Introduction
Osteosarcoma is a primary malignant bone tumor found
in children and adolescents (1).
Osteosarcoma is characterized by a high metastatic spread tendency,
poor prognosis and low patient survival rate. The current
treatments for osteosarcoma include surgery, radiation,
chemotherapy or a combination of radiotherapy and chemotherapy.
However, the 5-year survival of patients remains 5–20% (2).
Programmed cell death (PCD) is genetically regulated
cell death, involving a series of activation, expression and
regulation of genes, which include the following forms: apoptosis,
autophagy and necrosis (3). In
recent years, autophagy has received wide attention as having an
important role in tumorigenesis and tumor therapy. Autophagy, type
II programmed cell death, which is activated by numerous stresses
such as nutrient starvation, hypoxia (4), intracellular reactive oxygen species
(ROS) levels, bacteria and virus infection or chemical drugs and
oxidative stress, is an evolutionally conserved lysosomal activity
to degrade and recycle long-lived proteins and damaged cytoplasmic
organelles in order to maintain cell homeostasis (5). Moderate autophagy acts as
self-protection against cytotoxicity. However, consequent excessive
autophagy may lead to 'autophagic cell death (type II PCD)'
(6). The relationship between
autophagy and apoptosis is complex and may often appear
contradictory, but this relationship is critical to the overall
fate of the cell (7).
ROS are recognized as oxygen-containing, reactive
and short-lived molecules, that act as secondary messengers in
various signal transductions (8).
ROS include the superoxide anion (O2•−), the
hydroxyl radical (HO) and hydrogen peroxide
(H2O2). When at physiological levels, ROS
have been recognized to modulate cell signaling molecules. However,
aberrantly high ROS expression is intimately associated with
disease and is commonly observed in cancer (9). Upregulation of intracellular ROS
levels may then increase mitochondrial dysfunction and consequent
oxidization of proteins, lipids and DNA, redox imbalance and
oxidative stress. Various anticancer treatments have been shown to
promote ROS-induced autophagy which contributes to the development
of cell drug resistance and to the induction of apoptosis or both,
and several models between ROS and autophagy in cancer treatment
have been proposed (10).
Tanshinone IIA (TanIIA) is one of the major
lipophilic components isolated from the root of Salvia
miltiorrhiza, a widely used herbal medicine (11). It is traditionally used in the
clinic to expand coronary blood vessels, for anti-lipid
peroxidation and cerebrovascular diseases (12,13).
TanIIA has been extensively studied for its effects on a variety of
cancer cell types, including, but not limited to breast (14), lung (15), prostate (16) and bladder cancer (17), through a variety of detailed
mechanisms including antiangiogenic effects via inhibition of
HIF-1α, repression of IL-6/Stat3 signaling, modulation of apoptotic
regulators, regulation of epigenetic modifications and miRs, and
inhibition of glucose metabolism (18–22).
In recent years, research found that TanIIA induces autophagy in
cancer cells (23–25), and plays a pro-survival or pro-death
role in cancer therapy.
Moreover, Zhang et al (26) confirmed that TanIIA induced
apoptosis in osteosarcoma MG-63 cells, but there are no studies on
whether TanIIA triggers autophagy in MG-63 cells and the underlying
mechanism. Our experiments aimed to ascertain the following:
firstly, whether TanIIA induces autophagy in MG-63 cells; and
secondly, to define the relationship between ROS, autophagy and
apoptosis induced by TanIIA in human MG-63 cells.
Materials and methods
Reagents
TanIIA was purchased from the National Institute for
the Control of Pharmaceutical and Biological Products (Beijing,
China), and was then diluted with Dulbecco's modified Eagle's
medium (DMEM) (Gibco, Grand Island, NY, USA) to the desired working
concentration before each experiment. Fetal bovine serum (FBS) was
purchased from Hangzhou Sijiqing Biological Engineering Material
Co., Ltd. (Hangzhou, Zhejiang, China). Chloroquine (CQ), and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and N-acetyl cysteine were purchased from Sigma (St. Louis,
MO, USA). Hoechst 33258 was purchased from Promega (Madison, WI,
USA). Lipofectamine 2000 transfection reagent was obtained from
Invitrogen (Carlsbad, CA, USA). The Annexin V-FITC Apoptosis
Detection Kit I was purchased from Bender Biosciences (San Diego,
CA, USA). Microtubule-associated protein 1 light chain 3 (LC3) and
rabbit monoclonal anti-GAPDH antibodies, and the ECL
chemiluminescence western blotting kit were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture and viability assay
The MG-63 cells (Shanghai Institute of Cell Biology,
Chinese Academy of Sciences Shanghai, China) were maintained in
cultured DMEM containing 10% FBS, 100 µg/ml of penicillin
and 100 µg/ml of streptomycin at 37°C in a 5% CO2
incubator.
MTT assay
The osteosarcoma MG-63 cells were seeded into a
96-well flat bottom microtiter plates at a density of
1×104 cells/well overnight, and then treated with
various concentrations of TanIIA (0, 2.5, 5, 10 and 20 mg/l),
respectively. To each well a total of 20 µl MTT solution (5
g/l) was added and the cells were incubated for 4 h. We used an
automatic multiwell spectrophotometer to calculate the absorbance
value/well at 570 nm. All MTT assays were performed in triplicate.
The inhibitory rate for the proliferation of the MG-63 cells was
calculated according to the formula: (1 − experimental absorbance
value/control absorbance value) × 100%. IC50 values (50%
inhibition concentration) were then calculated using the
Statistical Package for the Social Sciences (SPSS, Inc., Chicago,
IL, USA).
Detection of apoptosis
Annexin V-FITC/propidium iodide (PI) double staining
assay was used to measure apoptosis of the MG-63 cells. After
washing the cells with phosphate-buffered saline (PBS) three times,
each group of cells was stained following the manufacturer's
instructions. The number of apoptotic cells was detected by flow
cytometry and analyzed using CellQuest™ software. Each group was
repeatedly measured three times.
Hoechst 33258 staining
The cells were fixed with 3.7% paraformaldelyde for
30 min at room temperature, and stained with 10 mg/l Hoechst 33258
at 37°C for 15 min. The MG-63 cells were observed under a
fluorescence microscope equipped with a UV filter. Hoechst 33258
freely permeates cell membranes, and apoptotic cells were
identified by the presence of condensed or fragmented nuclei that
stained bright blue.
Green fluorescent protein (GFP)-LC3 dot
assay
The MG-63 cells were transfected with GFP-LC3
plasmids with Lipofectamine 2000 following the manufacturer's
instructions. At 24 h after transfection, the cells were treated
with TanIIA for the indicated periods. Leica DM2500 fluorescence
microscope was used to analyze the number of LC3-II puncta. The
induction of autophagy was quantified by counting the percentage of
cells in each group that contained LC3 aggregates.
Western blot analysis
Cells were washed in PBS, and resuspended in RIPA
buffer at room temperature. After three freeze/thaw cycles and
incubation on ice for 30 min, the lysate was centrifuged at 140,009
× g for 10 min at 4°C. Protein concentration was measured with the
Bradford protein assay reagent using bovine serum albumin as a
standard. Equal amounts of total protein extracts were separated on
12% SDS-PAGE gel and transferred to polyvinylidene fluoride (PVDF)
membranes. The membranes were blocked in Tris-buffered
saline-Tween-20 (TBST) with 5% non-fat milk for 1 h, and incubated
overnight at 4°C with the designated primary antibodies and with
the secondary antibodies for 2 h at room temperature.
Transmission electron microscopy
The MG-63 cells were fixed overnight at 4°C in 2%
paraformaldehyde, 2.5% glutaraldehyde in 0.1 M sodium cacodylate
buffer (pH 7.2), before being postfixed with 1% OsO4 for
1 h. The cells were then dehydrated in a graded ethanol series and
embedded in Agar 100 epoxy resin. Ultrathin sections were mounted
on Cu grids and stained first with uranyl acetate followed by lead
citrate.
RNA interference
The cells were transfected with 60 nM of specific or
non-targeting siRNA using Lipofectamine 2000 according to the
manufacturer's instructions. The cells were treated with indicating
doses of TanIIA for the following experiments. The siRNAs were
obtained from GenePharma, Ltd. (Shanghai, China). The si-Beclin-1
targeting sequence was: 5′-UUCAACACUCUUCAGCUCAUCAUCC-3′; and the
scrambled siRNA sequence was: 5′-UUCUCCGAACGUGUCACGUTT-3′.
ROS detection
Following TanIIA treatment, the intracellular ROS
level was detected by fluorescence microscope using
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) 75 staining.
Briefly, the MG-63 cells were incubated with 5 mM DCFH-DA for 30
min in the dark, and washed with serum-free medium three times. The
fluorescence was excited at the wavelength of 485 nm and the
corresponding emission wavelength was 520 nm. ROS fluorescence
intensity was examined under a Leica DM2500 fluorescence microscope
with an analysis software system.
Phase contrast microscopy
The MG-63 cells were seeded at 37°C overnight, and
then treated with the indicated concentration of TanIIA,
respectively. After 48 h, changes in MG-63 cell morphology were
dynamically observed under a phase contrast microscope. Autophagy
was manifested as cell membrane integrity and cytoplasmic
characteristic vacuolation.
Statistical analysis
All data are presented as the mean ± SD. The
differences between the groups were analyzed using the Student's
t-test, Statistical analyses were performed using SPSS version 16.0
software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
TanIIA inhibits MG-63 cell
proliferation
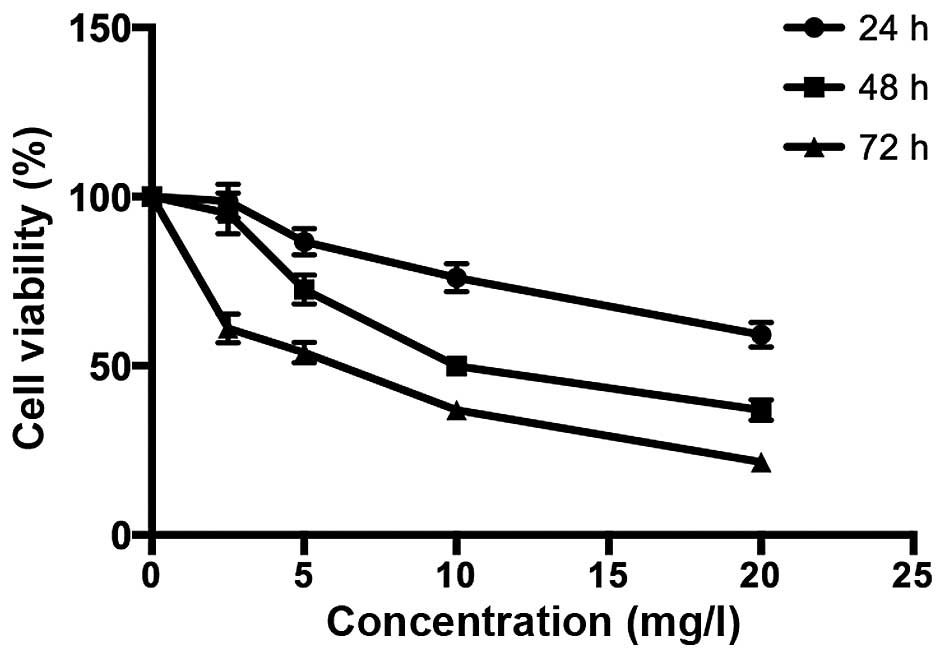
As shown in Fig. 1,
the MTT assay was used to assess the suppression of the
proliferation of MG-63 cells in a dose- and time-dependent manner
after treatment with various concentrations of TanIIA (0, 2.5, 5,
10 and 20 mg/l) for the indicated time periods. In addition, the
half maximal inhibitory concentration value of TanIIA treatment at
48 h was 8.8 mg/l.
TanIIA induces osteosarcoma MG-63 cell
autophagy
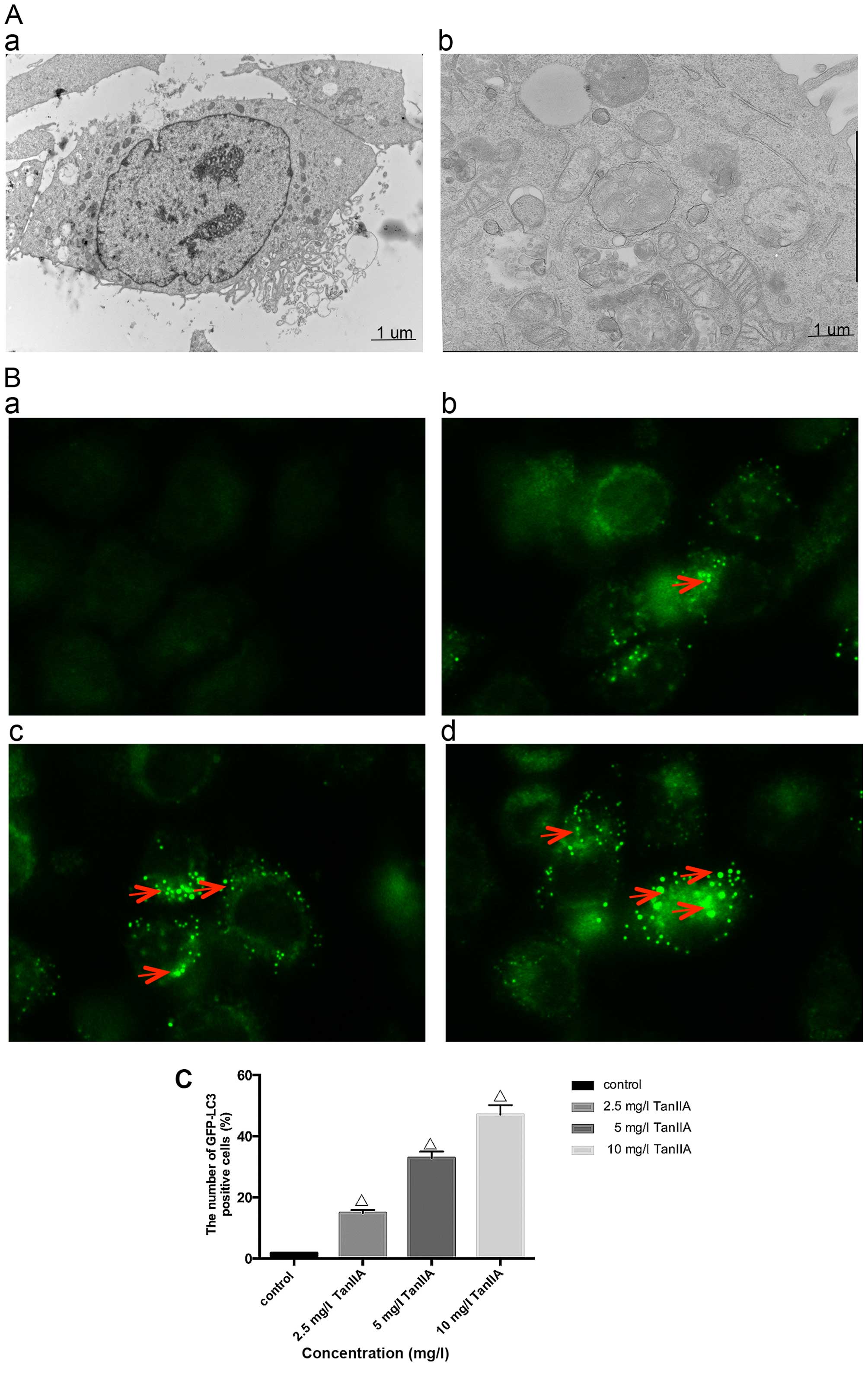
In order to determine whether TanIIA induces
autophagy, we examined using transmission electron microscopy, the
ultrastructure of MG-63 cells treated for 24 h with or without
TanIIA. As shown in Fig. 2A–a, the
control MG-63 cells showed entire nuclei and endoplasmic reticulum,
numerous mitochondria and Golgi. In the TanIIA-treated MG-63 cells
(Fig. 2A–b), several clearly
visible double-membrane autophagic bodies of different sizes
engulfing many cellular organelles, including non-degradable
organelles and macromolecules as well as the typical monolayer of
autophagosomes could be observed. Swollen mitochondria were noted
as well. To further confirm that TanIIA induced autophagy in the
MG-63 cells, GFP-LC3 transient green fluorescence transfection was
used to assay autophagy induction. When autophagy is at the basic
level, GFP-LC3 fluorescence plasmids are dispersed. When autophagy
is activated and autophagosomes are increased, GFP-LC3 puncta
accumulate. As shown in Fig. 2B and
C, compared with the control group which had almost no obvious
fluorescent particles, the faintly visible fluorescent granules
emerged at a lower dose of 2.5 mg/l TanIIA. Higher TanIIA
concentrations, increased not only the number but also the
brightness of the green fluorescent particles.
Studies suggest that the level of LC3II/LC3I is a
hallmark of the degree of autophagy. Beclin-1, part of a lipid
kinase complex, plays a pivotal role in coordinating the
cytoprotective function of autophagy and in opposing the cellular
death process of apoptosis (27).
In order to further confirm autophagy in the TanIIA-treated MG-63
cells, the expression of LC3II/LC3I and Beclin-1 was analyzed by
western blotting; the results of which were similar to those of
GFP-LC3 (Fig. 2D and E).
Autophagy is a dynamic process, and when inhibited
at the final degradation phase, the LC3II/LC3I level is also
elevated. Thus, autophagy flux should be monitored simultaneously.
Pharmacological inhibitors and a genetic approach were used. CQ,
which blocks autophagosome lysosome fusion and sequestrates
autophagosomes (28), was used to
inhibit autophagic flux. As shown in Fig. 2F and G, following pretreatment with
the autophagy inhibitor CQ, the levels of LC3II/LC3I and Beclin-1
were upregulated compared with the TanIIA alone group. We further
inhibited autophagy by silencing the Beclin-1 gene, Transfection
with si-Beclin-1 resulted in a marked reduction in Beclin-1 and
LC3II/LC3I protein expression, while the scrambled siRNA-control
group showed no change (Fig. 2H and
I). All the above results revealed that Beclin-1-dependent
autophagy flux was induced starting from 2.5 mg/l TanIIA.
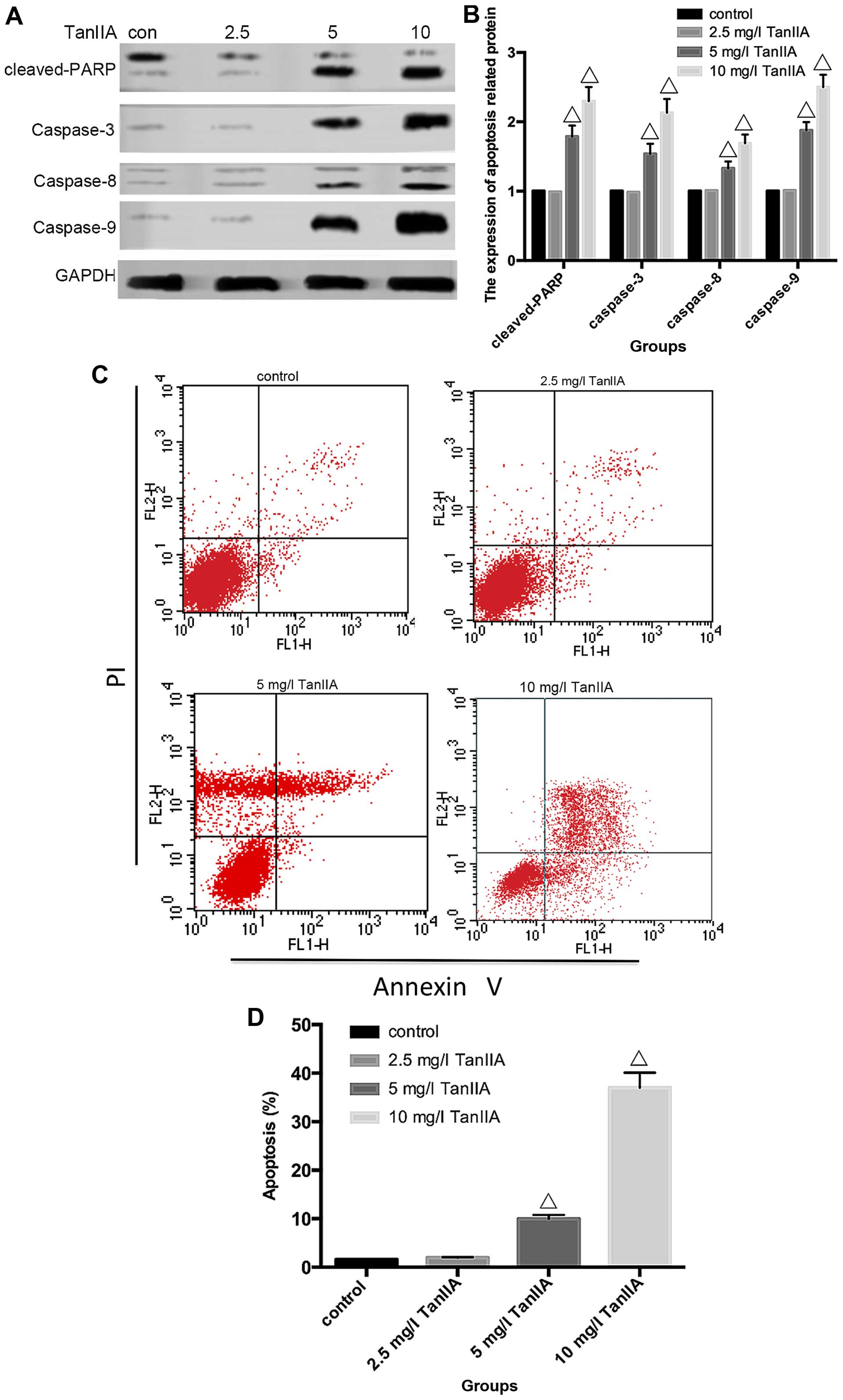
TanIIA triggers MG-63 cell apoptosis
Apoptosis can be induced either by extrinsic stimuli
through cell surface death receptors or by intrinsic stimuli
through the mitochondrial signaling pathway. The extrinsic pathway
is initiated by cell surface death receptor stimulation and
activation of caspase-8, while the intrinsic pathway involves
cytochrome c release from mitochondria and subsequent
caspase-9 activation (29,30). Activation of caspase-8 and -9
enhances executioner caspase-3 activation and ultimately
upregulation of cleaved-PARP. In order to determine which pathway
is induced by TanIIA in MG-63 cells, caspase-3, -8 and -9 as well
as cleaved-PARP were assessed by western blotting. As shown in
Fig. 3A and B, compared with the
control group, neither cleaved-caspase-3, -8 and -9 nor
cleaved-PARP were detected in the 2.5 mg/l TanIIA group. However,
caspase-3, -8 and -9, and cleaved-PARP were gradually increased in
a concentration-dependent (5 and 10 mg/l TanIIA) manner. To further
confirm these findings, caspase inhibitors were employed. The
apoptosis rates of the 2.5 mg/l TanIIA and control groups were not
significantly altered, when compared with that of the 5 and 10 mg/l
TanIIA groups pretreated with the caspase-specific inhibitors. As
expected (Fig. 3G), we observed a
moderate suppressive effect of z-IETD-fmk and z-LEHD-fmk on the
TanIIA-induced apoptotic rate while z-VAD-fmk had a more apparent
inhibitory effect. All the data implied that TanIIA induced
caspase-dependent apoptosis by activating both the extrinsic and
intrinsic pathways.
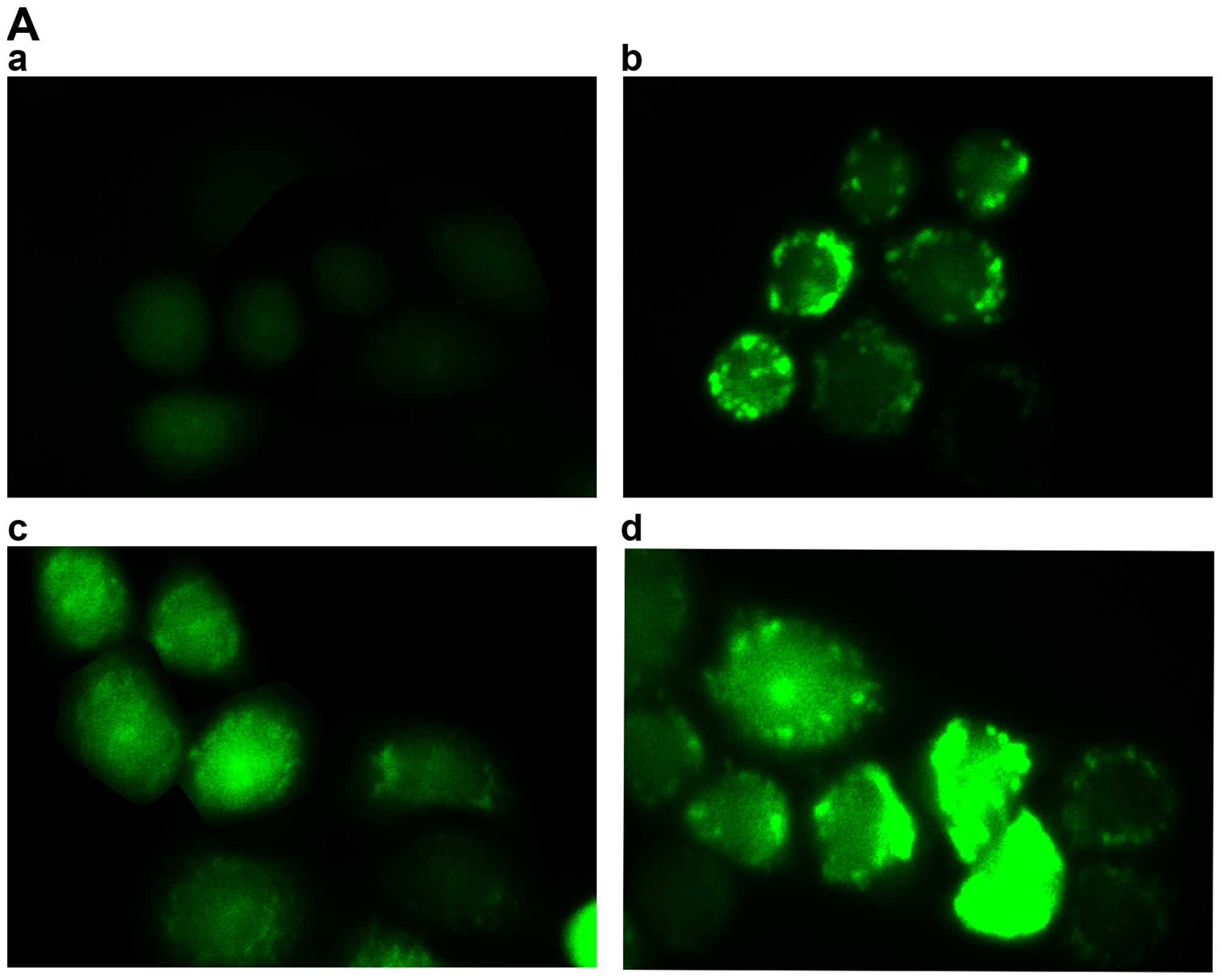
To further clarify whether TanIIA induces apoptosis,
flow cytometry and Hoechst 33258 were used. As shown in Fig. 3C and D, compared with the control
group, no obvious change in the 2.5 mg/l TanIIA group was noted,
while the apoptotic rates of the other groups were variously
increased (P<0.05). Similar results were found for the Hoechst
33258 experimental results. Normal nuclei emit light blue
fluorescence. In case of cell death, markedly induced chromatin
condensation or fragmentation, shows bright blue (yellow arrow).
DNA fragmentation ratio of the TanIIA-treated cells was
predominantly elevated, compared with the control and 2.5 mg/l
TanIIA groups, in a dose-dependent manner (Fig. 3E and F).
Inhibition of autophagy induces a
double-dose effect on TanIIA-induced apoptosis
Numerous studies have confirmed that antitumor drugs
regulate autophagy and apoptosis to exert a therapeutic effect. Our
data generally showed that both apoptosis and autophagy were
activated in a concentration-dependent manner in the MG-63 cells
following exposure to TanIIA. In our experiments, however, TanIIA
presented sectional effects; the lower and non-cytotoxic
concentration (2.5 mg/l) showed just autophagy activation and in
contrast, no caspase activation nor apoptosis induction was
recognized which was in agreement with the MTT assay (Fig. 1). Moreover, following treatment with
higher concentrations of TanIIA (5 and 10 mg/l), both apoptosis and
autophagy were found to occur in the MG-63 cells.
To further elucidate the role of autophagy in
TanIIA-treated MG-63 cells, CQ was employed. As shown in Fig. 4A, the MTT assay indicated that under
a condition of low-dose (2.5 mg/l), TanIIA no obvious change in
cell viability was showed in the control, CQ and TanIIA groups, but
a slight decreased in the CQ + 2.5 mg/l TanIIA group. However, cell
viability increased once autophagy was inhibited with CQ + TanIIA
groups at high doses (5 and 10 mg/l) TanIIA (Fig. 4B and C), that is to say, less
apoptotic cells were found in the TanIIA (5 and 10 mg/l) and TanIIA
+ CQ group.
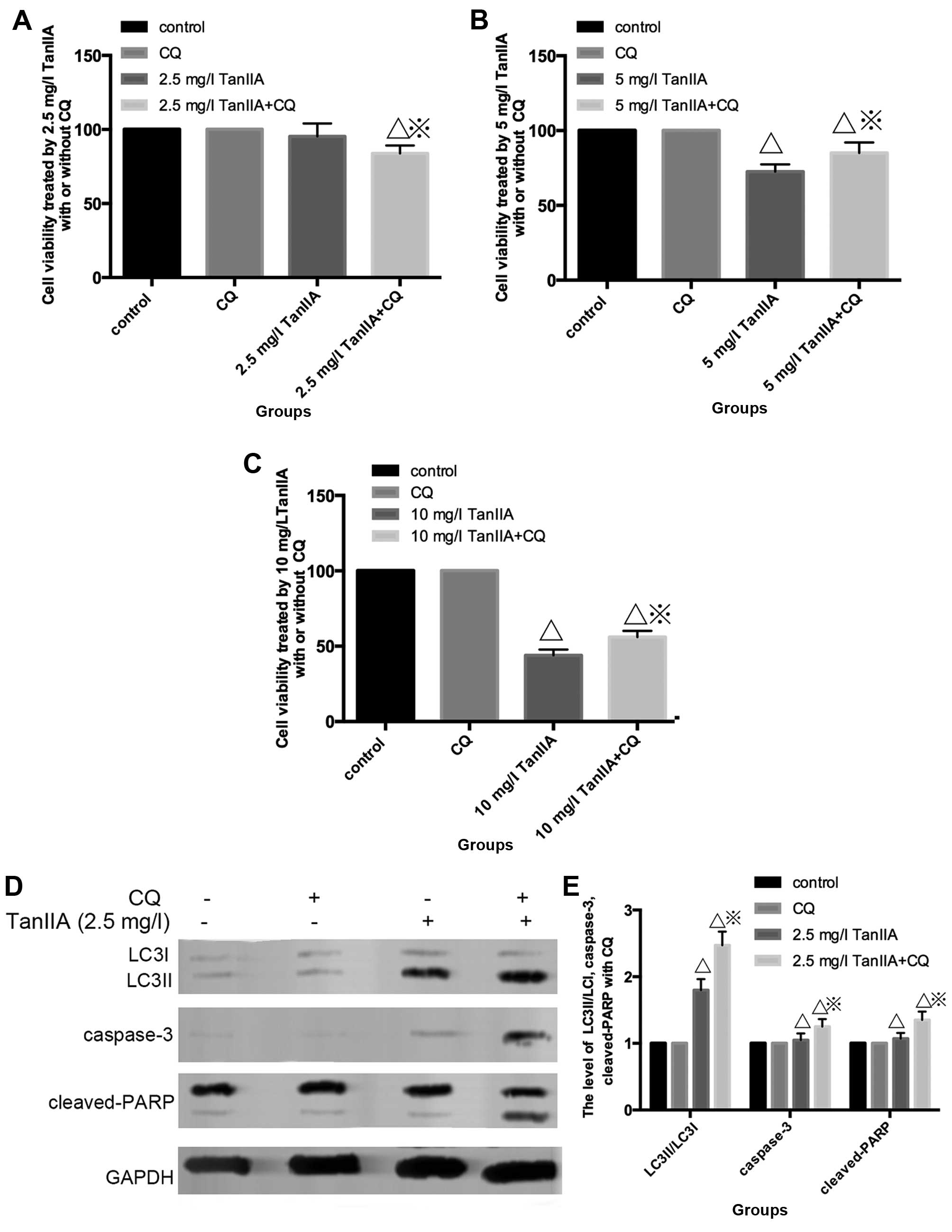 | Figure 4Inhibition of autophagy has a double
effect on TanIIA-induced apoptosis. (A and B) Effect of a low-dose
(2.5 mg/l) of TanIIA on cell viability as determined by MTT assay
with/without pretreatment of CQ. ΔP<0.05 vs. the
control group, ※P<0.05 vs. the TanIIA group. (B and
C) Effects of high-doses (5 and 10 mg/l) of TanIIA on cell
viability, respectively, as determined by MTT assay with/without
pretreatment of CQ. ΔP<0.05 vs. the control group,
※P<0.05 vs. the TanIIA group. (D and E) Apoptosis-
and autophagy-related proteins in MG-63 cells following treatment
with 2.5 mg/l TanIIA was explored through western blotting with or
without CQ. ΔP<0.05 vs. the control group,
※P<0.05 vs. the TanIIA group. (F–I) Levels of
caspase-3, cleaved-PARP and LC3II/LC3I were assessed in MG-63 cells
following treatment with 5 and 10 mg/l TanIIA by western blotting
in the presence or absence of CQ; ΔP<0.05 vs. the
control group, ※P<0.05 vs. the TanIIA group. (J and
K) Levels of LC3II/LC3I, cas-pase-3, cleaved-PARP were assessed in
MG-63 cells following treatment with 5 mg/l TanIIA for 0–72 h;
ΔP<0.05 vs. the control group. (L and M) Levels of
LC3II/LC3I, caspase-3, cleaved-PARP were assessed in MG-63 cells
following treatment with 5 mg/l TanIIA for 0–72 h with or without
CQ; ΔP<0.05 vs. the control group. |
Fig. 4D and E shows
that cleaved-PARP, caspase-3 and LC3II/LC3I were modestly increased
at 2.5 mg/l TanIIA + CQ compared with 2.5 mg/l TanIIA alone.
However, decreased cleaved-PARP, cleaved-caspase-3 but LC3II/LC3I
was still augmented at a higher dose of TanIIA + CQ compared with a
higher dose (5 and 10 mg/l) of TanIIA alone (Fig. 4F–I). Based on these observations, it
seemed then that MG-63 cells first underwent cytoprotective
autophagy and later, due to excessive exposure and cellular damage,
protective autophagy was altered to autophagic cell death involved
in apoptosis.
In order to confirm this point, we monitored the
induction of autophagy and apoptosis over time by performing
time-course analysis. MG-63 cells were treated with 5 mg/l TanIIA
for 0–72 h. Fig. 4J and K shows
that LC3II/LC3I accumulated although modestly, starting at 6 h but
became more evident at 12 h. However, caspase-3 and cleaved-PARP
appeared at 24 h, indicated that autophagy preceded apoptosis in
the TanIIA-treated cells. In addition, following pretreatment with
CQ (Fig. 4L and M), LC3II/LC3I was
more significantly increased. Moreover, caspase-3 and cleaved-PARP
were faintly upregulated at 6 and 12 h, but decreased at 24 h when
compared with the TanIIA alone group. Coincident with the western
blot results, the MTT results demonstrated that at 12 h, cell
viability was downregulated once autophagy was inhibited. On the
contrary, cell viability was increased following pretreatment with
CQ at 24 h.
Therefore, we speculated that the TanIIA-treated
MG-63 cells presented stage effects: low-dose 2.5 mg/l
TanIIA/shorter time-period induced moderate autophagy to offset a
small rate of apoptosis whereby the autophagic pathway could
possibly degrade the damaged cellular components, suggesting
pro-survival autophagy. In contrast, it is possible that high
TanIIA concentrations (5 and 10 mg/l) or a longer treatment period
induced a significant level of autophagy and damage that was beyond
repair, that is to say, a pro-death role of autophagy.
Effect of ROS on TanIIA induces autophagy
and apoptosis of MG-63 cells
There is mounting evidence pointing to the
involvement of ROS in the upstream of autophagy besides apoptosis
(31). We found that the level of
ROS was increased in a dose-dependent manner, compared with the
control group (Fig. 5A and B);
which indicated that TanIIA induced ROS generation in the MG-63
cells. In order to examine whether TanIIA-induced autophagy and
apoptosis was attributed to the generation of ROS in MG-63 cells we
examined the effect of NAC, a ROS scavenger on cell viability,
cleaved-PARP, caspase-3 and LC3II/LC3I. The ROS scavenger NAC
almost completely eliminated the ability of TanIIA to induce
autophagy and apoptosis, and decreased LC3-II/LC3I,
cleaved-caspase-3 and cleaved-PARP levels (Fig. 5C and D) which suggests that the
activation of intracellular ROS plays an essential role in
TanIIA-induced autophagy.
Discussion
Tumor occurrence is a multifactorial, multi-stage
and complex process, closely related to abnormal regulation of
programmed cell death (PCD). Cell apoptosis is one of the classical
mechanisms of PCD and has been extensively researched as the main
strategy in the treatment of cancer. The relationship of autophagy
and apoptosis is a research 'hotspot'. The use of autophagy to
control tumor development and the corresponding treatment
intervention strategy, is one of the means to study the prevention
and control cancer.
In our study, we found that when compared with the
control group, the cell viability of MG-63 cells incubated with 2.5
mg/l TanIIA was not statistically significantly altered. However,
at the range of 2.5–20 mg/l TanIIA, osteosarcoma MG-63 cell
proliferation was inhibited in dose- and time-dependent manner.
Similar to Gao et al (23), we also observed characteristic
autophagosomes which embraced extra-large proteins and organelles
under transmission electron microscope. The autophagy-related
protein LC3II/LC3I and Beclin-1 expression were upregulated and
GFP-LC3 green fluorescence plasmids accumulated starting from 2.5
mg/l TanIIA. In addition, following pretreatment with CQ and
Si-Beclin-1, the level of LC3II/LC3I as well as Beclin-1 was
altered. All the above experiments confirmed that TanIIA triggered
Beclin-1-dependent autophagy in the osteosarcoma MG-63 cells.
In line with Zhang et al (26), we revealed that TanIIA induced
apoptosis by activating both extrinsic and intrinsic pathways.
However, at the dose of 2.5 mg/l TanIIA, caspase-3, -8 and -9, and
cleaved-PARP, had no appreciable upregulation. When the TanIIA dose
was increased to 5 and 10 mg/l, evidence of apoptosis was confirmed
by flow cytometry and Hoechst 33258. In addition, following
pretreatment with the caspase inhibitors, the apoptotic rate was
decreased to varying degrees.
The role of TanIIA-induced autophagy in cancer
treatment, has been debated: Gao et al (23) and Li et al (24) found that TanIIA-induced autophagy
failed to benefit cytotoxicity in their experiments. However, Yun
et al (25) reported that
TanIIA-induced autophagic cell death benefited cancer treatment in
leukemic cells. The role of autophagy was positive or negative for
tumor growth or tumor therapy, according to the developmental stage
of the tumor as well as the tissue or cells targeted. In recent
years, autophagy has been recognized as another target for tumor
therapy in addition to apoptosis. The relationship between
autophagy and apoptosis is regarded as the key to the survival of
cells.
Several studies have demonstrated that both
apoptosis and autophagy occur concomitantly in the same cells under
certain circumstances. In our experiment, autophagy also played a
double role. Following treatment with 2.5 mg/l TanIIA, increased
LC3II/LC3I verified generation of autophagy, but there was no
evidence of apoptosis generation. In addition, for high-doses of
TanIIA (5 and 10 mg/l), not only 5 mg/l TanIIA, but also 10 mg/l
TanIIA, LC3II/LC3I was significantly upregulated and statistical
significance of apoptosis induction was observed. Al Dhaheri et
al (32) found that lower and
non-cytotoxic doses of carnosol induced (25 mM) autophagy alone,
which was the same as a low-dose of tetrandrine (33) in human hepatocellular carcinoma.
Nonetheless, pretreatment with autophagy inhibitor CQ, and a
low-dose 2.5 mg/l TanIIA, increased LC3II/LC3I, caspase-3 and
cleaved-PARP, and the cell viability was slightly lower than the
2.5 mg/l TanIIA alone group. The results of 5 and 10 mg/l TanIIA
treatment were contrary to 2.5 mg/l TanIIA. Decreased apoptosis was
noted and cell viability increased. Moreover, we used 5 mg/l TanIIA
to treat the MG-63 cells for 0–72 h. LC3II/LC3I was slightly higher
at 6 h, significantly increased at 12 h, and apoptosis occurred at
24 h. In addition, pre-incubation with CQ regulated the expression
of caspase-3, and cleaved-PARP was regulated at 6 and 12 h, and
decreased at 24 h.
The above experimental results unveiled that
regardless of the TanIIA dose or treatment period, autophagy
occurred earlier than apoptosis. In addition, Shrivastava et
al (34) concluded that the
ratio of cannabidiol-induced autophagy and apoptosis changed at
various times. Strikingly, following pretreatment with CQ,
apoptotic cells appeared at 2.5 mg/l TanIIA and at shorter
treatment periods; and for both high-dose TanIIA or a longer
treatment time, the apoptotic rate decreased. This showed that for
low-dose and shorter treatment time, moderate autophagy played a
pro-survival role against little apoptosis in the MG-63 cells.
Excessive autophagy caused by higher doses and longer treatment
time, participated in or contributed to cell death, thus played a
pro-death role.
Evidence suggests that many antitumor drugs
emphasize the pivotal role of ROS production in inducing autophagy
and cell death in cancer therapy (35,36).
Similarly, in the present study, we observed that TanIIA enhanced
ROS generation in the MG-63 cells in a dose- and time-dependent
manner. Recent studies have shown that the extent of ROS levels
produced in response to anticancer agents elicited different
effects on the cancer cells. While low level ROS generation was
shown to induce autophagy, excessive ROS accumulation triggered
both apoptosis and cell death (37). In agreement with these studies, we
found that, depending (Fig. 5E) on
the working-dose and exposure periods, TanIIA elicited different
responses in the MG-63 cells. We found that low-dose TanIIA (2.5
mg/l) as well as shorter treatment periods resulted in a basic
level of ROS production and autophagy. We believe that in this
limited range, autophagy was induced to remove damaged organelles
as a self-defense survival mechanism.
A prolonged exposure as well as higher
concentrations of TanIIA (5 and 10 mg/l) led to excessive ROS
production, which ultimately resulted in higher levels of oxidative
damage that exceeded the cell repair capabilities that eventually
caused autophagic cell death and caspase-dependent apoptosis. These
findings which highlighted the key role of ROS accumulation are
supported by the following evidence: the abrogation of ROS
production by NAC, and a total decrease in autophagy and apoptosis.
Altogether, these data strongly demonstrated that ROS production in
response to TanIIA acted as an upstream effector for autophagy and
subsequent apoptosis induction. Similar mechanism has been
described by some reports, for example: Lin et al for
safingol (38), an anticancer drug
in phase I clinical trial, which has been shown to mediate a
concentration-dependent effect on MDA-MB-231 and HT29 cancer cells
(38). These authors showed that a
low concentration of safingol triggered autophagy as a damage
repair mechanism, while a higher concentration led to cell death.
Al Dhaheri et al (32) found
that low-dose carnosol induced moderate ROS and led to protective
autophagy, while high expression of ROS generated PCD.
In this experiment, we found for the first time a
sequential effect of TanIIA on osteosarcoma MG-63 cells. Its basic
function is the degree of oxidative stress, different from other
studies concerning TanIIA treatment (23–25).
However, there is no doubt that the underlying mechanisms of TanIIA
in osteosarcoma therapy requires further investigation; for
example, to ascertain whether DNA damage participates in
TanIIA-treated MG-63 cells. In addition, endoplasmic reticulum (ER)
stress is an important mechanism involved in the activation of
autophagy and apoptosis, and elucidation of the relationship
between ER stress and ROS requires investigation. In addition, the
ERK signaling pathway is closely associated with ER stress-mediated
autophagy, which was not investigated in the present study. Thus,
further exploration is needed to identify the interplay between
them.
In conclusion, our study provides experimental
evidence that ROS play a pivotal role in mediating the cytotoxic
effect of TanIIA on MG-63 cells. Together, these data suggest that
TanIIA may be a potential agent with which to treat osteosarcoma at
appropriate concentrations. When combined with ROS-generating
agonists, a synergistic effect is exhibited by executing different
modes of cell death.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar
|
|
2
|
PosthumaDeBoer J, Witlox MA, Kaspers GJ
and van Royen BJ: Molecular alterations as target for therapy in
metastatic osteosarcoma: A review of literature. Clin Exp
Metastasis. 28:493–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Edinger AL and Thompson CB: Death by
design: Apoptosis, necrosis and autophagy. Curr Opin Cell Biol.
16:663–669. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yorimitsu T and Klionsky DJ: Autophagy:
Molecular machinery for self-eating. Cell Death Differ. 12(Suppl
2): S1542–S1552. 2005. View Article : Google Scholar
|
|
5
|
Hale AN, Ledbetter DJ, Gawriluk TR and
Rucker EB III: Autophagy: Regulation and role in development.
Autophagy. 9:951–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Apel A, Zentgraf H, Büchler MW and Herr I:
Autophagy - A double-edged sword in oncology. Int J Cancer.
125:991–995. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oral O, Akkoc Y, Bayraktar O and Gozuacik
D: Physiological and pathological significance of the molecular
cross-talk between autophagy and apoptosis. Histol Histopathol.
31:479–498. 2016.
|
|
8
|
D'Autreaux B and Toledano MB: ROS as
signaling molecules: Mechanisms that generate specificity in ROS
homeostasis. Nat Rev Mol Cell Biol. 8:813–824. 2007. View Article : Google Scholar
|
|
9
|
Gough DR and Cotter TG: Hydrogen peroxide:
a Jekyll and Hyde signalling molecule. Cell Death Dis. 2:e2132011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cross CE, Halliwell B, Borish ET, Pryor
WA, Ames BN, Saul RL, McCord JM and Harman D: Oxygen radicals and
human disease. Ann Intern Med. 107:526–545. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu J, Huang H, Liu J, Pi R, Chen J and Liu
P: Tanshinone IIA protects cardiac myocytes against oxidative
stress-triggered damage and apoptosis. Eur J Pharmacol.
568:213–221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li X, Xu X, Wang J, Yu H, Wang X, Yang H,
Xu H, Tang S, Li Y, Yang L, et al: A system-level investigation
into the mechanisms of Chinese Traditional Medicine: Compound
Danshen Formula for cardiovascular disease treatment. PLoS One.
7:e439182012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li YI, Elmer G and Leboeuf RC: Tanshinone
IIA reduces macrophage death induced by hydrogen peroxide by
upregulating glutathione peroxidase. Life Sci. 83:557–562. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Wei Y, Yuan S, Liu G, Lu Y, Zhang
J and Wang W: Potential anticancer activity of tanshinone IIA
against human breast cancer. Int J Cancer. 116:799–807. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu F, Yu G, Wang G, Liu H, Wu X, Wang Q,
Liu M, Liao K, Wu M, Cheng X, et al: An NQO1-initiated and
p53-independent apoptotic pathway determines the anti-tumor effect
of tanshinone IIA against non-small cell lung cancer. PLoS One.
7:e421382012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu D, Lin TH, Zhang C, Tsai YC, Li S,
Zhang J, Yin M, Yeh S and Chang C: The selective inhibitory effect
of a synthetic tanshinone derivative on prostate cancer cells.
Prostate. 72:803–816. 2012. View Article : Google Scholar
|
|
17
|
Chiu SC, Huang SY, Chang SF, Chen SP, Chen
CC, Lin TH, Liu HH, Tsai TH, Lee SS, Pang CY, et al: Potential
therapeutic roles of tanshinone IIA in human bladder cancer cells.
Int J Mol Sci. 15:15622–15637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fu P, Du F, Chen W, Yao M, Lv K and Liu Y:
Tanshinone IIA blocks epithelial-mesenchymal transition through
HIF-1α down-regulation, reversing hypoxia-induced chemotherapy
resistance in breast cancer cell lines. Oncol Rep. 31:2561–2568.
2014.PubMed/NCBI
|
|
19
|
Lin C, Wang L, Wang H, Yang L, Guo H and
Wang X: Tanshinone IIA inhibits breast cancer stem cells growth in
vitro and in vivo through attenuation of IL-6/STAT3/NF-κB signaling
pathways. J Cell Biochem. 114:2061–2070. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Shi DY, Liu SL and Zhong L:
Tanshinone IIA induces growth inhibition and apoptosis in gastric
cancer in vitro and in vivo. Oncol Rep. 27:523–528. 2012.
|
|
21
|
Wang L, Zhang C, Guo Y, Su ZY, Yang Y, Shu
L and Kong AN: Blocking of JB6 cell transformation by tanshinone
IIA: Epigenetic reactivation of Nrf2 antioxidative stress pathway.
AAPS J. 16:1214–1225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li FQ, Zeng DK, Jia CL, Zhou P, Yin L,
Zhang B, Liu F and Zhu Q: The effects of sodium tanshinone IIa
sulfonate pretreatment on high glucose-induced expression of
fractalkine and apoptosis in human umbilical vein endothelial
cells. Int J Clin Exp Med. 8:5279–5286. 2015.PubMed/NCBI
|
|
23
|
Gao H, Sun W, Zhao W, Hao W, Leung CH, Lu
J and Chen X: Total tanshinones-induced apoptosis and autophagy via
reactive oxygen species in lung cancer 95D cells. Am J Chin Med.
43:1265–1279. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li CL, Han XC, Zhang H, Wu J and Li B: The
interplay between autophagy and apoptosis induced by tanshinone IIA
in prostate cancer cells. Tumour Biol. 37:7667–7674. 2016.
View Article : Google Scholar
|
|
25
|
Yun SM, Jung JH, Jeong SJ, Sohn EJ, Kim B
and Kim SH: Tanshinone IIA induces autophagic cell death via
activation of AMPK and ERK and inhibition of mTOR and p70 S6K in
KBM-5 leukemia cells. Phytother Res. 28:458–464. 2014. View Article : Google Scholar
|
|
26
|
Zhang Y, Wei RX, Zhu XB, Cai L, Jin W and
Hu H: Tanshinone IIA induces apoptosis and inhibits the
proliferation, migration, and invasion of the osteosarcoma MG-63
cell line in vitro. Anticancer Drugs. 23:212–219. 2012. View Article : Google Scholar
|
|
27
|
Cao Y and Klionsky DJ: Physiological
functions of Atg6/Beclin 1: A unique autophagy-related protein.
Cell Res. 17:839–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carew JS, Medina EC, Esquivel JA II,
Mahalingam D, Swords R, Kelly K, Zhang H, Huang P, Mita AC, Mita
MM, et al: Autophagy inhibition enhances vorinostat-induced
apoptosis via ubiquitinated protein accumulation. J Cell Mol Med.
14:2448–2459. 2010. View Article : Google Scholar :
|
|
29
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogenne.
25:4798–4811. 2006. View Article : Google Scholar
|
|
30
|
Tait SW and Green DR: Mitochondrial
regulation of cell death. Cold Spring Harb Perspect Biol.
5:a0087062013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kongara S and Karantza V: The interplay
between autophagy and ROS in tumorigenesis. Front Oncol. 2:1712012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Al Dhaheri Y, Attoub S, Ramadan G, Arafat
K, Bajbouj K, Karuvantevida N, AbuQamar S, Eid A and Iratni R:
Carnosol induces ROS-mediated beclin1-independent autophagy and
apoptosis in triple negative breast cancer. PLoS One.
9:e1096302014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gong K, Chen C, Zhan Y, Chen Y, Huang Z
and Li W: Autophagy-related gene 7 (ATG7) and reactive oxygen
species/extracellular signal-regulated kinase regulate
tetrandrine-induced autophagy in human hepatocellular carcinoma. J
Biol Chem. 287:35576–35588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shrivastava A, Kuzontkoski PM, Groopman JE
and Prasad A: Cannabidiol induces programmed cell death in breast
cancer cells by coordinating the cross-talk between apoptosis and
autophagy. Mol Cancer Ther. 10:1161–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dewaele M, Maes H and Agostinis P:
ROS-mediated mechanisms of autophagy stimulation and their
relevance in cancer therapy. Autophagy. 6:838–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Panda PK, Mukhopadhyay S, Das DN, Sinha N,
Naik PP and Bhutia SK: Mechanism of autophagic regulation in
carcinogenesis and cancer therapeutics. Semin Cell Dev Biol.
39:43–55. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen N and Karantza-Wadsworth V: Role and
regulation of autophagy in cancer. Biochem Biophys Acta.
1793:1516–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ling LU, Tan KB, Lin H and Chiu GN: The
role of reactive oxygen species and autophagy in safingol-induced
cell death. Cell Death Dis. 2:e1292011. View Article : Google Scholar : PubMed/NCBI
|