematoxylin was used to stain the cell nuclei. Sections were
observed under a light microscope, and positive staining was shown
by the development of brown particles.
Statistical analysis
All experiments were independently performed at
least three times. The values are presented as the means ± SD.
Differences were assessed by two-tailed Student's t-test. P<0.05
was considered to indicate a statistically significant result.
Results
MEG3 expression is increased after
Lv-MEG3 transfection in the HepG2 cells
In the present study, we described the generation of
a recombinant Lv-MEG3. The pcDNA3.1-MEG3 plasmid vectors and the
recombinant lentiviral vectors (Lv-MEG3) were identified using
sequencing technique. The particular sequence of MEG3 insertion
into vectors was the same as designed. The mRNA expression of MEG3
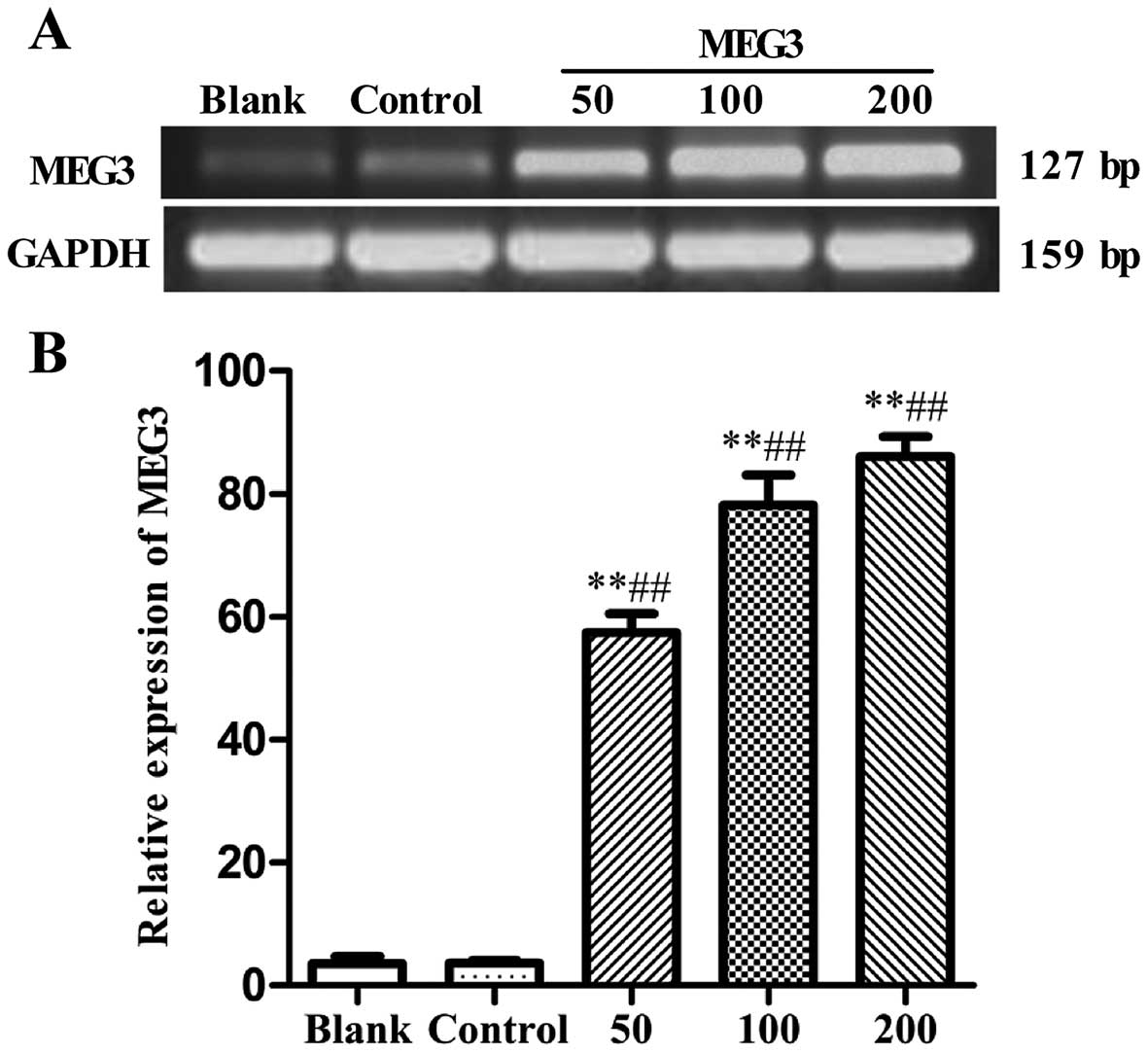
was observed by RT-PCR. As shown in Fig. 1, compared with the blank group or
empty control group (Lenti6.3-MIG), the mRNA expression of MEG3 was
significantly increased after Lv-MEG3 transfection at 50, 100 and
200 MOI (57.44±3.08, 78.13±4.91, and 86.09±3.17%, respectively, vs.
2.32±0.14, and 3.58±1.19%, both p<0.01), showing a marked
dose-dependent increase in MEG3 expression. This demonstrated that
Lv-MEG3 was successfully transfected and a high level of mRNA
expression of MEG3 was achieved in the HepG2 cells.
Ectopic expression of MEG3 inhibits
hepatoma cancer cell proliferation in vitro and in vivo
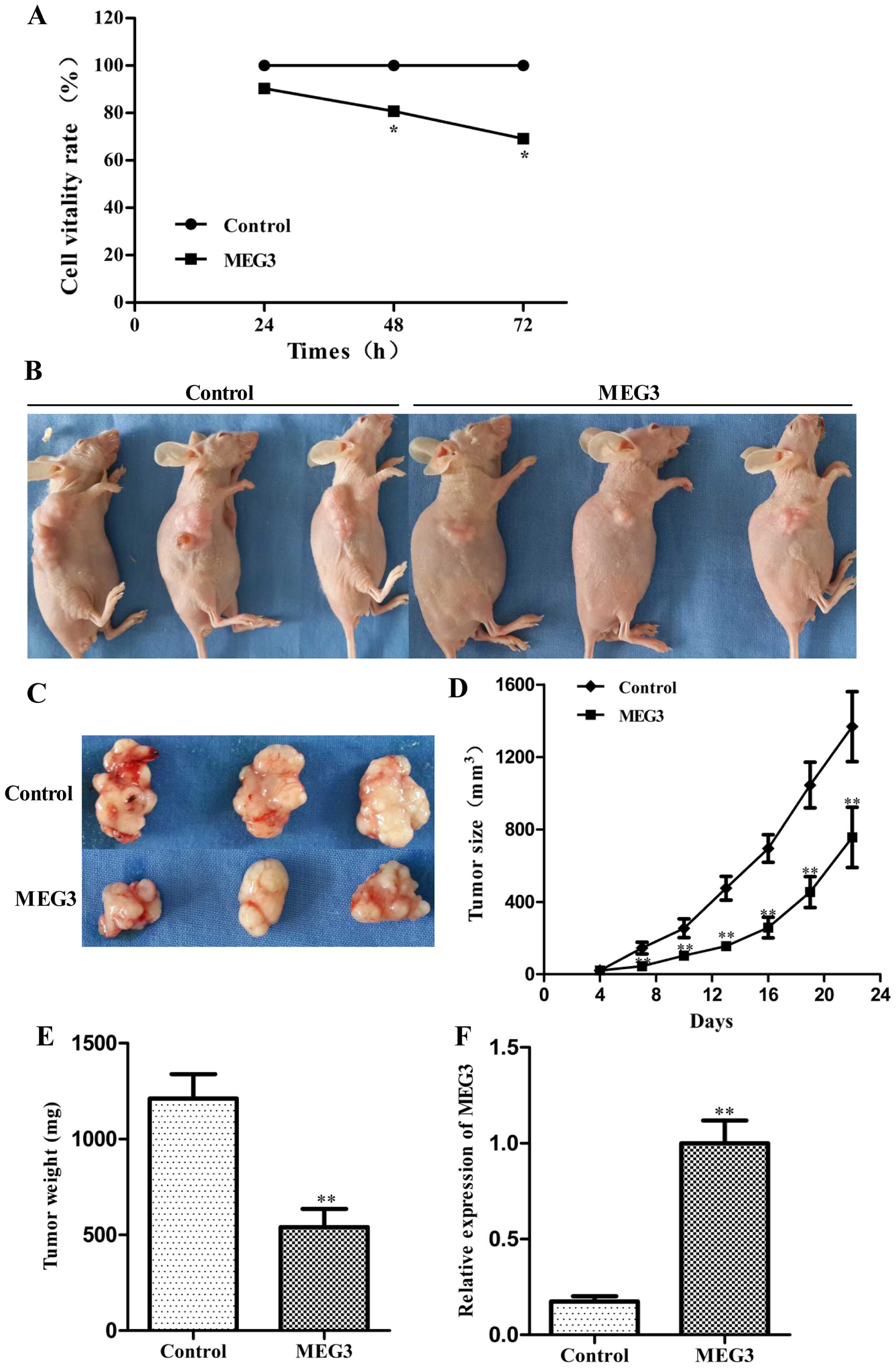
To determine whether MEG3 affects the proliferation
of HepG2 cells, we performed an MTT assay following transfection of
either Lv-MEG3 or Lenti6.3-MIG. Ectopic expression of MEG3
significantly decreased cell growth by 19.3 and 30.8% after 48 and
72 h, respectively, compared with the control group (Fig. 2A, p<0.05). Ectopic expression of
MEG3 inhibited cell pro liferation in a time-dependent manner.
These results suggest that overexpression of MEG3 inhibited HepG2
cell proliferation in vitro.
To explore whether MEG3 inhibits hepatoma growth
in vivo, the HepG2 cells transfected with Lv-MEG3 or
Lenti6.3-MIG were inoculated into nude mice respectively, and all
mice developed xenograft tumors at the injection site. Tumor growth
in the Lv-MEG3 group was significantly slower than that in the
control group on day 22 after injection (Fig. 2B–D). The average tumor weight in the
Lv-MEG3 transfection group was lower than that in control group
(Fig. 2E). RT-PCR analysis showed
that the mRNA expression level of MEG3 in the Lv-MEG3 transfection
group was significantly higher than that in the control group
(Fig. 2F). Taken together, these
results demonstrated that ectopic expression of MEG3 inhibited
hepatoma growth in nude mice.
Ectopic expression of MEG3 induces HepG2
cell apoptosis
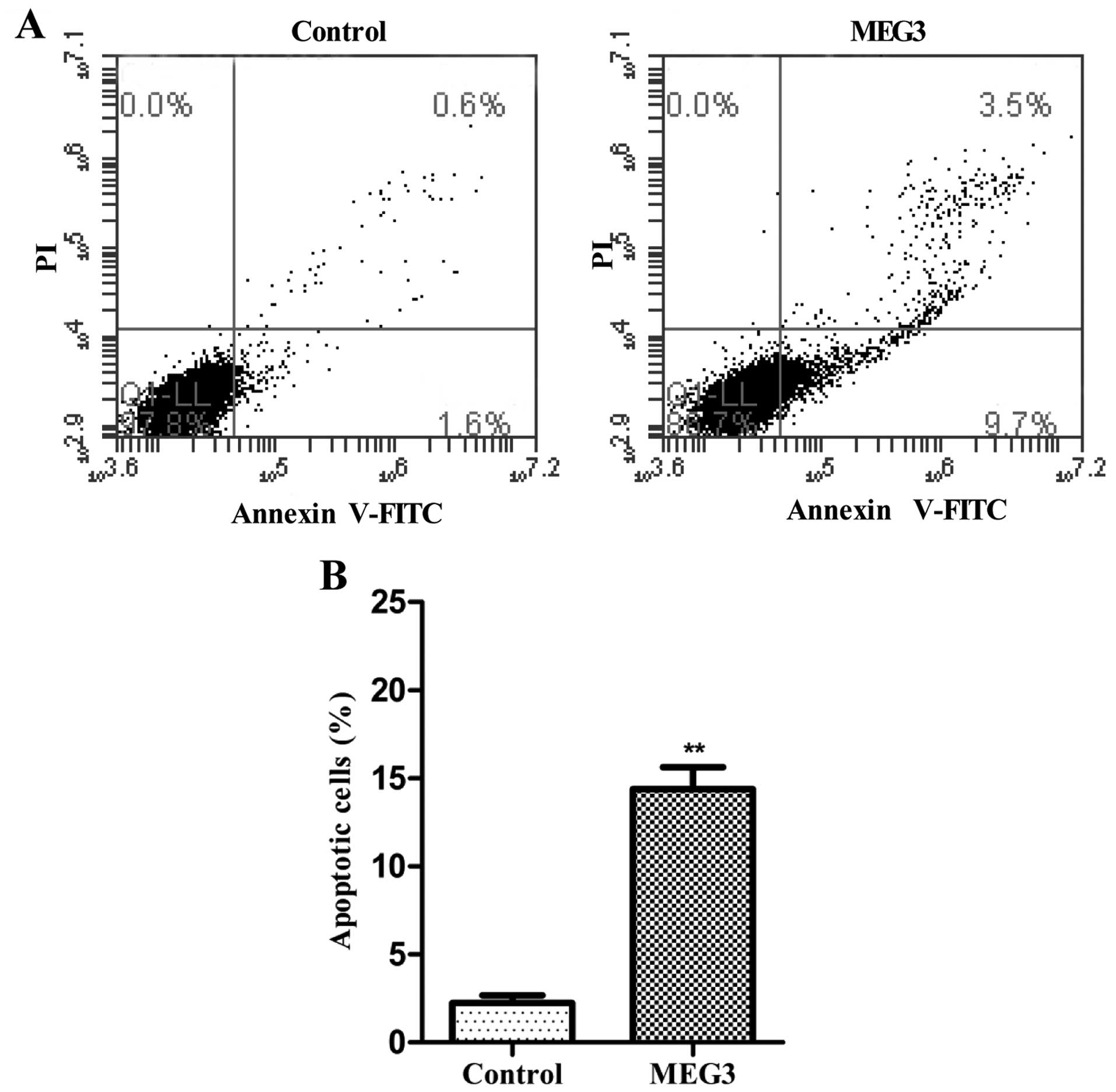
To determine whether apoptosis was a contributing
factor to cell growth arrest in the MEG3-transfected HepG2 cells,
we performed flow cytometric analysis after transfection with
Lv-MEG3 or Lenti6.3-MIG. Transfection with Lv-MEG3 increased the
fraction of apoptotic cells by ~14.7% in comparison with cells
transfected with the empty lentivirus (Lenti6.3-MIG, Fig. 3). This indicated that overexpression
of MEG3 induced HepG2 cell apoptosis in vitro.
Ectopic expression of MEG3 activates the
ER stress pathway
To verify whether the MEG3-induced cell apoptosis is
related to ER stress, ER stress-relative proteins were detected by
western blot analysis. Fig. 4 shows
that overexpression of MEG3 not only increased the expression of
GRP78, three key proteins of UPR (IRE1, PERK, ATF6), but also
increased CHOP, caspase-3 and p53 expression. The results showed
that ectopic expression of MEG3 induced HepG2 cell apoptosis
through the ER stress pathway.
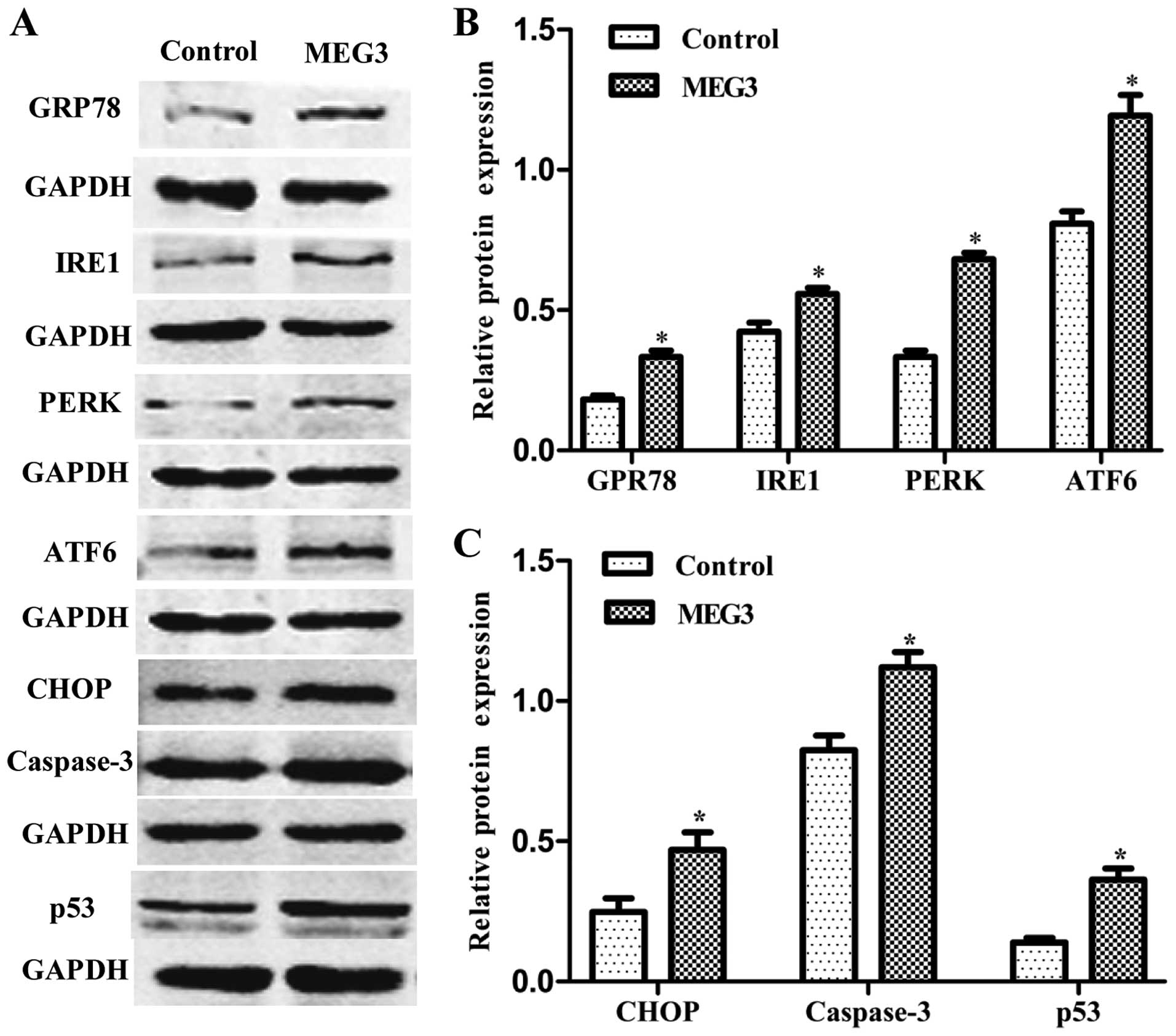 | Figure 4Effects of ectopic MEG3 on protein
expression of ER stress-related genes (GRP78, IRE1, PERK, ATF6,
CHOP, caspase-3 and p53). (A) Representative images of western blot
analysis. The total cellular proteins derived from MEG3-transfected
or Lenti6.3-MIG-transfected HepG2 cells were immunoblotted with a
panel of antibodies specific for GRP78, IRE1, PERK, ATF6, CHOP,
caspase-3, p53 and GAPDH. (B and C) The expression of each index
was normalized to the expression level of GAPDH, and the relative
change was expressed as a ratio or fold. Data represent the means ±
SD of three independent experiments. *P<0.05 vs.
control. MEG3, maternally expressed gene 3; ER, endoplasmic
reticulum; GRP78, 78-kDa glucose-regulated protein; IRE1,
inositol-requiring enzyme 1; PERK, RNA-dependent protein
kinase-like ER kinase; ATF6, activating transcription factor 6;
CHOP, C/EBP homologous protein. |
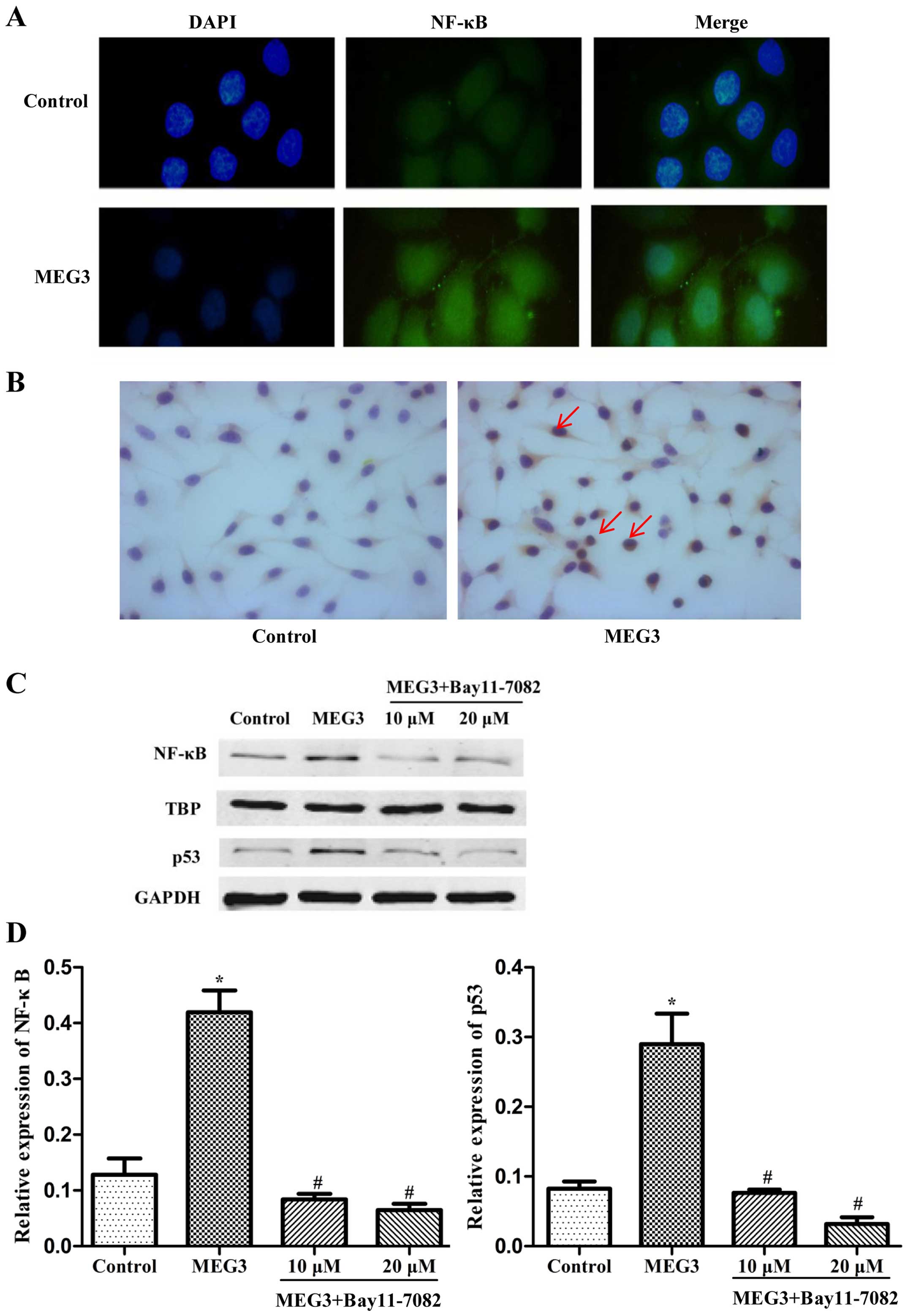
Ectopic expression of MEG3 activates
NF-κB, p53 and causes nuclear translocation of NF-κB protein
In order to further explore the possible
relationship between NF-κB and p53, we investigated the
distribution of NF-κB in the HepG2 cells transfected with MEG3 by
immunofluorescence and immunohistochemistry assay. As indicated in
Fig. 5A and B, a weak NF-κB signal
was detected in the control cells and NF-κB was obviously
translocated from the cytoplasm to the nucleus in the
MEG3-transfected cells. In order to further validate the results
using microscopy, total cell lysates collected from the transfected
HepG2 cells were fractionated to separate the cytoplasmic and
nuclear components, and western blot analysis was performed to
measure the NF-κB protein level only in the nuclear components. A
sharp accumulation of NF-κB in the nuclear fraction was detected in
the cells transfected with MEG3 (Fig.
5C). Moreover, inhibition of NF-κB by Bay11-7082 decreased the
protein expression of both NF-κB and p53 (Fig. 5D). These results demonstrated that
ER stress activates the p53 pathway and p53 expression is mediated
by NF-κB in MEG3-induced apoptosis.
Discussion
Accumulating evidence indicates that MEG3 plays an
important role in the formation and progression of HCC. In order to
further identify the biological function of MEG3, recombinant
lentivirus of MEG3 was constructed and transfected into hepatoma
HepG2 cells. The results showed that ectopic expression of MEG3
inhibited HepG2 cell proliferation in vitro (Fig. 2A). Moreover, overexpression of MEG3
decreased tumor growth in nude mice (Fig. 2B–D). FCM analysis further confirmed
that the ectopic expression of MEG3 increased apoptosis in the
HepG2 cells (Fig. 3), which shows
that MEG3 functions as a tumor suppressor.
In the present study, we observed that
overexpression of MEG3 significantly increased the relative protein
expression of the ER stress pathway. Many studies have indicated
that activation of IRE1, ATF6, and PERK represents the standard UPR
pathways. Once PERK, ATF6 and/or IRE1 are activated, they initiate
an early adaptive response to unfolded proteins that, if the stress
is short-lived, can facilitate clearance of the unfolded proteins
and cell survival; specifically, inhibition of protein translation,
transcriptional induction and increased expression of GRP78
(21). However, with prolonged
stress, additional responses are initiated, including
caspase-12/caspase-9/caspase-3, ERK/ATF-4/CHOP, IRE1/Ask1/JNK,
PERK/eIF2a/NF-κB and p53 pathways and these pathways can promote
cell apoptosis (22). CHOP is a
transcription factor and plays a critical role in ER
stress-mediated apoptosis (22).
Caspase-3 is a main executioner caspase. In the present study,
three key proteins of UPR (IRE1, ATF6, PERK) and chaperone GRP78
were obviously increased in the transfected HepG2 cells.
Furthermore, CHOP, NF-κB, caspase-3 and p53 were also upregulated
(Fig. 4). These experimental
results demonstrated that MEG3 triggered the ER stress pathway in
the HepG2 cells.
Our previous study revealed that MEG3 functions as a
tumor-suppressor gene by regulating p53 activation (10). NF-κB family members also play a role
in regulating the p53 gene in certain types of stress (23,24).
However, the regulatory mechanism of p53 activation in ER stress is
still unclear. Studies indicate that persistent ER stress can
trigger a switch in the UPR signaling pathways from pro-survival to
pro-apoptotic pathways by the PERK/eIF2a/NF-κB pathways (25). Activation of NF-κB regulates cell
death-associated gene expression (26–28).
In order to probe the effect of NF-κB on p53 activation, the
distribution and expression of NF-κB protein in the HepG2 cells
were detected. In normal states, NF-κB is sequestered in the
cytoplasm in an inactive form bound to one of many inhibitory
molecules (IκBs). Phosphorylated and ubiquitinated IκB is degraded
by the 26S proteasome, leading to the translocation of active NF-κB
to the nucleus where it binds to κB elements and regulates
transcription of genes mediating inflammation, carcinogenesis, and
pro-apoptotic or anti-apoptotic functions. In this study, the
results of the immunofluorescence and immunohistochemistry assays
showed that ectopic expression of MEG3 obviously caused NF-κB
trans-location from the cytoplasm to the nucleus and increased its
expression in nuclei (Fig. 5A and
B). Furthermore, western blot analysis demonstrated that the
majority of the NF-κB proteins resided in the nucleus and the
expression level of PERK was also increased in the MEG3-transfected
cells (Fig. 5C), indicating that
the PERK/eIF2a/NF-κB pathway was activated. Moreover, inhibition of
NF-κB by Bay11-7082 decreased p53 protein expression (Fig. 5C and D), showing that p53 expression
was regulated by NF-κB under ER stress. Our previous study
identified that MEG3 repressed MDM2 expression and induced p53
activation (10). Recently, Zhu
et al demonstrated that MEG3 can also interact directly with
the p53 DNA binding domain in hepatoma cells (29). In the present study, we observed
that overexpression of MEG3 activated the ER stress pathway and
increased p53 expression via the NF-κB pathway, which indicates
that multiple mechanisms participate in p53 activation in
MEG3-induced apoptosis.
The role of NF-κB in regulating cell survival or
death is complex. NF-κB mediates tumor promotion, angiogenesis,
metastasis, and resistance to chemotherapeutics (30). In a study of gastric carcinoma, we
demonstrated that NF-κB was constitutively active and associated
with advanced pathologic stage and tumor size (31). In adenosine-induced apoptosis, we
observed that NF-κB plays an anti-apoptotic role (32). However, in the present study,
unexpectedly, inhibition of NF-κB decreased p53 protein expression.
We hypothesize that the difference may be due to the types of
stress and the features of the cell lines. ER stress can operate in
parallel with multiple signaling mechanisms in different
situations, and the overall outcome in terms of cell survival or
apoptosis depends on the additive effects on downstream effectors.
The role of NF-κB on p53 regulation needs further investigation in
MEG3-mediated ER stress.
In summary, the present study demonstrated that
exogenous MEG3 impeded tumorigenesis both in vitro and in
vivo and induced hepatoma cell apoptosis. Moreover,
overexpression of MEG3 caused ER stress and resulted in the
activation of NF-κB and p53. Furthermore, inhibition of NF-κB
decreased p53 protein expression. These results showed that the ER
stress pathway may be involved in MEG3-induced apoptosis and that
NF-κB signaling is required for p53 activation in ER stress.
Acknowledgments
This study was supported by the Guangdong Natural
Science Foundation in China (no. 2014A030313470) and the
Collaborative and Creative Center, Molecular Diagnosis and
Personalized Medicine, Shantou University, Guangdong, China. This
study was also supported by the Department of Education, Guangdong
Government under the Top-tier University Development Scheme for
Research and Control of Infectious Diseases.
References
|
1
|
Wapinski O and Chang HY: Long noncoding
RNAs and human disease. Trends Cell Biol. 21:354–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang P, Ren Z and Sun P: Overexpression of
the long non-coding RNA MEG3 impairs in vitro glioma cell
proliferation. J Cell Biochem. 113:1868–1874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li Z, Li C, Liu C, Yu S and Zhang Y:
Expression of the long non-coding RNAs MEG3, HOTAIR, and MALAT-1 in
non-functioning pituitary adenomas and their relationship to tumor
behavior. Pituitary. 18:42–47. 2015. View Article : Google Scholar
|
|
4
|
Miyoshi N, Wagatsuma H, Wakana S,
Shiroishi T, Nomura M, Aisaka K, Kohda T, Surani MA, Kaneko-Ishino
T and Ishino F: Identification of an imprinted gene, Meg3/Gtl2 and
its human homologue MEG3, first mapped on mouse distal chromosome
12 and human chromosome 14q. Genes Cells. 5:211–220. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou Y, Zhang X and Klibanski A: MEG3
noncoding RNA: A tumor suppressor. J Mol Endocrinol. 48:R45–R53.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bando T, Kato Y, Ihara Y, Yamagishi F,
Tsukada K and Isobe M: Loss of heterozygosity of 14q32 in
colorectal carcinoma. Cancer Genet Cytogenet. 111:161–165. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu
WQ, Xie WP and Hou YY: Long non-coding RNA MEG3 inhibits NSCLC
cells proliferation and induces apoptosis by affecting p53
expression. BMC Cancer. 13:4612013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Braconi C, Kogure T, Valeri N, Huang N,
Nuovo G, Costinean S, Negrini M, Miotto E, Croce CM and Patel T:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is associated
with meningioma pathogenesis and progression. Cancer Res.
70:2350–2358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu LX, Deng W, Zhou XT, Chen RP, Xiang
MQ, Guo YT, Pu ZJ, Li R, Wang GF and Wu LF: The mechanism of
adenosine-mediated activation of lncRNA MEG3 and its antitumor
effects in human hepatoma cells. Int J Oncol. 48:421–429. 2016.
|
|
11
|
Schröder M: Endoplasmic reticulum stress
responses. Cell Mol Life Sci. 65:862–894. 2008. View Article : Google Scholar
|
|
12
|
Wu LF, Ye YQ, Huang GY, Li HB, Li GP, Pu
ZJ, Wei BL and Feng JL: Involvement of endoplasmic reticulum stress
in adenosine-induced human hepatoma HepG2 cell apoptosis. Oncol
Rep. 26:73–79. 2011.PubMed/NCBI
|
|
13
|
Giampietri C, Petrungaro S, Conti S,
Facchiano A, Filippini A and Ziparo E: Cancer microenvironment and
endoplasmic reticulum stress response. Mediators Inflamm.
2015:4172812015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu LF, Guo YT, Zhang QH, Xiang MQ, Deng W,
Ye YQ, Pu ZJ, Feng JL and Huang GY: Enhanced antitumor effects of
adenoviral-mediated siRNA against GRP78 gene on adenosine-induced
apoptosis in human hepatoma HepG2 cells. Int J Mol Sci. 15:525–544.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista
DL, Gejman R, Ansell PJ, Zhao J, Weng C and Klibanski A: Activation
of p53 by MEG3 non-coding RNA. J Biol Chem. 282:24731–24742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang PM, Lin YT, Shun CT, Lin SH, Wei TT,
Chuang SH, Wu MS and Chen CC: Zebularine inhibits tumorigenesis and
stemness of colorectal cancer via p53-dependent endoplasmic
reticulum stress. Sci Rep. 3:32192013.PubMed/NCBI
|
|
20
|
Kilkenny C, Browne W, Cuthill IC, Emerson
M and Altman DG; NC3Rs Reporting Guidelines Working Group: Animal
research: Reporting in vivo experiments: The ARRIVE guidelines. Br
J Pharmacol. 160:1577–1579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bromati CR, Lellis-Santos C, Yamanaka TS,
Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anhê GF
and Bordin S: UPR induces transient burst of apoptosis in islets of
early lactating rats through reduced AKT phosphorylation via
ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr
Comp Physiol. 300:R92–R100. 2011. View Article : Google Scholar
|
|
23
|
Wu H and Lozano G: NF-kappa B activation
of p53. A potential mechanism for suppressing cell growth in
response to stress. J Biol Chem. 269:20067–20074. 1994.PubMed/NCBI
|
|
24
|
Furlong EE, Rein T and Martin F: YY1 and
NF1 both activate the human p53 promoter by alternatively binding
to a composite element, and YY1 and E1A cooperate to amplify p53
promoter activity. Mol Cell Biol. 16:5933–5945. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kitamura M: Control of NF-κB and
inflammation by the unfolded protein response. Int Rev Immunol.
30:4–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaneko M, Niinuma Y and Nomura Y:
Activation signal of nuclear factor-kappa B in response to
endoplasmic reticulum stress is transduced via IRE1 and tumor
necrosis factor receptor-associated factor 2. Biol Pharm Bull.
26:931–935. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang HY, Wek SA, McGrath BC, Scheuner D,
Kaufman RJ, Cavener DR and Wek RC: Phosphorylation of the alpha
subunit of eukaryotic initiation factor 2 is required for
activation of NF-kappaB in response to diverse cellular stresses.
Mol Cell Biol. 23:5651–5663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamazaki H, Hiramatsu N, Hayakawa K,
Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW,
et al: Activation of the Akt-NF-kappaB pathway by subtilase
cytotoxin through the ATF6 branch of the unfolded protein response.
J Immunol. 183:1480–1487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu J, Liu S, Ye F, Shen Y, Tie Y, Zhu J,
Wei L, Jin Y, Fu H, Wu Y, et al: Long noncoding RNA MEG3 interacts
with p53 protein and regulates partial p53 target genes in hepatoma
cells. PLoS One. 10:e01397902015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pikarsky E, Porat RM, Stein I, Abramovitch
R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E and
Ben-Neriah Y: NF-kappaB functions as a tumour promoter in
inflammation-associated cancer. Nature. 431:461–466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu L, Pu Z, Feng J, Li G, Zheng Z and Shen
W: The ubiquitin-proteasome pathway and enhanced activity of
NF-kappaB in gastric carcinoma. J Surg Oncol. 97:439–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu LF, Li GP, Su JD, Pu ZJ, Feng JL, Ye YQ
and Wei BL: Involvement of NF-kappaB activation in the apoptosis
induced by extracellular adenosine in human hepatocellular
carcinoma HepG2 cells. Biochem Cell Biol. 88:705–714. 2010.
View Article : Google Scholar : PubMed/NCBI
|