Introduction
Bladder cancer (BC) is one of the most common
cancers of the urinary system worldwide (1). Approximately 90% of BCs are urothelial
cell carcinomas with an epithelial origin (2). Muscle-invasive BC occurs in ~1/3 of
patients and is associated with a poor prognosis, with a 5-year
patient survival rate of 50% (2,3).
Elucidating the mechanisms underlying BC invasion and metastasis is
indispensable for the development of effective therapies for this
disease.
The transforming growth factor-β (TGF-β) signaling
pathway plays an important role in carcinoma development (4,5). This
signaling pathway induces epithelial-mesen-chymal transition (EMT)
and promotes cell invasiveness and metastasis in multiple cancers,
including BC (6-8). TGF-β binds to cell surface
transmembrane serine/threonine kinase receptors and transduces
signals principally through Smad proteins, which induce cellular
responses by directly activating the expression of EMT
transcription factors (9). Several
mechanisms are involved in the regulation of TGF-β signaling, such
as positive regulation by stimulatory factors and negative
regulation by negative feedback mechanisms (10).
SnoN (also known as SKI-like proto-oncogene, SKIL),
a member of the Ski family, is a negative regulator of TGF-β
signaling (11). SnoN acts as a
Smad corepressor in the nucleus by interacting with Smad complexes
and recruiting other core-pressors to inhibit Smad transcriptional
activities (11). In the cytoplasm,
SnoN blocks TGF-β signals by sequestering Smad proteins and
preventing their translocation to the nucleus (12). TGF-β also tightly regulates SnoN
levels in a biphasic manner: short stimulation with TGF-β causes
rapid and transient SnoN protein degradation via the
ubiquitin-proteasome system, whereas longer TGF-β treatment
increases SnoN levels by inducing SnoN gene expression (13). SnoN thus participates in a negative
feedback loop to regulate TGF-β signaling (14). Recent studies suggest that the
ability of SnoN to repress TGF-β signaling is regulated by
SUMOylation, a post-translational modification that is catalyzed by
a small ubiquitin-like modifier (SUMO)-activating E1 enzyme, a
SUMO-conjugating E2 enzyme, and a SUMO E3 ligase (15-17).
Investigation of the role of SnoN in cancer revealed both
pro-oncogenic and anti-oncogenic activities (18). However, the role of SnoN in BC
remains to be elucidated.
In the present study, the expression of SnoN did not
differ between BC tissues and adjacent normal tissues and between
BC cell lines and normal cells, whereas its function in repressing
TGF-β was significantly attenuated compared with that in the normal
control. TIF1γ, a newly identified SUMO E3 ligase, promoted SnoN
SUMOylation and restored the ability of SnoN to repress TGF-β
signaling. The present study demonstrated that the TIF1γ-SnoN1
pathway has an inhibitory effect on TGF-β-induced EMT and invasion
in BC.
Materials and methods
Tissue samples and cell culture
A total of 33 bladder tumor tissues and matched
adjacent normal tissues were collected from Qilu Hospital of
Shandong University (Qingdao, China) between 2010 and 2013 with
informed consent. All specimens were frozen in liquid nitrogen
immediately and subsequently confirmed by pathological analysis.
The study was approved by the Ethics Committee of Shandong
University (Jinan, China).
The human bladder cancer cell lines T24 and TCCSUP
and the normal urothelial epithelial cell line SV-HUC-1 were
purchased from the Cell Bank of the Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). The cells were grown
in complete growth medium at 37°C with 5% CO2, as
recommended by the manufacturer.
Real-time PCR assay
Total RNA was extracted from tissues and cells using
the TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA) and reverse-transcribed using oligo(dT) primers and
SuperScript II transcriptase (Invitrogen Life Technologies). The
cDNAs were subjected to quantitative PCR using the following
primers: SnoN forward 5′-CTCACAAAGACAGAGGCAAGTA-3′ and reverse,
5′-CCTCAAGTGAGACATCTGGATAAG-3′; TIF1γ forward,
5′-CAGCTCCTGGTTATACTCCTAATG-3′ and reverse
5′-GAGTCGAAGCTGTGCTAAGT-3′; and Power SYBR Green PCR Master Mix
(Invitrogen Life Technologies) on an Applied Biosystems 7300
Real-Time PCR system (Applied Biosystems, Grand Island, NY, USA).
β-actin was used as the reference gene and gene expression was
quantified using the 2-ΔΔCt method (19).
Western blotting
Proteins were extracted from cultured cells using
lysis buffer (Beyotime, Nantong, China) and then quantified with
the bicinchoninic acid protein assay kit (Pierce Biotechnology,
Rockford, IL, USA). Equal amounts of protein were resolved by 10%
SDS-PAGE and then transferred to nitrocellulose membranes. After
blocking with 5% non-fat milk, the membranes were incubated
overnight with the following primary antibodies: mouse anti-SnoN,
rabbit anti-TIF1γ (both from Santa Cruz Biotechnology, Dallas, TX,
USA), mouse anti-E-cadherin, mouse anti-N-cadherin (both from Cell
Signaling Technology, Danvers, MA, USA) and mouse anti-fibronectin
(Santa Cruz Biotechnology). Subsequently, the membranes were next
incubated with horseradish peroxidase-conjugated secondary
antibodies and target proteins were detected using an enhanced
chemiluminescence system (Pierce Biotechnology, Inc., Rockford, IL,
USA).
p3TP-lux luciferase reporter assay
TGF-β-dependent transcriptional activation was
detected with the p3TP-lux luciferase reporter, which consists of
firefly luciferase under the control of three consecutive
12-O-tetradecanoylphorbol-1 3-acetate (TPA) response
elements (20). Cells were
transiently transfected with the p3TP-lux reporter plasmid
(Addgene, Cambridge, MA, USA) using FuGENE6 (Roche Diagnostics,
Indianapolis, IN, USA) according to the manufacturer's
instructions. The phRL-TK vector (Promega, Madison, WI, USA) was
co-transfected to determine transfection efficiency. After 24 h,
the cells were treated with or without TGF-β (Biolegend, San Diego,
CA, USA) for the indicated times. Cells were then lysed and
luciferase activity was measured using the Dual-Luciferase reporter
assay system (Promega) according to the manufacturer's
instructions. p3TP-lux luciferase activity was normalized to that
of the control phRL-TK vector.
Lentivirus-mediated overexpression and
RNA interference
To construct the overexpression lentivirus plasmid,
the coding DNA sequence of SnoN or TIF1γ was PCR amplified from
cDNA of cultured normal epithelial cells, and cloned into the
pHBLV-CMVIE-IRES-Puro lentiviral vector (Hanbio, Shanghai, China).
The recombinant lentivirus (Lv-SnoN or Lv-TIF1γ) was produced by
co-transfection of 293T cells with the plasmids psPAX2 and pMD2G
using LipoFiter (Hanbio). To knock down SnoN, a lentivirus with a
SnoN shRNA sequence (Lv-shSnoN) was purchase from Santa Cruz
Biotechnology. The empty lentivector or control shRNA lentiviral
particles (Santa Cruz Biotechnology) were used as the negative
control (Lv-NC). Cells were exposed to the lentivirus-containing
supernatant for 24 h in the presence of polybrene (Sigma-Aldrich,
St. Louis, MO, USA). Stable trans-fectants were selected with
puromycin (2 mg/ml) and verified by western blotting and real-time
PCR.
Immunoprecipitation assay
Cells were lysed in TNTE buffer (50 mM Tris-HCl, pH
7.4, 150 mM NaCl and 1 mM EDTA) containing 0.5% Triton X-100 plus
protease and phosphatase inhibitors. N-ethylmaleimide (NEM,
20 mM), an isopeptidase inhibitor, or vehicle alone was included in
the lysis buffer where indicated. Cell lysates were centrifuged at
15,000 × g for 10 min at 4°C and the supernatant was subjected to
immunoprecipitation using mouse anti-SnoN antibody (Santa Cruz
Biotechnology). Immunoprecipitated proteins were then separated by
SDS-PAGE followed by immunoblotting using rabbit anti-SnoN and
rabbit anti-SUMO antibodies (Santa Cruz Biotechnology), and
visual-ized as described for western blotting.
Transwell invasion assay
Cell invasion was assessed using the Transwell
chamber invasion assay. Cells (1×105) were added to the
top chamber with Matrigel-coated membranes (8-µm pore size;
Millipore, Bedford, MA, USA). Medium with 10% fetal bovine serum
was added to the lower chamber as a chemoat-tractant. TGF-β1 (10
ng/ml) or vehicle alone was added to the upper and lower chambers.
After 48 h, cells that had invaded to the lower surface of the
membrane were stained with 0.1% crystal violet and counted in five
random fields.
Statistical analyses
The data from independent experiments repeated at
least three times are presented as the mean ± standard error of the
mean (SEM). Statistical significance (p<0.05) was determined by
the Student's t-test or analysis of variance followed by
Bonferroni' post hoc tests.
Results
SnoN regulation of the TGF-β pathway is
absent in BC
The expression levels of SnoN were examined in BC
tissues and cell lines. Real-time PCR assessment of 33 BC and
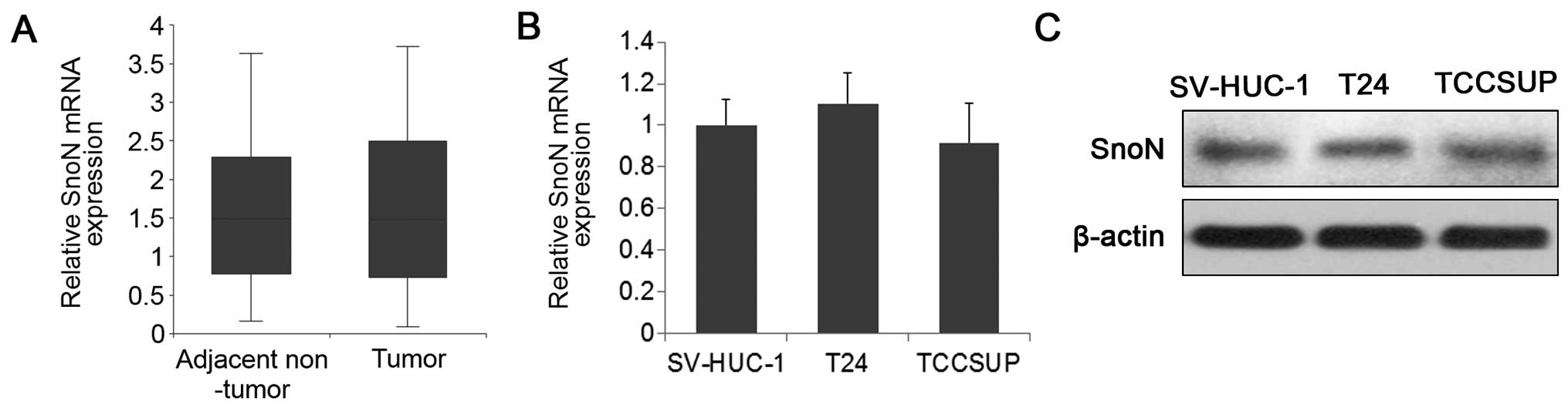
adjacent normal tissue samples showed that SnoN expression did not
differ significantly between the BC and normal tissues (Fig. 1A). Similar results were obtained
when comparing SnoN expression levels between BC cells and normal
urothelial epithelial cells (Fig. 1B
and C). Since SnoN is an important participant of a negative
feedback loop regulating the TGF-β pathway, we next examined the
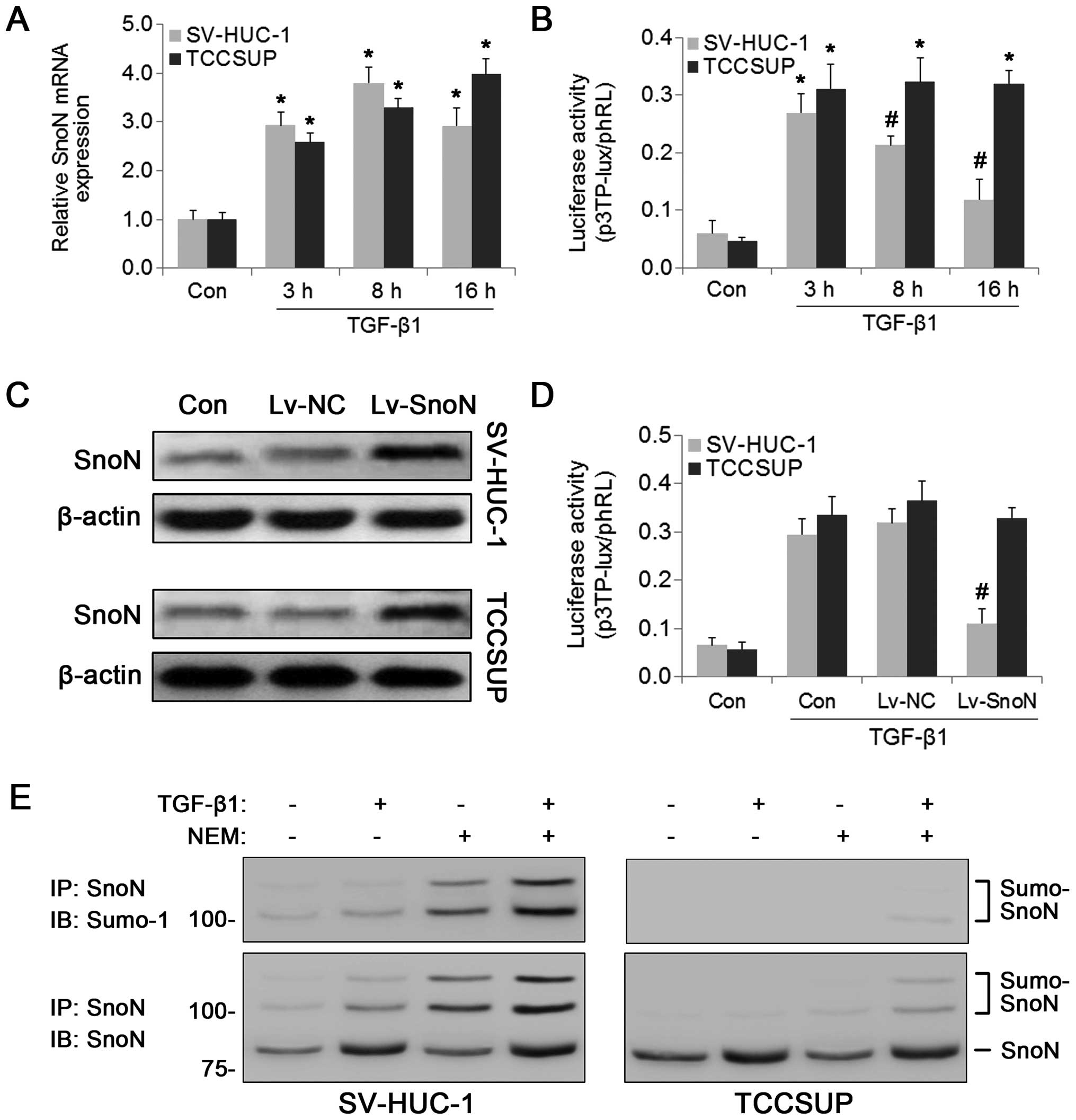
effect of TGF-β on SnoN expression. Consistent with a previous
study (13), TGF-β positively
regulated SnoN mRNA expression in both normal and BC cells at least
in the first 8 h (Fig. 2A).
However, at 16 h after TGF-β treatment, SnoN expression began to
decline in the SV-HUC-1 cells, whereas high SnoN expression levels
were maintained in the TCCSUP cells. TGF-β-dependent
transcriptional activation was examined next using the p3TP-lux
luciferase reporter assay. As shown in Fig. 2B, the luciferase activities of
p3TP-lux were significantly increased in response to TGF-β
treatment for 3 h in both cell lines; however, p3TP-lux activity
was suppressed at 8 and 16 h compared with that at 3 h in the
normal urothelial epithelial cell line SV-HUC-1, but not in the BC
cells. These results suggest that the negative regulation of TGF-β
signaling by SnoN was blocked in the BC cells. To confirm the role
of SnoN in the regulation of the TGF-β pathway in BC cells, SnoN
was overexpressed in TCCSUP and SV-HUC-1 cells using a lentiviral
vector (Fig. 2C). As shown in
Fig. 2D, TGF-β-dependent
transcriptional activity was reduced by SnoN overexpression in the
SV-HUC-1 cells but not in the TCCSUP cells. Previous studies
indicated that post-translational modification by SUMOylation may
contribute to the ability of SnoN to regulate transcription
(16,21). Therefore, we examined the
SUMOylation status of SnoN using immunoprecipitation assays. In the
presence of the SUMO-protease inhibitor NEM, SUMOylated SnoN was
detected in the SV-HUC-1 cells in the presence or absence of TGF-β
treatment (Fig. 2E). However, the
SUMO immunoreactive protein bands were undetectable in the TCCSUP
cells in the absence of TGF-β treatment and detected at low levels
in the presence of TGF-β (Fig. 2E).
These results indicated that the regulatory function of SnoN in the
TGF-β pathway was absent in BC cells, which could be attributed to
the weak SUMOylation of SnoN.
Restoration of TIF1γ expression represses
the TGF-β pathway in BC
Next, we investigated the mechanisms underlying the
abnormal SUMOylation of SnoN in BC cells. TIF1γ is a member of
multiple families (22) and was
recently reported to function as a SUMO E3 ligase that promotes the
SUMOylation of SnoN (15). Because
TIF1γ-induced SUMOylation is required for SnoN to suppress
TGF-β-induced EMT in mouse mammary epithelial cells (15), we examined whether TIF1γ affects
SnoN SUMOylation and TGF-β signaling in BC cells. The expression
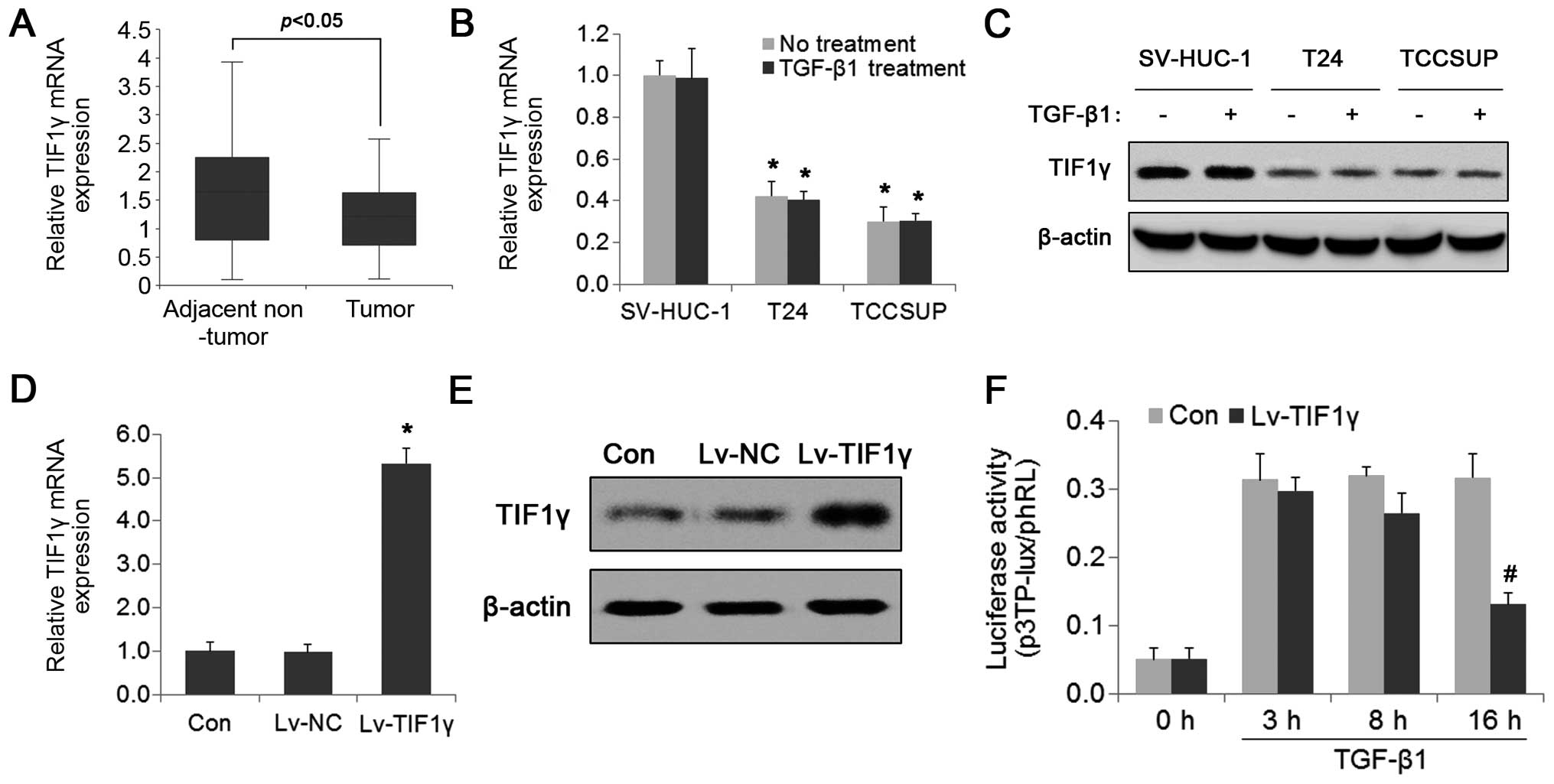
levels of TIF1γ in BC tissues and cells were first evaluated. As
shown in Fig. 3A, TIF1γ mRNA
expression was significantly downregulated in the BC tissues
compared with that noted in the adjacent normal control tissues.
Similar results were obtained when comparing the mRNA and protein
expression of TIF1γ between BC cells and normal epithelial cells
(Fig. 3B and C). Unlike SnoN, TIF1γ
expression was not significantly affected by TGF-β (Fig. 3B and C). To further assess the
effect of TIF1γ on TGF-β signaling, TIF1γ was stably overexpressed
in the TCCSUP cells by lentivirus and the expression levels were
assessed by real-time PCR and western blotting (Fig. 3D and E). The p3TP-lux luciferase
reporter assay showed that luciferase activity was significantly
reduced in the TIF1γ-overexpressing TCCSUP cells after 16 h of
TGF-β1 treatment compared with that at 3 h (Fig. 3F). A similar trend was observed in
the SV-HUC-1 cells (Fig. 2B),
suggesting that restoring TIF1γ recovered the negative regulation
of the TGF-β pathway in BC cells.
SnoN is necessary for TIF1γ-mediated
negative regulation of the TGF-β pathway in BC cells
To investigate whether the suppressive effect of
TIF1γ on the TGF-β pathway is mediated by SnoN SUMOylation, SnoN
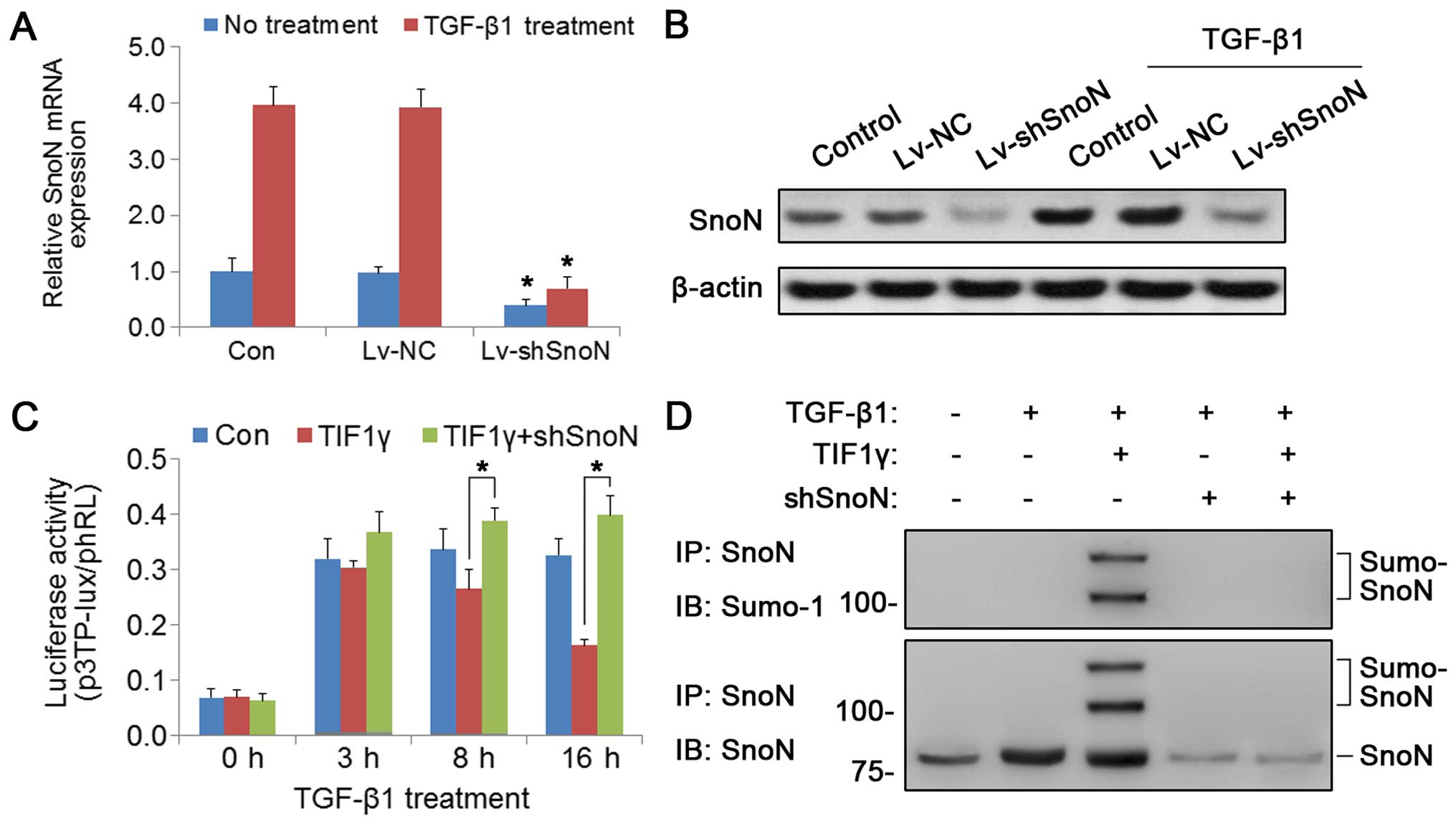
expression was knocked down in the TIF1γ-overexpressing TCCSUP
cells by lentiviral transient transfection. Real-time PCR and
western blotting confirmed that the expression of SnoN was markedly
reduced after lentiviral transfection (Lv-shSnoN) (Fig. 4A and B). Next, the effect of SnoN
knockdown on TGF-β-dependent transcriptional activation was
examined. As shown in Fig. 4C, SnoN
silencing resulted in the recovery of p3TP-lux activity in the
TIF1γ-overexpressing cells. Assessment of the effect of TIF1γ on
SnoN SUMOylation by immunoprecipitation showed that TIF1γ
overexpression increased the levels of SUMOylated SnoN, and this
effect was abrogated by SnoN knockdown (Fig. 4D). All things considered, these
results indicated that TIF1γ promoted the SUMOylation of SnoN,
which was necessary for the inhibitory effect of TIF1γ on the TGF-β
pathway in BC cells.
TIF1γ restores the effect of SnoN on
inhibiting TGF-β-induced EMT and invasion in BC
TGF-β induces EMT and promotes tumor metastasis in
BC (7,23). To test whether the TIF1γ-SnoN
SUMOylation pathway plays a role in TGF-β-induced EMT, the effects
of TIF1γ overexpression and/or SnoN silencing on the expression of
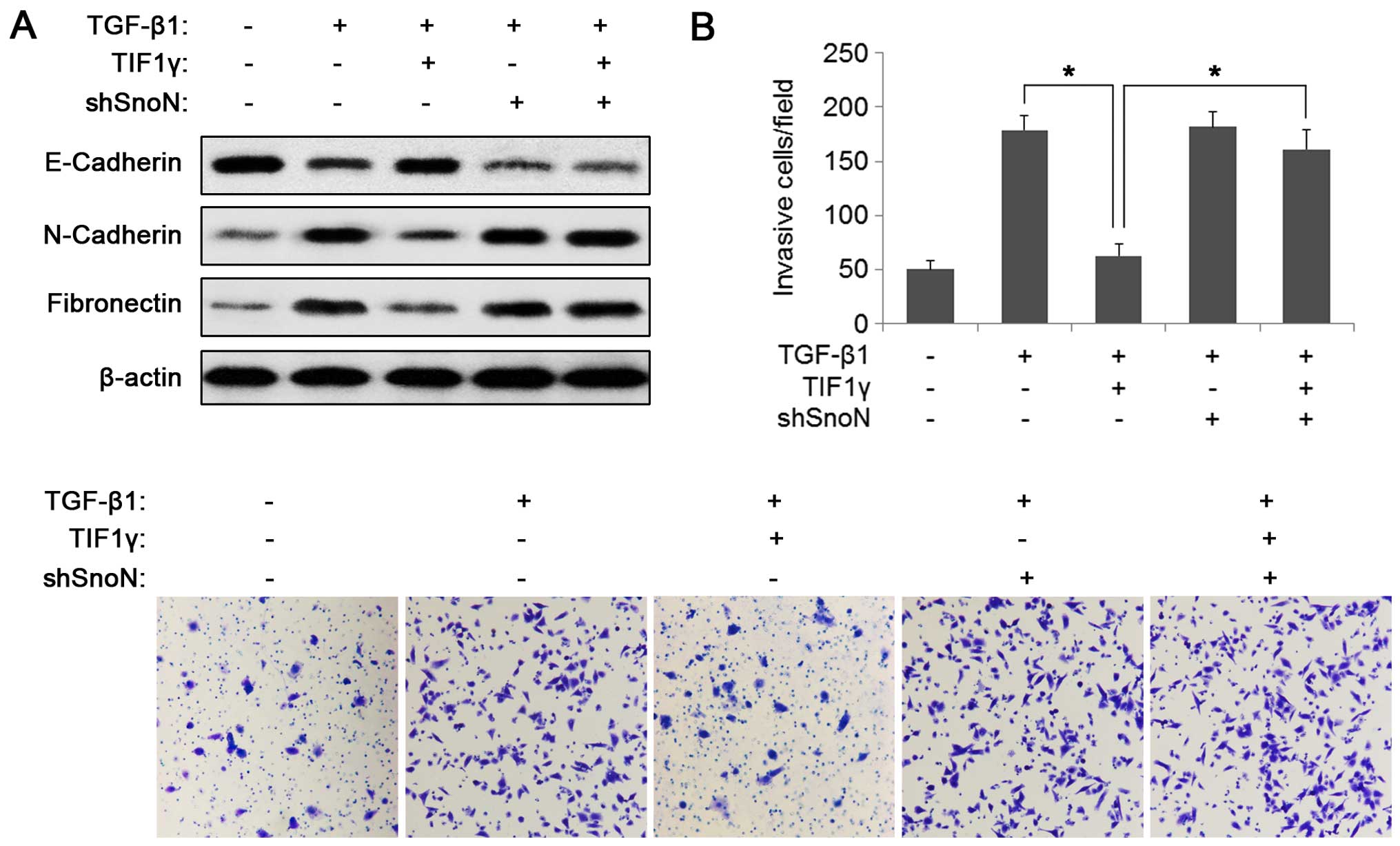
EMT markers were examined. As shown in Fig. 5A, TGF-β-induced changes of EMT
markers (decreased expression of E-cadherin and increased
expression of N-cadherin and fibronectin) were attenuated by TIF1γ
overexpression. Consistent with the SnoN-mediated suppression of
TIF1γ on the TGF-β pathway, SnoN knockdown abrogated the effect of
TIF1γ on TGF-β-induced EMT (Fig.
5A). The roles of TIF1γ and SnoN in TGF-β-induced cell invasion
using the Transwell assay were then examined. As shown in Fig. 5B, TIF1γ overexpression significantly
reduced TGF-β-induced cell invasion and knockdown of SnoN blocked
this ability of TIF1γ. These data suggest that the inhibitory
effects of TIF1γ on TGF-β-induced EMT and invasion are mediated by
SnoN in BC. TIF1γ thus restored the function of SnoN as an
inhibitor of TGF-β-induced EMT and invasion in BC.
Discussion
TGF-β signaling is an important pathway that
regulates many cell functions and is implicated in diverse
physiological and pathological events. To ensure its proper
physiological function, TGF-β signaling is tightly regulated at
different levels in different cells and tissues (24). Dysregulation of TGF-β signaling
induces EMT and contributes to tumor progression (6). In the present study, we demonstrated
that the loss of the regulatory function of SnoN in TGF-β signaling
is a potential mechanism whereby TGF-β induces EMT and promotes
tumor metastasis in BC.
SnoN can be induced by TGF-β1 and is a negative
regulator of TGF-β1 signaling, which suggests that a negative
feedback mechanism modulates TGF-β1 signaling (11). Alterations in SnoN expression in
certain cancers are associated with tumorigenesis and the prognosis
of patients (25–27). Our results showed no differences in
SnoN expression between BC tissues or cells and adjacent normal
tissues or normal urothelial epithelial cells. However, TGF-β
induced the expression of SnoN for a longer period of time in BC
cells than in normal epithelial cells. These results together with
the findings that TGF-β-dependent transcriptional activity
gradually declined from its peak in normal epithelial cells, but
not in BC cells, suggest that SnoN is dysfunctional in BC cells. In
line with this hypothesis, overexpressed SnoN had no effect on
TGF-β signaling in BC cells.
Post-translational modifications regulate protein
function, and SUMOylation is an important modification that affects
SnoN activity (16). SUMOylation
occurs via the covalent attachment of the protein SUMO to a lysine
residue on a substrate, and this process is catalyzed by the
sequential action of three sets of enzymes (17). Here, it was found that TIF1γ, a
newly identified SUMO E3 ligase, was significantly downregulated in
BC tissues and cells compared with normal controls. Restoring TIF1γ
significantly repressed TGF-β signaling after a specific period,
showing a similar trend to that in normal epithelial cells treated
by TGF-β1. TIF1γ (also referred to as Trim33) is a member of the
tripartite motif/RING finger, B-boxes, and a coiled-coil domain
(TRIM/RBCC) family and E3 ubiquitin-ligase family (28). TIF1γ functions as a suppressor of
the TGF-β superfamily signaling by inhibiting the formation of Smad
nuclear complexes (29,30). Recently, TIF1γ was shown to induce
the SUMOylation on SnoN by acting as a SUMO E3 ligase, and
SUMOylation is required for SnoN mediated abrogation of TGF-β1
signaling (15). The results here
showed that TIF1γ suppression of TGF-β1 signaling was dependent on
SnoN expression in BC cells, suggesting that TIF1γ plays a role as
a suppressor of TGF-β1 by restoring the regulatory function of SnoN
in BC. TIF1γ may play either a tumor-suppressor or -promoter role
in cancer (31,32). In BC cells, it was demonstrated that
TIF1γ could inhibit TGF-β-induced EMT and invasion in the presence
of normally expressed SnoN, implying that TIF1γ serves as a tumor
suppressor in BC.
In summary, this study demonstrated that the loss of
the function of SnoN as a suppressor of TGF-β resulted in the
dysregulation of TGF-β signaling in BC. TIF1γ, as a SUMO E3 ligase,
restored the function of SnoN, leading to the inhibition of
TGF-β-induced EMT and invasion in BC.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81202025).
References
|
1
|
Griffiths TR; Action on Bladder Cancer:
Current perspectives in bladder cancer management. Int J Clin
Pract. 67:435–448. 2013. View Article : Google Scholar
|
|
2
|
Kaufman DS, Shipley WU and Feldman AS:
Bladder cancer. Lancet. 374:239–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parekh DJ, Bochner BH and Dalbagni G:
Superficial and muscle-invasive bladder cancer: Principles of
management for outcomes assessments. J Clin Oncol. 24:5519–5527.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gold LI: The role for transforming growth
factor-beta (TGF-beta) in human cancer. Crit Rev Oncog. 10:303–360.
1999.
|
|
5
|
Fabregat I, Fernando J, Mainez J and
Sancho P: TGF-beta signaling in cancer treatment. Curr Pharm Des.
20:2934–2947. 2014. View Article : Google Scholar
|
|
6
|
Katsuno Y, Lamouille S and Derynck R:
TGF-β signaling and epithelial-mesenchymal transition in cancer
progression. Curr Opin Oncol. 25:76–84. 2013. View Article : Google Scholar
|
|
7
|
Fan Y, Shen B, Tan M, Mu X, Qin Y, Zhang F
and Liu Y: TGF-β-induced upregulation of malat1 promotes bladder
cancer metastasis by associating with suz12. Clin Cancer Res.
20:1531–1541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Geng J, Fan J, Ouyang Q, Zhang X, Zhang X,
Yu J, Xu Z, Li Q, Yao X, Liu X, et al: Loss of PPM1A expression
enhances invasion and the epithelial-to-mesenchymal transition in
bladder cancer by activating the TGF-β/Smad signaling pathway.
Oncotarget. 5:5700–5711. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saitoh M: Epithelial-mesenchymal
transition is regulated at post-transcriptional levels by
transforming growth factor-β signaling during tumor progression.
Cancer Sci. 106:481–488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miyazono K: Positive and negative
regulation of TGF-beta signaling. J Cell Sci. 113:1101–1109.
2000.PubMed/NCBI
|
|
11
|
Zeglinski MR, Hnatowich M, Jassal DS and
Dixon IM: SnoN as a novel negative regulator of TGF-β/Smad
signaling: A target for tailoring organ fibrosis. Am J Physiol
Heart Circ Physiol. 308:H75–H82. 2015. View Article : Google Scholar
|
|
12
|
Krakowski AR, Laboureau J, Mauviel A,
Bissell MJ and Luo K: Cytoplasmic SnoN in normal tissues and
nonmalignant cells antagonizes TGF-beta signaling by sequestration
of the Smad proteins. Proc Natl Acad Sci USA. 102:12437–12442.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tecalco-Cruz AC, Sosa-Garrocho M,
Vázquez-Victorio G, Ortiz-García L, Domínguez-Hüttinger E and
Macías-Silva M: Transforming growth factor-β/SMAD Target gene SKIL
is negatively regulated by the transcriptional cofactor complex
SNON-SMAD4. J Biol Chem. 287:26764–26776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stroschein SL, Wang W, Zhou S, Zhou Q and
Luo K: Negative feedback regulation of TGF-beta signaling by the
SnoN oncoprotein. Science. 286:771–774. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ikeuchi Y, Dadakhujaev S, Chandhoke AS,
Huynh MA, Oldenborg A, Ikeuchi M, Deng L, Bennett EJ, Harper JW,
Bonni A, et al: TIF1γ protein regulates epithelial-mesenchymal
transition by operating as a small ubiquitin-like modifier (SUMO)
E3 ligase for the transcriptional regulator SnoN1. J Biol Chem.
289:25067–25078. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hsu YH, Sarker KP, Pot I, Chan A,
Netherton SJ and Bonni S: Sumoylated SnoN represses transcription
in a promoter-specific manner. J Biol Chem. 281:33008–33018. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johnson ES: Protein modification by SUMO.
Annu Rev Biochem. 73:355–382. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pot I and Bonni S: SnoN in TGF-beta
signaling and cancer biology. Curr Mol Med. 8:319–328. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
20
|
Holz JD, Beier E, Sheu TJ, Ubayawardena R,
Wang M, Sampson ER, Rosier RN, Zuscik M and Puzas JE: Lead induces
an osteoarthritis-like phenotype in articular chondrocytes through
disruption of TGF-β signaling. J Orthop Res. 30:1760–1766. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Netherton SJ and Bonni S: Suppression of
TGFβ-induced epithelial-mesenchymal transition like phenotype by a
PIAS1 regulated sumoylation pathway in NMuMG epithelial cells. PLoS
One. 5:e139712010. View Article : Google Scholar
|
|
22
|
Peng H, Feldman I and Rauscher FJ III:
Hetero-oligomerization among the TIF family of RBCC/TRIM
domain-containing nuclear cofactors: A potential mechanism for
regulating the switch between coactivation and corepression. J Mol
Biol. 320:629–644. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Yang K, Mao Q, Zheng X, Kong D and
Xie L: Inhibition of TGF-beta receptor I by siRNA suppresses the
motility and invasiveness of T24 bladder cancer cells via
modulation of integrins and matrix metalloproteinase. Int Urol
Nephrol. 42:315–323. 2010. View Article : Google Scholar
|
|
24
|
Zhao B and Chen YG: Regulation of TGF-β
signal transduction. Scientifica (Cairo). 2014:8740652014.
|
|
25
|
Imoto I, Pimkhaokham A, Fukuda Y, Yang ZQ,
Shimada Y, Nomura N, Hirai H, Imamura M and Inazawa J: SNO is a
probable target for gene amplification at 3q26 in squamous-cell
carcinomas of the esophagus. Biochem Biophys Res Commun.
286:559–565. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang F, Lundin M, Ristimäki A, Heikkilä
P, Lundin J, Isola J, Joensuu H and Laiho M: Ski-related novel
protein N (SnoN), a negative controller of transforming growth
factor-beta signaling, is a prognostic marker in estrogen
receptor-positive breast carcinomas. Cancer Res. 63:5005–5010.
2003.PubMed/NCBI
|
|
27
|
Jahchan NS, Ouyang G and Luo K: Expression
profiles of SnoN in normal and cancerous human tissues support its
tumor suppressor role in human cancer. PLoS One. 8:e557942013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hatakeyama S: TRIM proteins and cancer.
Nat Rev Cancer. 11:792–804. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
He W, Dorn DC, Erdjument-Bromage H, Tempst
P, Moore MA and Massagué J: Hematopoiesis controlled by distinct
TIF1γ and Smad4 branches of the TGFβ pathway. Cell. 125:929–941.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dupont S, Mamidi A, Cordenonsi M,
Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ,
Soligo S, Morsut L, et al: FAM/USP9x, a deubiquitinating enzyme
essential for TGFbeta signaling, controls Smad4 monoubiquitination.
Cell. 136:123–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jain S, Singhal S, Francis F, Hajdu C,
Wang JH, Suriawinata A, Wang YQ, Zhang M, Weinshel EH, Francois F,
et al: Association of overexpression of TIF1γ with colorectal
carcinogenesis and advanced colorectal adenocarcinoma. World J
Gastroenterol. 17:3994–4000. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ding ZY, Jin GN, Wang W, Chen WX, Wu YH,
Ai X, Chen L, Zhang WG, Liang HF, Laurence A, et al: Reduced
expression of transcriptional intermediary factor 1 gamma promotes
metastasis and indicates poor prognosis of hepatocellular
carcinoma. Hepatology. 60:1620–1636. 2014. View Article : Google Scholar : PubMed/NCBI
|