Introduction
Metastasis is implicated in cancer aggressiveness
and poor clinical outcome, which accounts for ~90% of all
cancer-related deaths (1,2). Metastatic cascade is a multistep
process composed of a series of events such as invasion,
intravasation, transport, extravasation, and colonization (3). Tumor metastatic progression is driven
not only by genetic aberrations intrinsic to malignant cells but by
pro-metastatic factors from the surrounding environment (4). Besides being enriched with genetically
altered cancer cells, the tumor microenvironment (TME) consists of
heterogeneous mixture of non-cancerous cells, including
fibroblasts, immune cells, and endothelial cells in conjunction
with extracellular matrix (ECM) called stroma (5,6). The
progression of cancer towards metastasis is regulated by a
time-evolving network of interactions between neoplastic cells and
the associated stroma. In addition to providing a physical support
for architecture, ECM represents an important player in mediating
communication between cells and signaling cascades involved in cell
migration, proliferation, survival and differentiation (7–9).
Matricellular proteins, a family of non-structural ECM proteins,
play important roles in modulating cell-cell and cell-matrix
interactions (10). Multiple
members of the matricellular protein family have been identified as
important regulators in conferring various aspects of cancer cell
behavior, such as epithelial-mesenchymal transition, angiogenesis,
cell migration, survival, proliferation, and ECM degradation
(11).
Osteopontin (OPN) is a multifunctional matricellular
protein produced by a broad range of cells including osteoclasts,
macrophages, T cells, kidneys, and vascular smooth muscle cells
(12). OPN modulates multiple
cellular processes, such as inflammation, wound healing, bone
formation and remodeling, as well as tumor growth and metastasis
(13,14). By interaction with αvβ3 integrins
and CD44, OPN signals a complex cascade promoting proliferation,
migration and invasion of tumor cells, inhibiting apoptosis, and
facilitating extracellular remodeling and angiogenic processes
(15–18). In line with these experimental
observations, the pro-tumorigenic and pro-metastatic activities of
OPN were suggested in numerous clinical laboratory analyses. It has
been shown, for example, that OPN plasma concentration correlates
well with tumor grade and progression in multiple cancers such as
breast and ovarian cancer (19).
Therefore, targeted inhibition of OPN represents a rational and
promising therapeutic strategy in oncology.
Brefelamide is an aromatic amide isolated from
Dictyostelium cellular slim molds. It was previously shown
that brefelamide inhibits the proliferation of human-derived 1321N1
astrocytoma cells (20), and the
antiproliferative effect was associated with a reduced
phosphorylation of epidermal growth factor receptor (EGFR) and
attenuated EGFR-mediated ERK signaling cascade (21). In this study, we further explored
the feasibility of brefelamide as an anticancer therapeutic agent
and found that brefelamide inhibited OPN gene expression and
consequently reduced the invasion of human lung
adenocarcinoma-derived A549 cells. The inhibition of OPN by
brefelamide appears to involve induction of Smad4 expression and
subsequent restoration of the TGF-β/Smad-mediated OPN
repression.
Materials and methods
Plasmids
The reporter vector pOPN1-luc, as well as its
mutants pOPN-lucmTIE1 an 2 have been described previously (22,23)
The plasmid expressing shRNA against Smad4 under the control of the
U6 promoter was constructed as previously described (23,24).
For construction of pcDNA3.1-OPN, full-length cDNA fragment of OPN
open reading frame was amplified with primers
5′-ataaagcttATGAGAATTGCAGTGATTTG-3′ and
5′-atatctagaTTAATTGACCTCAGAAGATG-3′, digested with HindIII
and XbaI, and ligated into pcDNA3.1 vector (Invitrogen). The
sequence in the construct was confirmed by nucleotide
sequencing.
Cells
All cell lines were purchased from American Type
Culture Collection (ATCC) and maintained in Dulbecco's modified
Eagle's medium (DMEM, invitrogen) supplemented with 10% fetal
bovine serum (FBS) and 50 U/ml penicillin and streptomycin in a 5%
CO2 humidified atmosphere. The cell line, A549/OPN-luc,
was established by co-transfection of A549 cells with pOPN1-luc and
pPUR (Clontech Laboratories, Inc.), followed by selection in the
presence of 1 µg/ml puromycin (Sigma).
Chemicals
Brefelamide was synthesized as described previously
(20). It was dissolved in DMSO at
a concentration of 50 mmol/l and stored at −20°C. Aliquots of this
stock solution were subsequently diluted to the indicated
concentrations before the treatment of the cells. Cisplatin was
purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan)
and sorafenib was purchased from Cayman Chemicals (Ann Arbor, MI,
USA).
ELISA
For OPN detection, supernatants were collected from
culture after a 48-h treatment in the absence or presence of
brefelamide, and OPN levels in the culture medium were determined
with OPN ELISA kit (R&D systems).
Real-time RT-PCR
Cells were lysed with TRIzol reagent (Invitrogen),
and total RNA was extracted according to the manufacturer's
instructions. After treatment with RNase-Free DNase (Promega), the
DNA-Free RNA (250 ng) was used for the synthesis of the
first-strand cDNA at 42°C for 60 min using M-MLV Reverse
Transcriptase (Invitrogen). Real-time quantitative PCR using Power
SYBR Green PCR Master Mix was conducted for 40 cycles at 95°C for
15 sec and at 60°C for 1 min in a 96-well format on StepOnePlus™
Real-Time PCR System (both from Applied Biosystems). Primer
sequences were as follows: OPN forward, 5′-ACTCGTCTCAGGCCAGTTG-3′
and reverse, 5′-CGTTGGACTTGGAAGG-3′; Smad4 forward,
5′-GCATCGACAGAGACATACAG-3′ and reverse, 5′-AATCCATTCTGCTGCTGTCC-3′;
GAPDH forward, 5′-TGATGACATCAAGAAGGTGG-3′ and reverse,
5′-TCCTTGGAGGCCATGTGGGC-3′.
Transient transfection and luciferase
assay
Cells were seeded at 1×105 in 1 ml
medium/well of 12-well plates 24 h before transfection. Indicated
plasmid DNAs were transfected into cells with Effectene
Transfection Reagent (Qiagen). For each transient transfection,
pRL-TK vector (Promega) was co-transfected as an internal control
to normalize the transfection efficiency. The cells were harvested
at a 48-h post-transfection, and the cell lysates were prepared for
luciferase assay with Dual-Luciferase Reporter Assay System
(Promega) according to the manufacturer's instructions. Luciferase
activities were measured using a GloMax® 20/20
Luminometer (Promega).
Invasion assays
A549 cells were suspended in DMEM containing 0.1%
bovine serum albumin and seeded into cell culture inserts
constructed with an 8-µm porous membrane, which was
pre-coated with Matrigel (10 mg/ml; BD Biosciences). As a
chemoattractant, DMEM supplemented with 10% FCS was added outside
the inserts and incubated for 48 h at 37°C. The cells were labeled
with Calcein AM solution (BD Biosciences). The number of cells
which had migrated through the membrane was quantified using
SpectraMax M5 microplate reader (Molecular Devices). Three sets of
experiments were carried out, each one in triplicate.
Cell proliferation assay
Each cell line was plated in 96-well plates at a
concentration of 1×104 cells/well in a volume of 100
µl. Twenty-four hours later, brefelamide or another
anticancer reagent was added, individually or in combination, to
the cells at various concentrations. After an additional 72-h
culture, cell growth was measured with Cell Proliferation Reagent
WST-1 (Roche Diagnostics) according to the manufacturer's
instructions. The half maximal inhibitory concentration
(IC50) of compounds was determined from the
dose-response curves.
Statistics
All p-values were determined using Dunnett's test or
Tukey-Kramer test. The differences were considered significant at
p<0.05.
Results
Inhibition of OPN expression by
brefelamide
In an attempt to screen compounds that have the
potential to inhibit the transcription of OPN, we developed an
experimental system employing A549 human lung adenocarcinoma
epithelial cell line stably transfected with pOPN1-luc, in which
luciferase gene is directed under the control of human OPN promoter
(22). We monitored the luciferase
expression in A549/OPN-luc after treatment for 48 h with increasing
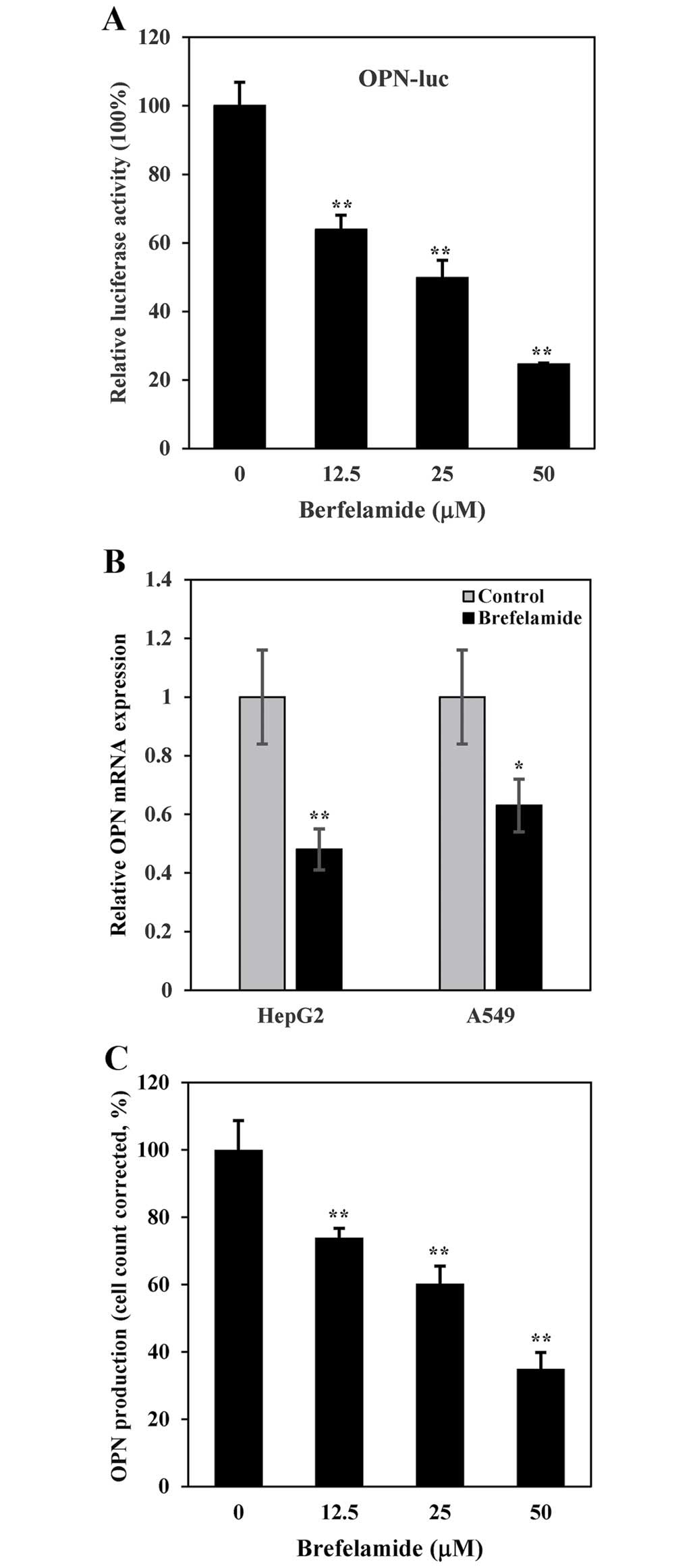
concentration of brefelamide. As shown in Fig. 1A, luciferase expression in
A549/OPN-luc cells was dose-dependently suppressed by the
brefelamide treatment. Similar results were obtained in real-time
RT-PCR, which showed a reduction of OPN mRNA in brefelamide-treated
HepG2 and A549 cells (Fig. 1B),
confirming a transcriptional repression of OPN by brefelamide. When
OPN production in the culture supernatant was measured by ELISA, we
observed a concentration-dependent reduction in OPN production in
A549 cells exposed to brefelamide (183±7, 115±6 and 57±2 ng/ml at a
concentration of 12.5, 25 and 50 µM, respectively), when
compared to that from DMSO (mock)-treated A549 cells (283±7 ng/ml)
(Fig. 1C).
Effect of brefelamide on cell
invasion
OPN is an extracellular matrix protein involved in
cell motility. We next performed Matrigel invasion assay to
investigate the functional impact of OPN suppression by brefelamide
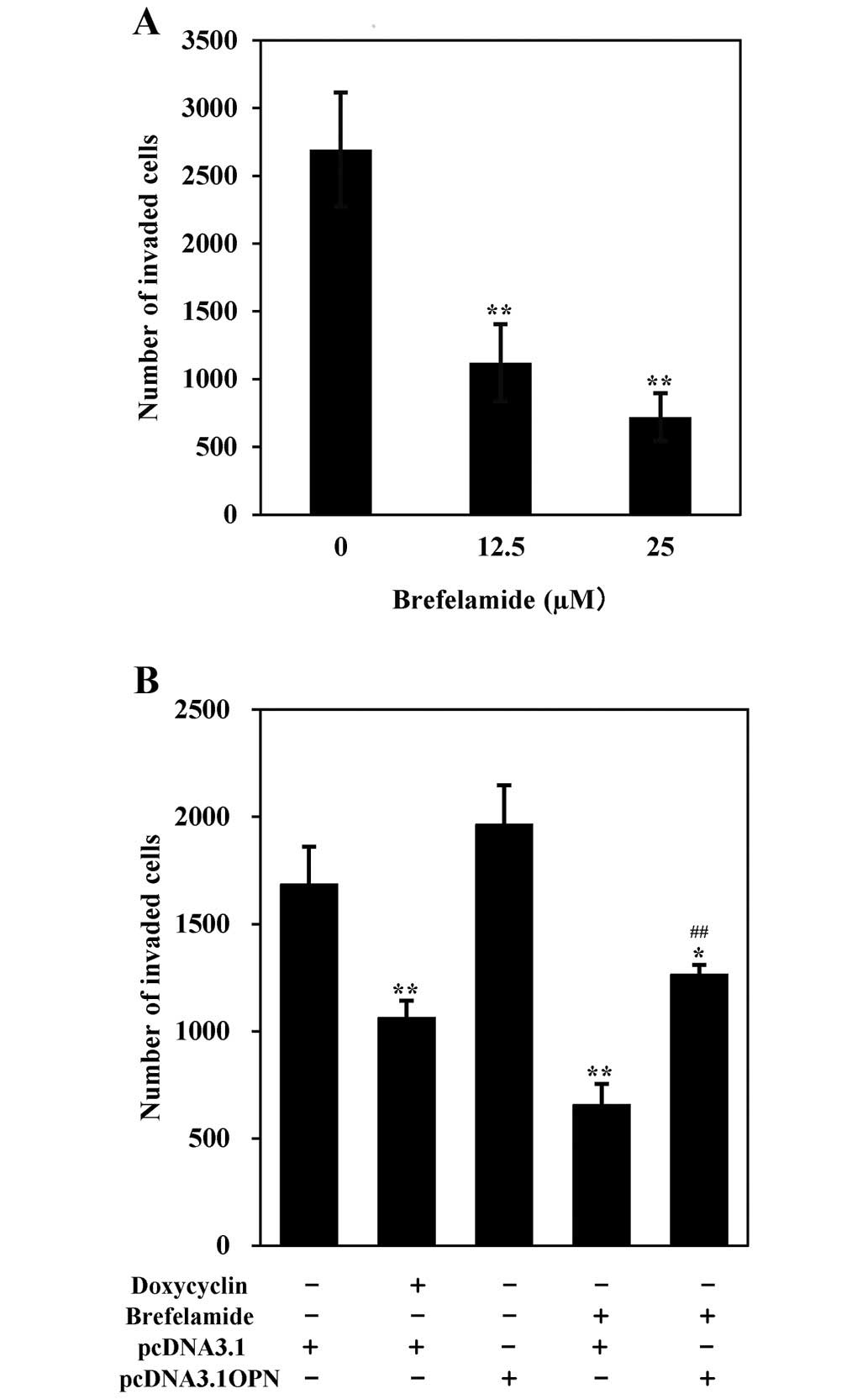
on A549 cells. As shown in Fig. 2A,
the cell numbers invaded through the Matrigel after an exposure of
brefelamide at the concentration of 12.5 and 25 µM were
significantly lower than that of untreated control (1120±285 and
719±176 vs. 2694±421, p<0.01), indicating that brefelamide
decreases the invasion activity of cells. Further, anti-invasive
activity of brefelamide was weakened in cells incubated with the
conditioned medium from pcDNA3.1-OPN transfectant, which contained
327 ng/ml of OPN as detected by ELISA (Fig. 2B). This finding indicates that
brefelamide inhibits cell invasion at least partially by its
suppression of OPN production. An inhibitory effect of brefelamide
on cell migration was also observed in wound healing assay (data
not shown). These data suggest that suppression of OPN expression
by brefelamide can inhibit the migratory and invasive activity of
the cancer cells, implying the potential of brefelamide as an
antimetastatic therapeutic agent.
Sensitization of cancer cells to
conventional anticancer drugs
Brefelamide has been reported to suppress the
proliferation of astrocytoma cells by inhibiting ERK
phosphorylation (21). We next
performed growth inhibition assay to investigate whether
brefelamide can enhance the chemosensitivity of cancer cells to
conventional anticancer agents such as cisplatin, etoposide and
sorafenib. The IC50 of cisplatin alone was 107.0 and
183.9 µM in A549 and HepG2 cells, respectively, which was
decreased to 48.8 and 53.0 µM when co-treated with
brefelamide (Table I). Reduction in
the IC50 value by combination with brefelamide was also
observed in the case of etoposide (IC50 from 55.1 to
25.4 µM in A549 cells) and sorafenib (IC50 from
2.3 to 0.5 µM in HepG2 cells). This indicates that
brefelamide treatment sensitized cancer cells to these anticancer
drugs, suggesting that brefelamide may provide a potential strategy
to overcome chemoresistance.
 | Table IReduction in the IC50
value (µM) of anticancer drugs used in combination with
brefelamide. |
Table I
Reduction in the IC50
value (µM) of anticancer drugs used in combination with
brefelamide.
| Cisplatin
| Etoposide
|
|---|
| Cells | Isolation | Combinationa | Isolation | Combinationa |
|---|
| A549 | >100 | 48.8 | 55.1 | 25.4 |
| Cisplatin
| Sorafenib
|
| Cells | Isolation | Combinationb | Isolation | Combinationb |
|
| HepG2 | >100 | 53 | 2.3 | 0.5 |
Induction of Smad4 by brefelamide
To gain some insights into the molecular mechanism
underlying the brefelamide-mediated OPN inhibition, we conducted
microarray analysis to clarify the different gene expression
profiles between brefelamide- and mock-treated A549 cells. We found
1,382 genes with expression altered ≥1.5-fold after exposure to
brefelamide, which were distributed into distinct functional groups
including those involved in proliferation and cell motility. One of
these genes, Smad4, whose expression was induced after brefelamide
treatment, was chosen for further study. We focused on Smad4 in
view of the previous publication demonstrating deletion of Smad4 in
metastatic-prone condition in the prostate cancer model was
associated with aberrant expression of OPN (25), and further, our recent study
demonstrating that OPN was a downstream target negatively regulated
by TGF-β/Smad signaling (23).
These observations prompted us to hypothesize that brefelamide
suppresses OPN expression via induction of Smad4. To investigate
this possibility, expression of Smad4 mRNA was determined by
real-time RT-PCR in cells cultured in the absence or presence of
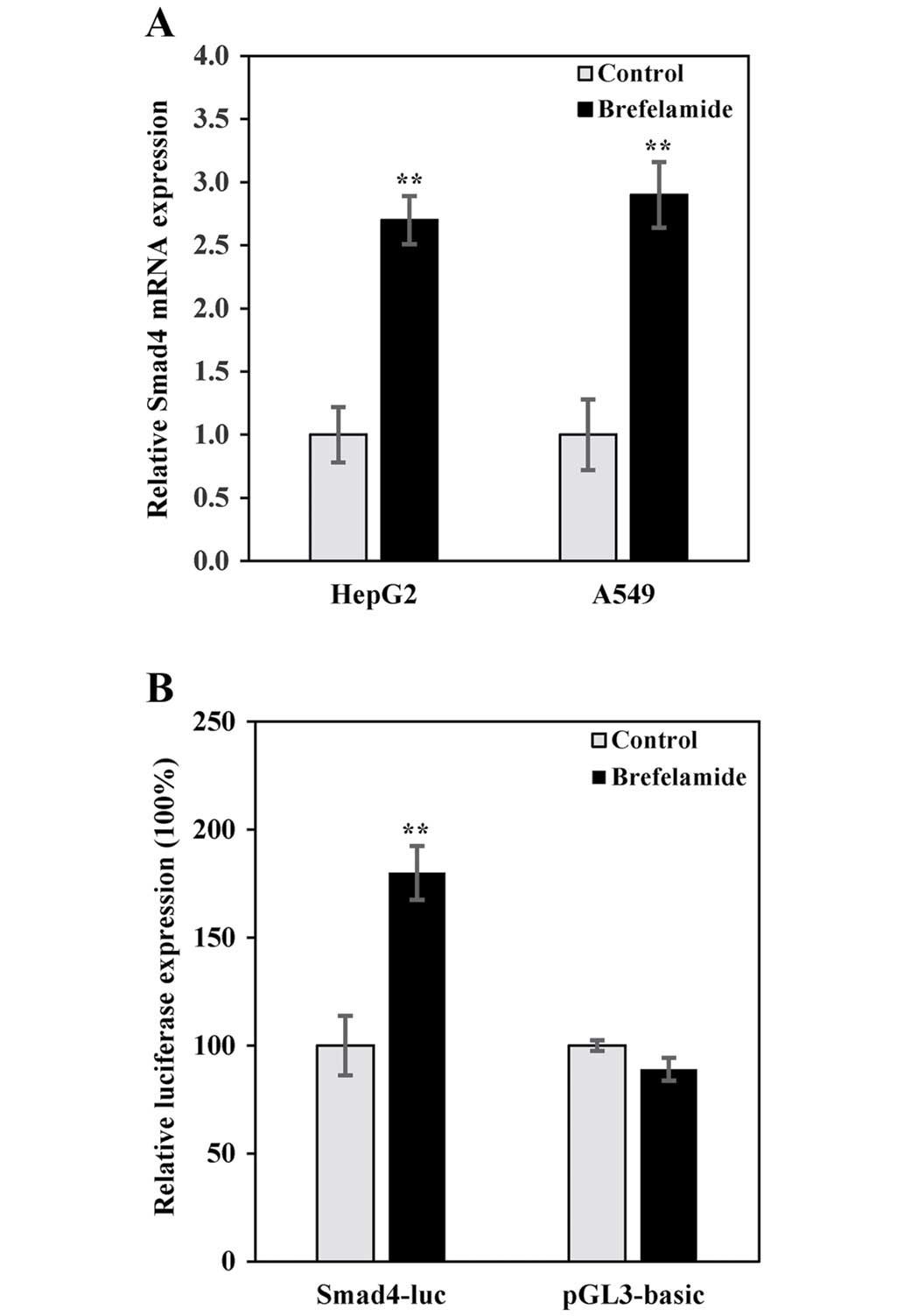
brefelamide. Consistent with that obtained from the microarray
analysis, endogenous Smad4 mRNA in HepG2 or A549 cells was enhanced
by brefelamide treatment (Fig. 3A).
Similar results were also obtained in the reporter assay with
pSmad4-luc, in which luciferase gene is directed under the control
of human Smad4 promoter. As shown in Fig. 3B, Smad4-directed luciferase
expression was increased in A549 cells treated with brefelamide,
while no effect was observed on empty vector (pGL3-basic),
confirming a transcriptional induction of Smad4 by brefelamide.
Involvement of TGF-β/Smad4 signaling in
brefelamide-mediated OPN suppression
To confirm a mechanistic role for Smad4 in
brefelamide-mediated OPN suppression, we next took a different
approach using shRNA-mediated silencing of the expression of Smad4.
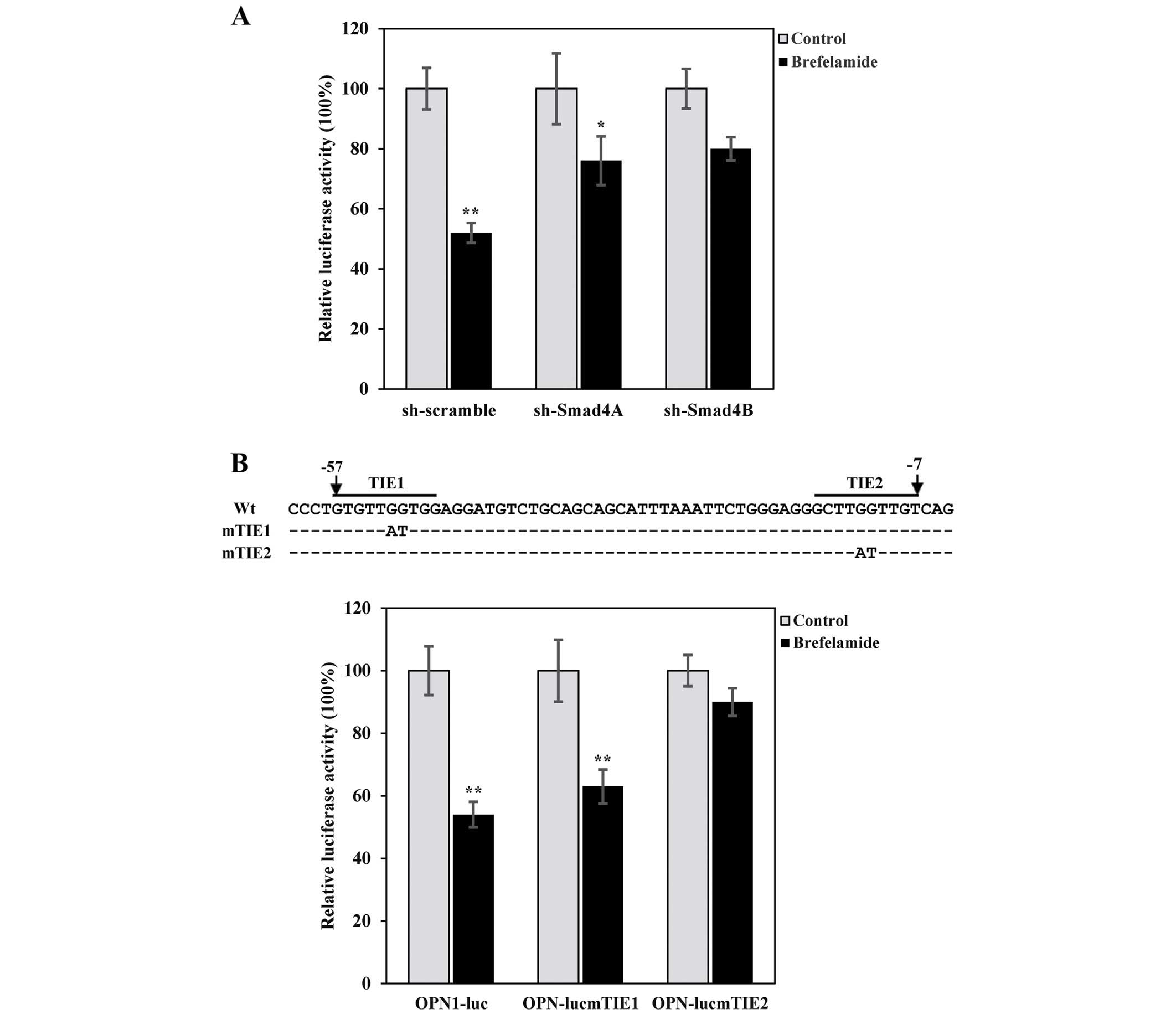
pOPN1-luc was transfected with construct expressing
control-(sh-scramble) or two independent sh-Smad4 (sh-Smad4A and
B), which resulted in a moderate knockdown of Smad4 as confirmed by
quantitative real-time PCR (23).
Consistent with the aforementioned results, OPN promoter activity
in cells transfected with sh-scramble was decreased in cells
treated with brefelamide, and notably, the extent of
brefelamide-mediated OPN suppression was partially attenuated in
cells transfected with either sh-Smad4A or B (Fig. 4A), suggesting that Smad4 is a
mediator involved in the inhibition of OPN by brefelamide.
Furthermore, brefelamide-induced OPN repression was largely
abrogated in reporter assay with pOPN-lucmTIE2 in which the
putative TGF-β inhibitory element was disrupted (23), whereas disruption of TIE1 did not
significantly alter OPN inhibition by brefelamide (Fig. 4B). These data provide an additional
line of evidence supporting that brefelamide inhibits OPN through
inducing Smad4 and subsequently restoring the negative regulation
of OPN by TGF-β/Smad signaling.
Discussion
Implication of OPN in the development and
progression of multiple tumor types suggests that targeted
inhibition of OPN may represent a promising therapeutic modality
against malignant diseases. A variety of strategies have been
endeavored to ablate OPN function, examples of such approaches
include silencing of OPN using RNAi technology, blocking OPN
activity using specific antibodies, RNA aptamer, and small-molecule
inhibitors (26–29). Transcriptional regulation of OPN is
complex and involves various transcription factors, including AP-1,
Ets, Myc, and v-Src (30,31). More recently, we demonstrated that
OPN is a downstream target negatively regulated by TGF-β signaling,
and deregulation of OPN by TGF-β is linked with loss of TGF-β
cytostatic responsiveness (23).
Here, we identify brefelamide as a novel OPN
inhibitor, and the molecular basis for OPN inhibition by
brefelamide appears to be associated with induction of Smad4 and
consequent restoration of OPN repression by TGF-β/Smad signaling.
Our conclusion was inferred based on the following observations: i)
brefelamide suppresses OPN expression, with subsequent inhibition
of cell invasion; ii) brefelamide induces Smad4 expression; iii)
elimination of a TIE-like element largely abolishes OPN suppression
by brefelamide; and iv) shRNA-mediated knockdown of Smad4 partially
abrogates OPN inhibition by brefelamide. Together, these findings
suggest that brefelamide inhibits OPN expression and OPN-mediated
invasion through restoration of Smad4-mediated TGF-β/OPN regulatory
axis.
A couple of reports linked aberrant OPN expression
to the activated ERK pathway, demonstrating that OPN is a
downstream target of ERK/AP-1 signaling pathway (32). Further, brefelamide was reported to
inhibit EGF-dependent activation of the ERK pathway (20). Thus, the OPN inhibition observed
here may occur as a consequence of brefelamide-mediated suppression
of the EGFR-dependent ERK activation. The microarray-based
comparison analysis, however, did not show significant change in
the expression level of ERK signaling-associated genes in
brefelamide-treated A549 cells (data not shown), and further,
brefelamide-mediated OPN inhibition was largely diminished in
pOPN-lucmTIE2, in which the identified AP-1-binding motifs were
intact. Thus, it was largely excluded, if not completely, that the
OPN suppression by brefelamide was attributable to its inhibitory
effect on ERK signaling pathway. Given the importance of cellular
context in conferring the sensitivity to ERK inhibition (33), it may not be surprising that
EGFR-ERK pathway was inhibited in brefelamide-treated astrocytoma
1321N1 cells, but not in lung adenocarcinoma A549 cells.
Consistent with the results from precedent
microarray analysis, real-time quantitative PCR and reporter assay
showed increased expression of Smad4 after treatment with
brefelamide. Previous studies have shown that EGF triggers GSK3
phosphorylation of Smad4 through activation of the ERK pathway,
subsequently resulting in the polyubiquitination and proteasomal
degradation of Smad4 (34).
Considering the property of brefelamide in inhibiting ERK signaling
pathway, it is suggested that, in addition to increasing Smad4
synthesis, brefelamide may enhance the stability of Smad4 protein
by inhibiting EGF/ERK-mediated Smad4 ubiquitination. The ability of
brefelamide to induce Smad4 activity is of potential clinical
relevance. Smad4 is a major tumor suppressor that is frequently
deleted or inactive in cancers of the pancreas, gastrointestine,
and skin (35–37). Decreased Smad4 expression has been
reported in various human cancers and the Smad4 expression level is
inversely correlated with tumor grade and TNM stage. Actually, it
was reported that re-establishment of Smad4 suppressed
Wnt/β-catenin signaling activity and increased the expression of
E-cadherin in human colon carcinoma cells, both of which were
suggested to be involved in the migration-suppressive function of
Smad4 (38). Also,
adenoviral-mediated restoration of Smad4 inhibited tumor growth,
invasion and angiogenesis in pancreatic adenocarcinoma cells.
Negative regulation of downstream targets including VEGF, MMP-2,
MMP-9, and especially ETS-1, by ectopically expressed Smad4 was
demonstrated as the mechanism underlying the observed antitumor
effects (39). It is thus probable,
although remains to be proven, that besides OPN, these downstream
targets of Smad4 may also be suppressed by brefelamide-mediated
Smad4 induction. Studies are underway to investigate this
hypothesis. If confirmed, breferlamide may be a promising
therapeutic candidate that targets the malignancies associated with
loss of Smad4 expression.
When used in combination with the conventional
anticancer drugs, brefelamide re-sensitized A549 and HepG2 cells to
cisplatin, and decreased the IC50 value of etoposide and
sorafenib in A549 and HepG2 cells, respectively. In addition to a
crucial role in cancer progression, invasion and metastasis,
overexpressed OPN has been reported to be involved in the
development of resistance to chemotherapy by inhibiting apoptosis
(40). Thus, brefelamide-mediated
sensitizing effect may occur as a consequence of OPN inhibition.
Indeed, silencing of OPN by siRNA significantly enhanced
chemotherapy sensitivity of breast cancer cells (41). Additionally, microarray analysis
revealed that p53 tumor suppressor and its downstream target genes
such as p21 were induced in brefelamide-treated A549 cells, and
further, our preliminary data indicated that p53-driven luciferase
reporter was activated by treatment with brefelamide. Both of which
suggested that brefelamide may activate p53-regulated pro-apoptotic
signaling pathways, consequently potentiating drug-mediated
apoptosis.
Different from gene silencing strategies, small
molecules are usually not specific for a single molecular target.
Simultaneously affecting a set of functionally linked genes
sometimes leads to enhanced efficacy. The conventional agents such
as cisplatin and doxorubicin, for example, have been identified to
affect the expression of multiple genes involved in cell
proliferation and apoptosis. It may also be the case that
brefelamide may affect other molecules besides OPN. Consistent with
this hypothesis, it was found that incubation with the
OPN-containing conditioned medium did not fully abolish the
anti-invasive activity of brefelamide (Fig. 2B), the partially retained inhibition
may be attributable to the brefelamide's action on other genes
regulating cell motility and invasion. Actually, microarray
analysis of brefelamide-treated A549 cells revealed altered
expression of genes associated with cell migration and invasion.
Nonetheless, the data presented here suggest that brefelamide
inhibits invasion of A549 cells at least partially through
downregulation of OPN. On the contrary, interaction of drugs with
molecules other than the intended target may cause an undesired
side-effect. Unintended side-effect is also an issue of concern in
the case of brefelamide. As judged by morphologic observation and
viability measurement, however, no discernible adverse effects were
observed in brefelamide-treated A549 and HepG2 cells. Further
independent experiments are required to delineate off-target-based
toxicological effects of brefelamide.
In conclusion, the results presented here indicated
that brefelamide suppressed OPN expression and OPN-mediated cell
invasion. This is likely mediated through inducing Smad4 expression
and consequently restoring Smad4-mediated TGF-β/OPN regulatory
axis. These data suggest the potential utility of brefelamide as
antimetastatic agent in patients with OPN-overexpressed
malignancies.
References
|
1
|
Weigelt B, Peterse JL and van't Veer LJ:
Breast cancer metastasis: Markers and models. Nat Rev Cancer.
5:591–602. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nguyen DX, Bos PD and Massagué J:
Metastasis: From dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Joyce JA and Pollard JW:
Microenvironmental regulation of metastasis. Nat Rev Cancer.
9:239–252. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Otranto M, Sarrazy V, Bonté F, Hinz B,
Gabbiani G and Desmoulière A: The role of the myofibroblast in
tumor stroma remodeling. Cell Adhes Migr. 6:203–219. 2012.
View Article : Google Scholar
|
|
6
|
Mao Y, Keller ET, Garfield DH, Shen K and
Wang J: Stromal cells in tumor microenvironment and breast cancer.
Cancer Metastasis Rev. 32:303–315. 2013. View Article : Google Scholar
|
|
7
|
Geiger B, Bershadsky A, Pankov R and
Yamada KM: Transmembrane crosstalk between the extracellular matrix
- cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2:793–805. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Discher DE, Janmey P and Wang YL: Tissue
cells feel and respond to the stiffness of their substrate.
Science. 310:1139–1143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Engler AJ, Sen S, Sweeney HL and Discher
DE: Matrix elasticity directs stem cell lineage specification.
Cell. 126:677–689. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roberts DD: Emerging functions of
matricellular proteins. Cell Mol Life Sci. 68:3133–3136. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chiodoni C, Colombo MP and Sangaletti S:
Matricellular proteins: From homeostasis to inflammation, cancer,
and metastasis. Cancer Metastasis Rev. 29:295–307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brown LF, Berse B, Van de Water L,
Papadopoulos-Sergiou A, Perruzzi CA, Manseau EJ, Dvorak HF and
Senger DR: Expression and distribution of osteopontin in human
tissues: Widespread association with luminal epithelial surfaces.
Mol Biol Cell. 3:1169–1180. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rangaswami H, Bulbule A and Kundu GC:
Osteopontin: Role in cell signaling and cancer progression. Trends
Cell Biol. 16:79–87. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anborgh PH, Mutrie JC, Tuck AB and
Chambers AF: Role of the metastasis-promoting protein osteopontin
in the tumour microenvironment. J Cell Mol Med. 14:2037–2044. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tuck AB, Arsenault DM, O'Malley FP, Hota
C, Ling MC, Wilson SM and Chambers AF: Osteopontin induces
increased invasiveness and plasminogen activator expression of
human mammary epithelial cells. Oncogene. 18:4237–4246. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin YH and Yang-Yen HF: The
osteopontin-CD44 survival signal involves activation of the
phosphatidylinositol 3-kinase/Akt signaling pathway. J Biol Chem.
276:46024–46030. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Philip S, Bulbule A and Kundu GC:
Osteopontin stimulates tumor growth and activation of promatrix
metalloproteinase-2 through nuclear factor-kappa B-mediated
induction of membrane type 1 matrix metalloproteinase in murine
melanoma cells. J Biol Chem. 276:44926–44935. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leali D, Dell'Era P, Stabile H, Sennino B,
Chambers AF, Naldini A, Sozzani S, Nico B, Ribatti D and Presta M:
Osteopontin (Eta-1) and fibroblast growth factor-2 cross-talk in
angiogenesis. J Immunol. 171:1085–1093. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Furger KA, Menon RK, Tuck AB, Bramwell VH
and Chambers AF: The functional and clinical roles of osteopontin
in cancer and metastasis. Curr Mol Med. 1:621–632. 2001. View Article : Google Scholar
|
|
20
|
Kikuchi H, Saito Y, Sekiya J, Okano Y,
Saito M, Nakahata N, Kubohara Y and Oshima Y: Isolation and
synthesis of a new aromatic compound, brefelamide, from
dictyostelium cellular slime molds and its inhibitory effect on the
proliferation of astrocytoma cells. J Org Chem. 70:8854–8858. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Honma S, Saito M, Kikuchi H, Saito Y,
Oshima Y, Nakahata N and Yoshida M: A reduction of epidermal growth
factor receptor is involved in brefelamide-induced inhibition of
phosphorylation of ERK in human astrocytoma cells. Eur J Pharmacol.
616:38–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Yamada O, Matsushita Y,
Chagan-Yasutan H and Hattori T: Transactivation of human
osteopontin promoter by human T-cell leukemia virus type 1-encoded
Tax protein. Leuk Res. 34:763–768. 2010. View Article : Google Scholar
|
|
23
|
Zhang J, Yamada O, Kida S, Matsushita Y
and Hattori T: Down-regulation of osteopontin mediates a novel
mechanism underlying the cytostatic activity of TGF-β. Cell Oncol
(Dordr). 39:119–128. 2016. View Article : Google Scholar
|
|
24
|
Zhang J, Yamada O, Sakamoto T, Yoshida H,
Iwai T, Matsushita Y, Shimamura H, Araki H and Shimotohno K:
Down-regulation of viral replication by adenoviral-mediated
expression of siRNA against cellular cofactors for hepatitis C
virus. Virology. 320:135–143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang
J, Perry SR, Labrot ES, Wu X, Lis R, et al: SMAD4-dependent barrier
constrains prostate cancer growth and metastatic progression.
Nature. 470:269–273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boissy P, Andersen TL, Abdallah BM, Kassem
M, Plesner T and Delaissé JM: Resveratrol inhibits myeloma cell
growth, prevents osteoclast formation, and promotes osteoblast
differentiation. Cancer Res. 65:9943–9952. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chakraborty G, Jain S, Patil TV and Kundu
GC: Down-regulation of osteopontin attenuates breast tumour
progression in vivo. J Cell Mol Med. 12(6A): 2305–2318. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao J, Dong L, Lu B, Wu G, Xu D, Chen J,
Li K, Tong X, Dai J, Yao S, et al: Down-regulation of osteopontin
suppresses growth and metastasis of hepatocellular carcinoma via
induction of apoptosis. Gastroenterology. 135:956–968. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mi Z, Guo H, Russell MB, Liu Y, Sullenger
BA and Kuo PC: RNA aptamer blockade of osteopontin inhibits growth
and metastasis of MDA-MB231 breast cancer cells. Mol Ther.
17:153–161. 2009. View Article : Google Scholar
|
|
30
|
Hijiya N, Setoguchi M, Matsuura K, Higuchi
Y, Akizuki S and Yamamoto S: Cloning and characterization of the
human osteopontin gene and its promoter. Biochem J. 303:255–262.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang D, Yamamoto S, Hijiya N, Benveniste
EN and Gladson CL: Transcriptional regulation of the human
osteopontin promoter: Functional analysis and DNA-protein
interactions. Oncogene. 19:5801–5809. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim HJ, Lee MH, Park HS, Park MH, Lee SW,
Kim SY, Choi JY, Shin HI, Kim HJ and Ryoo HM: Erk pathway and
activator protein 1 play crucial roles in FGF2-stimulated premature
cranial suture closure. Dev Dyn. 227:335–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Joseph EW, Pratilas CA, Poulikakos PI,
Tadi M, Wang W, Taylor BS, Halilovic E, Persaud Y, Xing F, Viale A,
et al: The RAF inhibitor PLX4032 inhibits ERK signaling and tumor
cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad
Sci USA. 107:14903–14908. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Demagny H, Araki T and De Robertis EM: The
tumor suppressor Smad4/DPC4 is regulated by phosphorylations that
integrate FGF, Wnt, and TGF-β signaling. Cell Reports. 9:688–700.
2014. View Article : Google Scholar
|
|
35
|
Alazzouzi H, Alhopuro P, Salovaara R,
Sammalkorpi H, Järvinen H, Mecklin JP, Hemminki A, Schwartz S Jr,
Aaltonen LA and Arango D: SMAD4 as a prognostic marker in
colorectal cancer. Clin Cancer Res. 11:2606–2611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peng B, Fleming JB, Breslin T, Grau AM,
Fojioka S, Abbruzzese JL, Evans DB, Ayers D, Wathen K, Wu T, et al:
Suppression of tumorigenesis and induction of p15(ink4b) by
Smad4/DPC4 in human pancreatic cancer cells. Clin Cancer Res.
8:3628–3638. 2002.PubMed/NCBI
|
|
37
|
Zhang B, Halder SK, Kashikar ND, Cho YJ,
Datta A, Gorden DL and Datta PK: Antimetastatic role of Smad4
signaling in colorectal cancer. Gastroenterology. 138:969–980.
2010. View Article : Google Scholar
|
|
38
|
Tian X, Du H, Fu X, Li K, Li A and Zhang
Y: Smad4 restoration leads to a suppression of Wnt/beta-catenin
signaling activity and migration capacity in human colon carcinoma
cells. Biochem Biophys Res Commun. 380:478–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Duda DG, Sunamura M, Lefter LP, Furukawa
T, Yokoyama T, Yatsuoka T, Abe T, Inoue H, Motoi F, Egawa S, et al:
Restoration of SMAD4 by gene therapy reverses the invasive
phenotype in pancreatic adenocarcinoma cells. Oncogene.
22:6857–6864. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tajima K, Ohashi R, Sekido Y, Hida T, Nara
T, Hashimoto M, Iwakami S, Minakata K, Yae T, Takahashi F, et al:
Osteopontin-mediated enhanced hyaluronan binding induces multidrug
resistance in mesothelioma cells. Oncogene. 29:1941–1951. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pang H, Cai L, Yang Y, Chen X, Sui G and
Zhao C: Knockdown of osteopontin chemosensitizes MDA-MB-231 cells
to cyclo-phosphamide by enhancing apoptosis through activating p38
MAPK pathway. Cancer Biother Radiopharm. 26:165–173. 2011.
View Article : Google Scholar : PubMed/NCBI
|