Introduction
Cancer stem cells (CSCs) are a type of tumor cell
that can self-renew and are pluripotent. They have been closely
linked to tumor occurrence, development, metastasis, recurrence and
drug resistance. Over the past several decades, the use of specific
stem cell surface markers to identify stem cells has been widely
used in CSC research, and several molecular markers have been
thought to be relatively precise stem cell markers in colorectal
cancer, including CD44, CD133, CD24, Lgr5 and EpCAM (1–6).
However, the use of a single stem cell marker to purify stem cells
has been controversial; thus, using two markers in combination,
such as CD44 and CD133, is considered a more accurate approach to
purify and define stem cells (7,8).
CD200 is a type I membrane glycoprotein with two
extracellular domains, a single transmembrane region, and a
cytoplasmic tail with no known signaling motifs. It is a member of
the highly conserved immunoglobulin family, and it is expressed in
myeloid cells, such as macrophages, dendritic and mast cells, and
eosinophils. Interactions between CD200 and its receptor CD200R act
as an immune tolerance signal, which reduces myeloid cell activity
and change their migration ability (9–12). In
recent years, high CD200 expression was found in colon cancer,
myeloma, breast and brain cancer, melanoma and normal mesenchymal
stem cells. Moreaux et al (13) previously observed significant CD200
overexpression in colon, head and neck and renal carcinoma,
malignant mesothelioma and testicular cancer, MGUS myeloma and
chronic lymphocytic leukemia compared to normal cell or tissue
counterparts. Moreover, CD200 expression plays important roles in
tumor progression of various types of cancers (14,15).
Based on these findings, CD200 has been proposed as a new CSC
marker in colon cancer. However, further experiments have not been
performed to confirm its accuracy as a colorectal CSC marker. In
our previous study, xenograft transplantation assays confirmed that
CD200+ cells were more tumorigenic than
CD200− cells. Continuous monitoring of xenograft tumor
growth revealed that at the same cellular dosage, CD200+
cells initiated tumor growth much faster than CD200−
tumors, resulting in a larger tumor mass. These data provided
preliminarily evidence that CD200 is a reliable colon cancer stem
cell (CCSC) marker. In the present study, we performed in
vitro experiments to further confirm CD200 as a stem cell
surface marker.
Since CSCs are thought to initiate tumor formation,
development, metastasis and recurrence, they must be able to escape
tumor immune killers. It was previously hypothesized that during
the process from immune clearance to immune balance or during
immune escape, CSCs express specific surface molecules to secrete
immune inhibitory factors, protect against tumor-specific immune
responses and increase immune inhibition responses to escape from
immune killers. Since CSCs and tumor immune protection are causes
of tumor recurrence and metastasis, we hypothesized that CSCs
reduce tumor immune responses by expression of specific markers.
There are clear links between CSC marker expression and immune
tolerance. Numerous previous studies have shown that tumor cells
overexpressing CD200 can better escape from immune damage and evade
the immune system (14,16). Therefore, CD200 is thought to be a
crucial regulator of CSC immune escape, and it likely links
colorectal CSCs (CCSCs) to immune evasion. Therefore, an in-depth
understanding of CD200 and CD200-related genes is important to
explore tumor immunosuppression mechanisms and tumor targeting
therapy.
To address these issues, we compared the FACS-sorted
CD200+ population with the CD200− population
for their self-renewal abilities in vitro, which is a key
feature of CSCs. Moreover, long-term cultured CD200+
cells were assayed for invasion ability, another defining
characteristic of CCSCs. We also investigated the expression
pattern of genes commonly modulated by CD200 in human COLO 205
colorectal cancer cells. After clustering and molecular function
analysis, biological process and related pathway analysis, we chose
several important functional genes and pathways with specific
connections to CD200 as targets for further research.
Materials and methods
Cell culture
The human colon cancer cell lines, COLO 205, LoVo,
SW620, SW480, SW1116 and HT29 were obtained from the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China).
The cells were cultured in RPMI-1640 medium (HyClone, Logan, UT,
USA) culture medium supplemented with 10% fetal bovine serum (FBS),
1% penicillin and streptomycin in a humidified incubator under 5%
CO2 and 95% air at 37°C. Cells were grown to logarithmic
phase in serum medium, the upper medium containing cells in
suspension were removed, and the adherent cells were digested using
0.25% trypsin + 0.02% EDTA into a single cell suspension. Cells
were replated and cultured in serum-free medium/F12 (DMEM/F12;
HyClone) supplemented with epidermal growth factor (EGF; 20 ng/ml),
basic fibroblast growth factor (bFGF; 20 ng/ml), leukemia
inhibitory factor (LIF; 10 ng/ml) (all from PeproTech, Rocky Hill,
NJ, USA), and insulin (4 U/l; Sigma, St. Louis, MO, USA) onto
ultralow attachment T25 slide dishes (Corning, Tewksbury, MA, USA).
At least 1,000 cells/ml were used for colonosphere formation. Cells
were maintained at 37°C in a humidified incubator under 5%
CO2 and 95% air.
Flow cytometry and FACS
CD200, CD133 and CD44 expression was analyzed by
flow cytometry. The cells were diluted into a single cell
suspension, washed twice and suspended in 100 µl assay
buffer [phosphate-buffered saline (PBS) 0.5% BSA, 2 mM EDTA, pH
7.2] with 10 µl APC-conjugated anti-CD200 or 10 µl
PE-conjugated anti-CD133 antibodies (Miltenyi Biotec, Bergisch
Gladbach, Germany) or 20 µl FITC-conjugated anti-CD44
antibody (BD Biosciences, San Jose, CA, USA). Mouse PE-IgG2b,
APC-IgG2b (both from Miltenyi Biotec) and FITC-IgG2b antibodies
(BD) were used as isotype controls. Cells were incubated in the
dark at 4°C for 20 min, washed twice with 1 ml assay buffer, and
centrifuged at 300 g for 10 min. The cells were resuspended in
assay buffer for analysis and sorting on a FACSCalibur flow
cytometer or subjected to fluorescence-activated cell sorting
(FACS) using a BD FACsAria II sorter (both from BD Biosciences).
Side scatter and forward scatter profiles were used to eliminate
cell doublets. Cell purity was analyzed after sorting and was
typically >95%.
Colonosphere formation assay
To evaluate the self-renewal potential of
CD200+ and CD200− subpopulations in COLO 205
cells in vitro, FACS sorted cells were collected and
enzymatically digested with Accutase (Millipore, Billerica, MA,
USA) to obtain a single cell suspension (100 cells/ml). A total of
100 µl was added to each well of a 96-well plate (Corning).
The cells were cultured in serum-free medium/F12 (DMEM/F12)
supplemented with 20 ng/ml EGF, 20 ng/ml bFGF, 10 ng/ml LIF (all
from PeproTech) and 4 U/l insulin (Sigma). All growth factors were
added to the culture medium every 3 days. Cells were cultured for
14 days, and the number of floating colonospheres containing >50
cells was counted in each well. Colonosphere forming efficiency
(CFE) was calculated in each well as the number of
colonospheres/10. In addition, each generation of colonospheres was
diluted into a single-cell suspension and re-seeded to test CFE for
each indicated passage. The colonosphere formation assay was
confirmed with at least 3 replicates for each cell population.
Matrigel-coated Transwell assay
A Transwell assay was conducted to evaluate the
invasiveness of CD200+ and CD200− cells. Each
cell population was added to an 8.0-µm pore Matrigel-coated
Transwell (BD) insert at a cell density of 5×104/insert
in DMEM/F12 basic medium. The lower chambers were filled with
DMEM/F12 medium supplemented with 10% FBS as a chemoattractant, and
the cells were cultured for 24 h. Cells that passed through the
membrane to the lower chamber were stained with crystal violet. A
light microscope (Olympus IX71) was used to calculate the number of
invaded cells per high power field. At least 10 microscopic fields
were randomly selected for each experiment.
Microarray analysis
Gene expression profiling of CD200+ and
CD200− COLO 205 cells was performed using the Affymetrix
Human U133 Plus2.0 GeneChip according to the manufacturer's
protocol (Affymetrix, Santa Clara, CA, USA). Chips were scanned
with a GeneChip® Scanner 3000 (Cat#00-00212;
Affymetrix). The raw data were read with the Command Console
Software 3.1 (Affymetrix). Quality qualified data were normalized
with the Gene Spring Software 11.0 (Agilent Technologies, Santa
Clara, CA, USA). The MAS5.0 algorithm was used. NetAffx (http://www.affymetrix.com) was used to obtain
information of each probe on the Human U133 Plus2.0 chip.
Differentially expressed genes were identified through data
analysis of the SAS system. Fold-change was used to calculate the
expression differences of the signal value between the two sets
after homogenization (fold-change = 2mean2-mean1) A gene
expression value of 2 was set as the ratio cut-off of 2, as it is
commonly adopted for microarray data analysis. The Database for
Annotation, Visualization and Integrated Discovery (DAVID;
http://niaid.abcc.ncifcrf.gov/) and the
SAS Analysis Systemb(http://sas.ebioservice.com/) were used for further
analysis of the selected differentially expressed genes.
RT-PCR
Total cellular mRNA was extracted from FACs-sorted
cells with TRIzol reagent (Invitrogen, Grand Island, NY, USA)
according to the manufacturer's protocol. mRNA expression was
determined by RT-PCR using SYBR® Premix Ex Taq™
(Takara, Otsu, Shiga, Japan) on a LightCycler 480 real-time genetic
analyzer (Roche), with the indicated amplicon length. The PCR
conditions were as follows: initial denaturation at 95°C for 30
sec, followed by 40 cycles of denaturation at 95°C for 5 sec,
annealing at 60°C for 20 sec and extension at 65°C for 15 sec, and
finally followed by melting curve analysis. GAPDH expression served
as an internal control.
Sodium butyrate treatment
COLO 205 cells were diluted into a single-cell
suspension, and then, 1×106 cells were seeded in 10-cm
plastic dishes at day 0. Sodium butyrate (NaBT; Wako, Osaka, Japan)
was added on day 1 at concentrations of 0, 3, 5, 8 or 12 mM, and
incubated for 24 h. Enzyme-linked immunosorbent assay (ELISA) with
SensoLyte™ pNPP alkaline phosphatase assay kit (AnaSpec, Inc.,
Fremont, CA, USA) was used to detect changes in alkaline
phosphatase levels according to the manufacturer' protocol.
Western blotting
Whole cell lysates were prepared as previously
described (17). Samples were
subjected to sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) using a 10% resolving polyacrylamide gel
and transferred onto a Hybond-P polyvinylidene fluoride (PVDF)
membrane (Amersham Biosciences, Piscataway, NJ, USA). Membranes
were blocked in 5% bovine serum albumin (BSA)/0.1% Tween-20 in PBS
solution (TPBS) for 60 min and then incubated overnight at room
temperature with β-catenin, Wnt3a, pLRP6, DVL2, Wnt5a, LRP6 or
Naked1 rabbit mAbs (Cell Signaling Technology, Danvers, MA, USA) at
a concentration of 1:1,000. The membrane was washed 3 times in 0.1%
TPBS for 5 min each, then, incubated for 1 h at room temperature
with a horseradish peroxidase-linked secondary antibody (Sigma)
(1:10,000; anti-rabbit IgG) diluted in 5% BSA/PBS containing 0.1%
Tween-20. After 3 washes, the membrane was visualized using
enhanced chemiluminescence reagents (Thermo Scientific, Waltham,
MA, USA). The monoclonal anti-β-actin antibody (Sigma) was used at
1:2,000 as a loading control.
Statistical analysis
Data are reported as means ± SEM of triplicate
experiments. Statistical analysis (analysis of variance) was
performed using SPSS 16.0 (SPSS, Inc., Chicago, IL, USA) and
P<0.05 was considered to indicate a statistically significant
result.
Results
Isolation of CD200+ colorectal
cells from human colon cancer cell lines
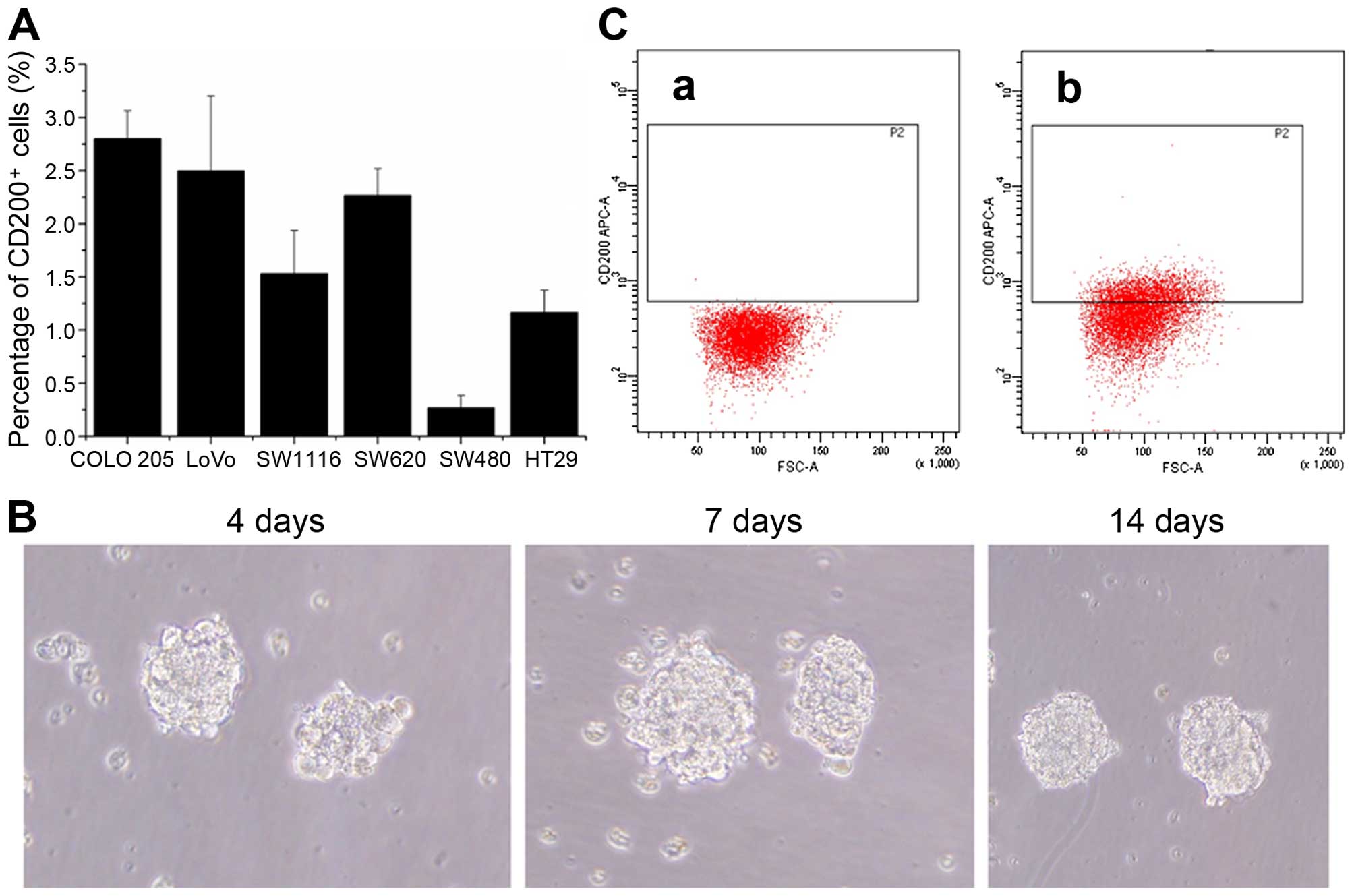
We preliminarily screened 6 human colon cancer cell
lines (COLO 205, LoVo, SW620, SW480, SW1116 and HT29) for CD200
expression by flow cytometry. The proportion of CD200+
cells in all 6 colorectal cancer cell lines was <3% (Fig. 1A). However, when we verified the
cell purity, it was <90%, with yields ranging from 40–60%. Since
our previous study showed that COLO 205 colonospheres contain
cancer stem-like cells under serum-free culture conditions
(18), we used COLO 205 cells in
the present study in serum-free culture experiments (Fig. 1B). We examined the ratio of
CD200+ cells by flow cytometry at different time points
after transfer to serum-free medium. We found that the ratio of
CD200+ COLO 205 cells was highest in colonospheres
cultured for 2 weeks in serum-free medium, and its expression was
high enough to accurately distinguish and sort CD200+
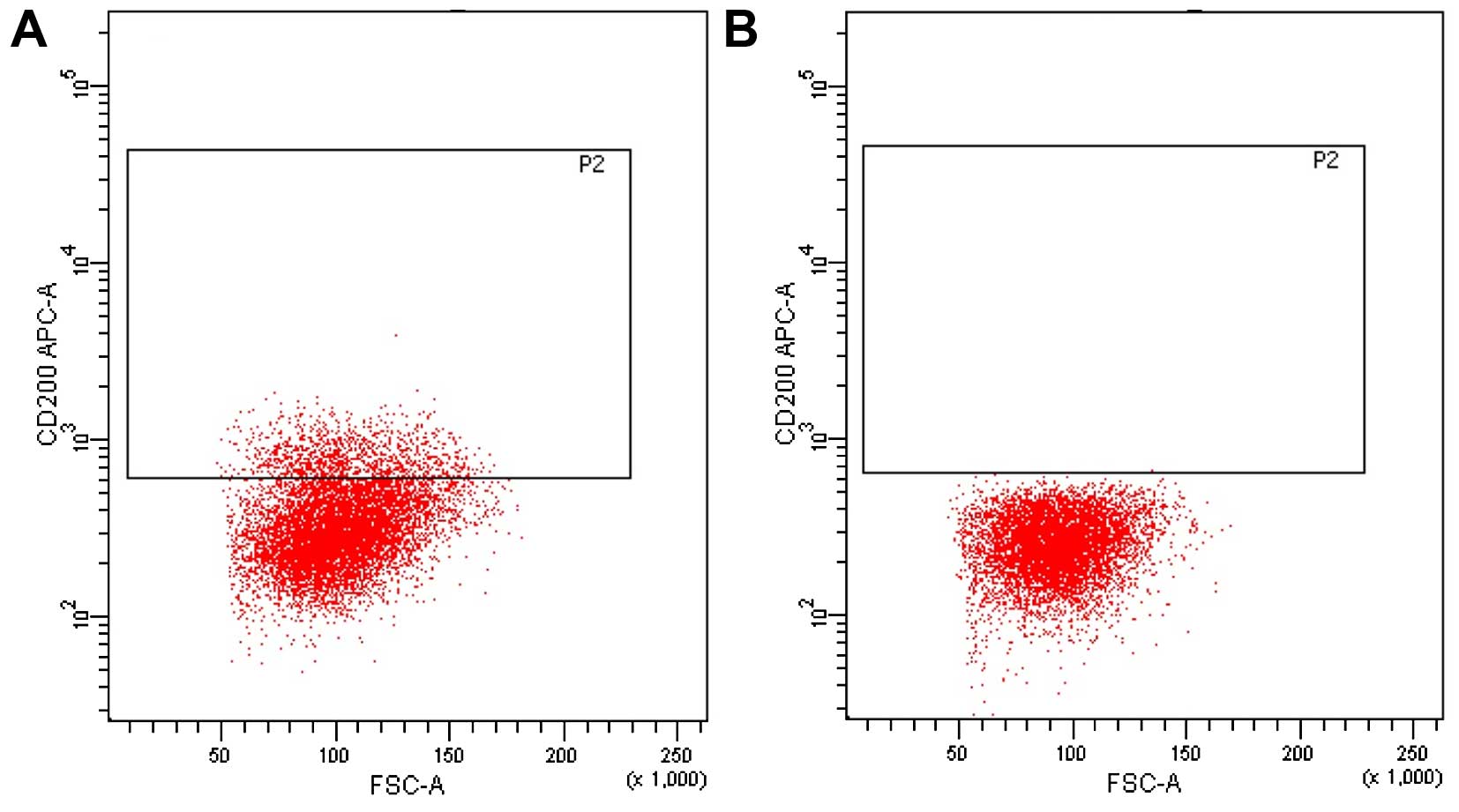
cells from C200− cells. We sorted and collected 10% of
the cells with strongest positive signal and 10% of the cells with
the strongest negative signal by FACS. We verified the purity of
the sorted CD200+ and CD200− cells by flow
cytometry, and it was always >95% for both cell fractions
(Fig. 1C).
CD200+ COLO 205 cells have
strong self-renewal and invasive abilities
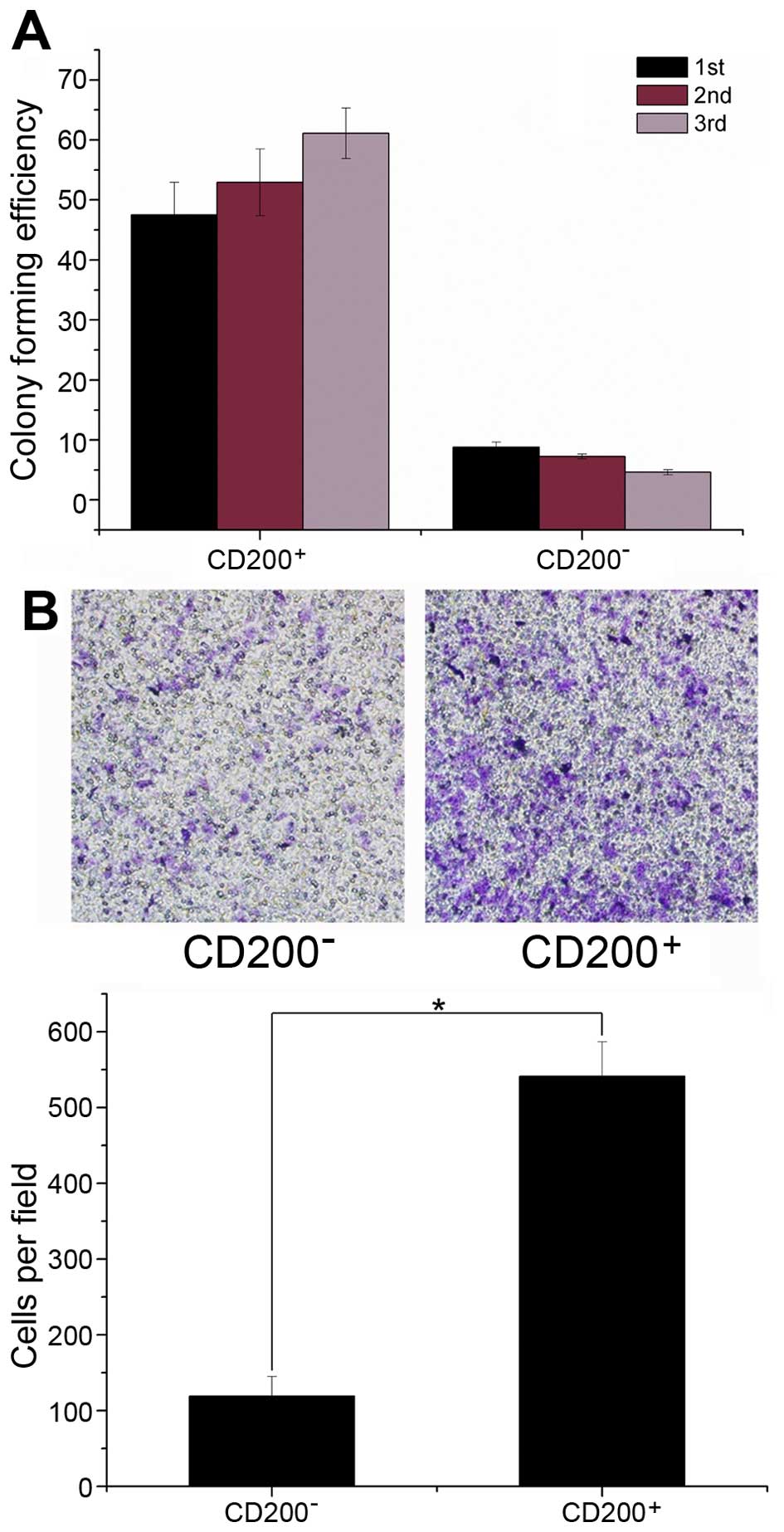
We performed colonosphere formation assays using
sorted CD200+ and CD200− cells to determine
the self-renewing capability of each fraction. We found that
CD200+ cells more efficiently generated colonospheres
upon serial passage compared to CD200− cells (Fig. 2A). For primary sphere formation,
46.6±6.4% of CD200+ cells formed spheres compared to
9.3±2.1% of CD200− cells. Moreover, the CFE of
CD200+ cells increased upon passaging, while the CFE of
CD200− cells decreased (Fig.
2A). These results suggest that CD200+ COLO 205
cells have a stronger self-renewing capacity compared to
CD200− cells. In parallel, we performed a Transwell
migration assay, which is an in vitro indicator of
metastatic potential, to evaluate the effects of CD200 expression
on migration ability of COLO 205 cells. As shown in Fig. 2B, after 48 h of incubation, the
number of CD200+ cells that passed through the membrane
was significantly higher than the number of CD200−
cells.
Microarray analysis of CD200+
COLO 205 cell gene expression compared to CD200−
cells
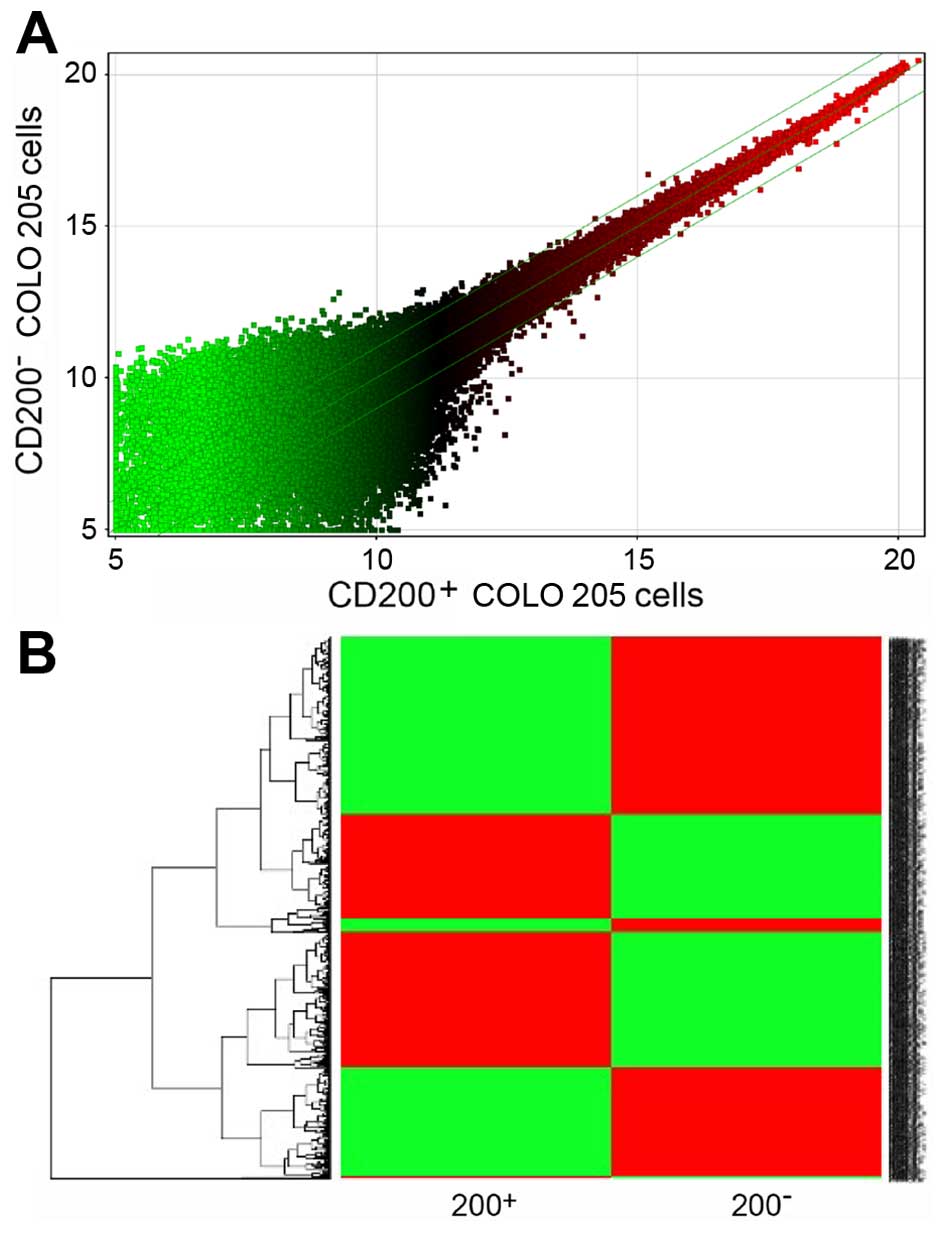
To identify potential genes associated with the
distinct cellular behavior of the 2 populations, we generated gene
expression profiles of CD200+ COLO 205 and
CD200− COLO 205 cells. The gene chip results showed that
a total of 958 genes were upregulated or downregulated at least
2-fold in CD200+ COLO 205 cells; 433 genes were
upregulated and 525 genes were down-regulated (Fig. 3A). Gene Pathway analysis revealed
that the differentially expressed genes were distributed in
multiple pathways, including metabolic, cancer, immune response,
apoptosis and several classical signaling pathways. Integrating
data from Gene Ontology with the published data, we identified
several genes that may be related to the tumor-initiating ability
of CD200+ COLO 205 cells. Genes associated with tumor
proliferation and invasion, such as GLI2, RAC3 and PRKCB were
upregulated. These genes have been reported to be involved in
proliferation and metastasis regulation in various cancers
(19–21). CD200+ cells also showed
increased PPARD expression, a PPARS family member that regulates
lipid metabolism (22),
proliferation (23) and the
inflammatory response (24).
In contrast, genes with tumor-suppressor activities,
such as CACNA1G, RPS6KA6 and the pro-apoptotic gene FAS were
downregulated in CD200+ cells compared to
CD200− cells. Among these, CACNA1G (Cav3.1) encodes a
T-type, low-voltage activated calcium channel, which along with
FAS, may contribute to the promotion of apoptosis and the
repression of tumor proliferation (25,26).
CACNA1G is considered a target of abnormal methylation and
silencing in colorectal and lung cancer, and several other tumors
(27). RPS6KA6 may be an important
tumor-suppressor gene by modulating senescence induction and
regulating cell proliferation (28).
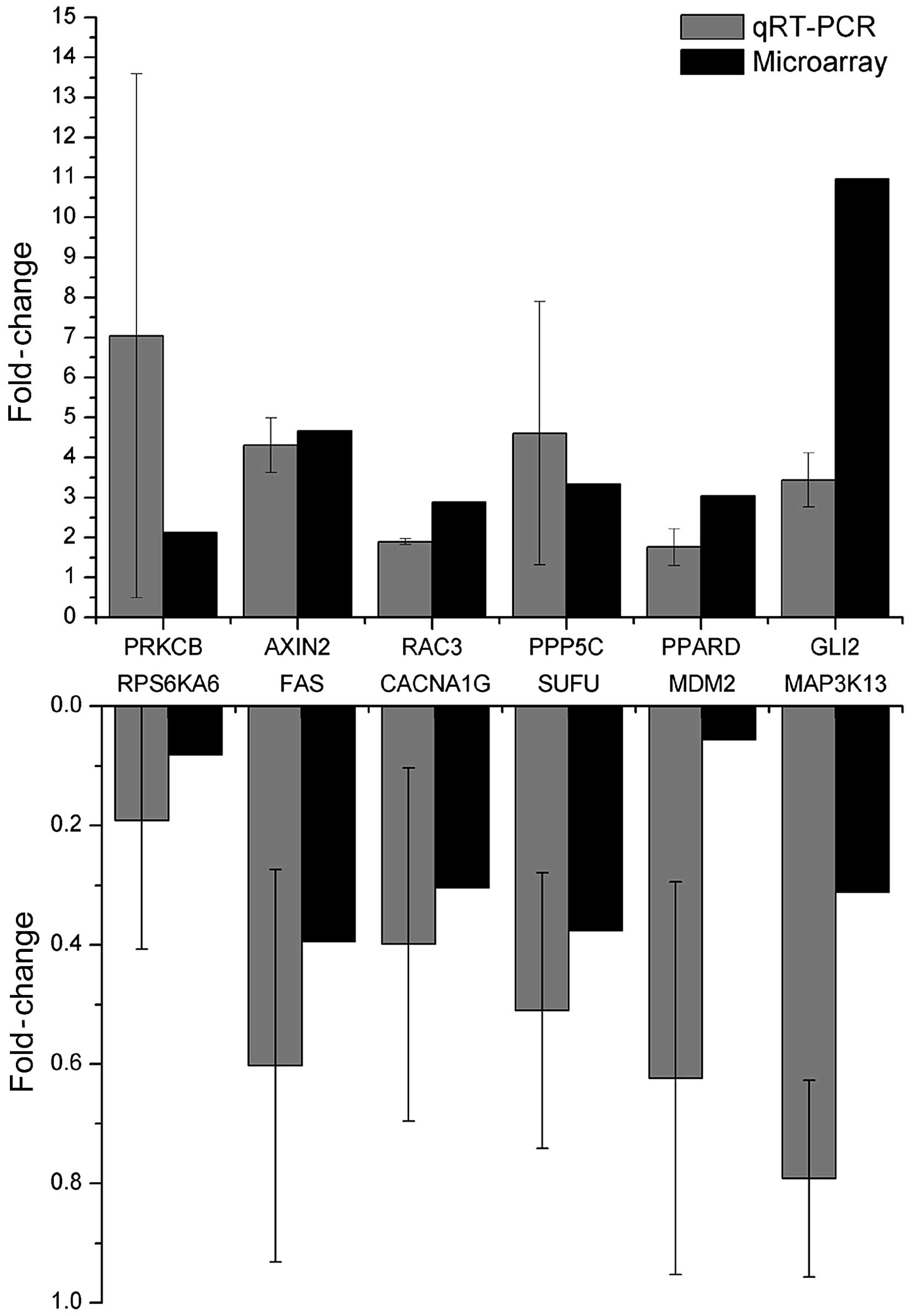
We validated the expression of a subset of the genes
upregulated (GLI2, RAC3, PRKCB, PPARD, PPP5C and FGF14) or
downregulated (FAS, RPS6KA6, CACNA1G, SUFU, MDM2 and MAP3K13) in
CD200+ COLO 205 cells by qRT-PCR. We selected candidate
genes that represented diverse biological functions that could be
involved in proliferation, invasion and stemness, and that reflects
the extent of differential gene expression. Notably, qRT-PCR assays
confirmed the same trends observed in the microarray analyses
(Fig. 4).
CD44+CD133+
population is enriched in CD200+ cells and shares a
similar gene expression profile
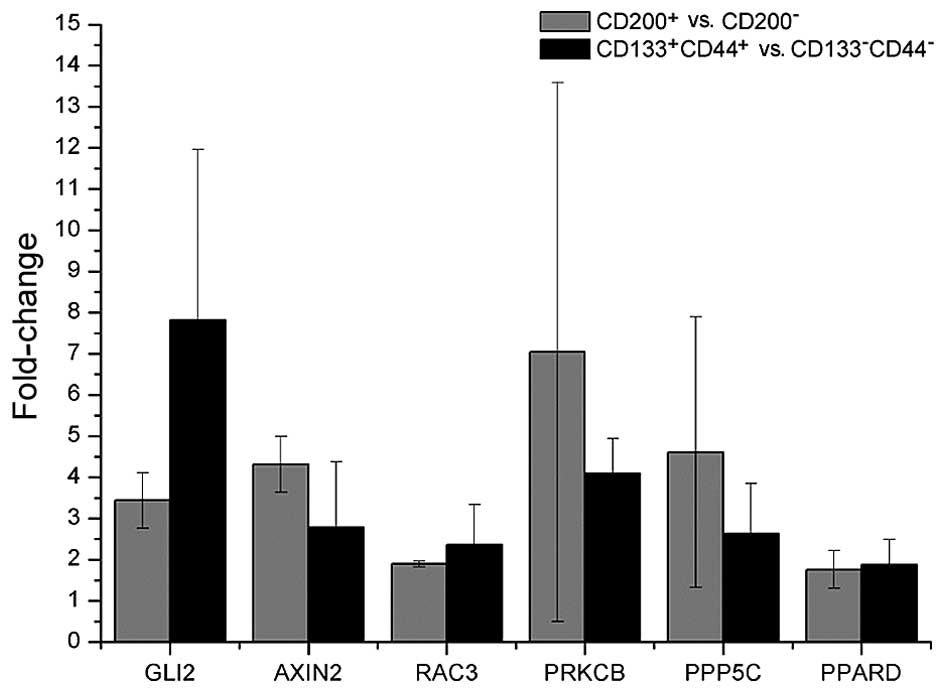
To investigate whether there was a correlation
between the CD44+CD133+ population and CD200
expression, we sorted CD44+CD133+ and
CD44−CD133− COLO 205 cells by FACS. Flow
cytometric analysis showed that CD200 expression was always higher
in the CD44+CD133+ population compared to the
CD44−CD133− population (Fig. 5). To further explore the
relationship between CD200 expression and
CD44+CD133+ cells, we examined the expression
of genes that were upregulated in the CD200+ cells in
the CD44+CD133+ and
CD44−CD133− cells by qRT-PCR. The results
showed that some of the genes upregulated in the CD200+
cells were also upregulated in the
CD44+CD133+ fraction compared to the
CD44−CD133− fraction (Fig. 6).
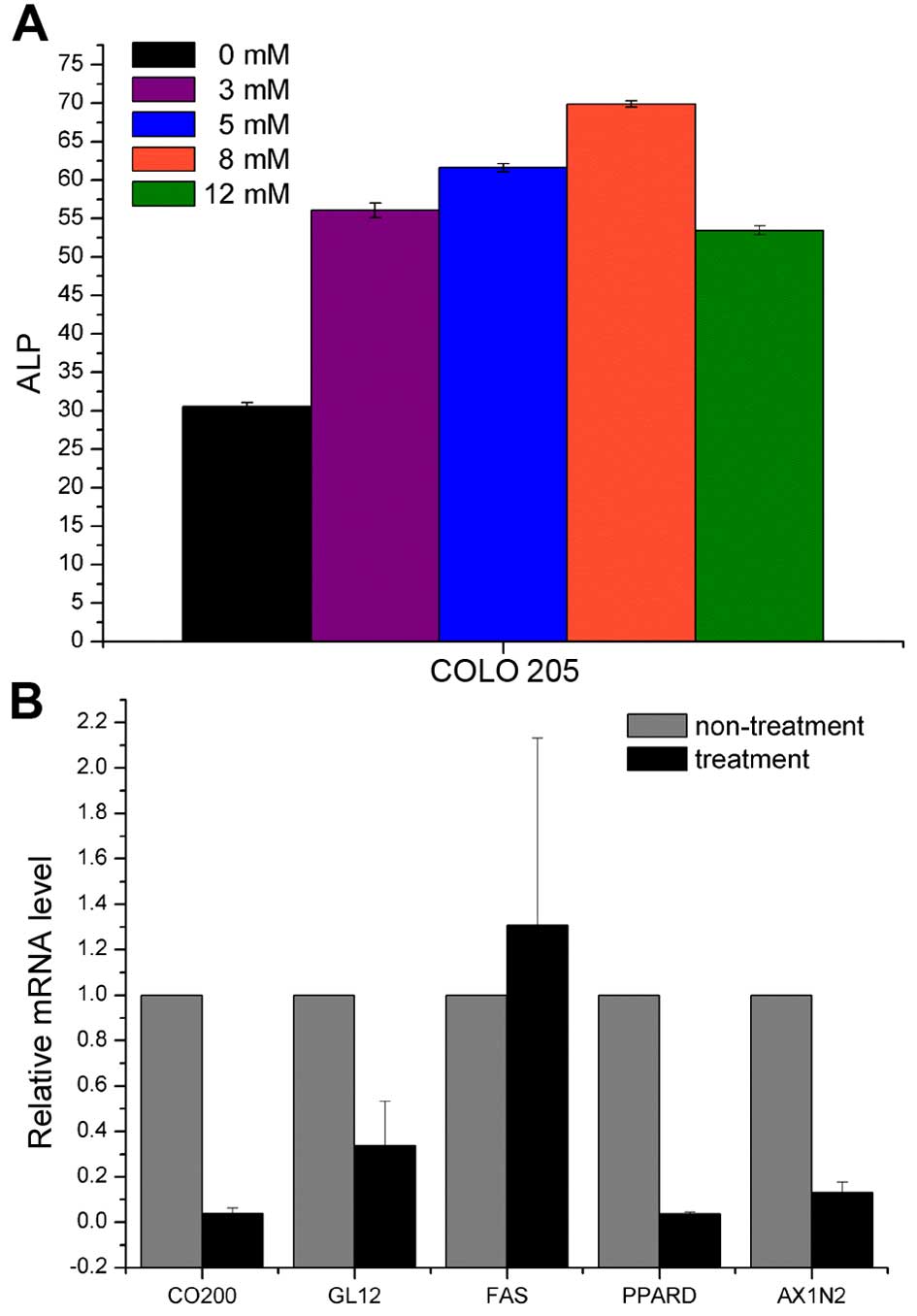
NaBT-induced differentiation alters
CD200, GLI2, FAS, PPARD and AXIN2 expression
NaBT is known to induce colonic epithelial cell
differentiation in colon cancer (29,30).
Thus, we examined the expression of several stem cell markers
(GLI2, FAS, PPARD and AXIN2) and CD200 before and after NaBT
treatment of COLO 205 cells. We estimated the efficiency of NaBT
treatment by ELISA for the well-known differentiated colonocyte
marker alkaline phosphatase (ALP). ALP expression was highest after
treatment with 8 mM NaBT (Fig. 7A).
NaBT treatment significantly decreased CD200, GLI2, PPARD and AXIN2
expression. In contrast, FAS expression increased upon NaBT
treatment (Fig. 7B). These findings
confirm that CD200, GLI2, PPARD and AXIN2 are useful markers of
undifferentiated or primitive colon cancer cells.
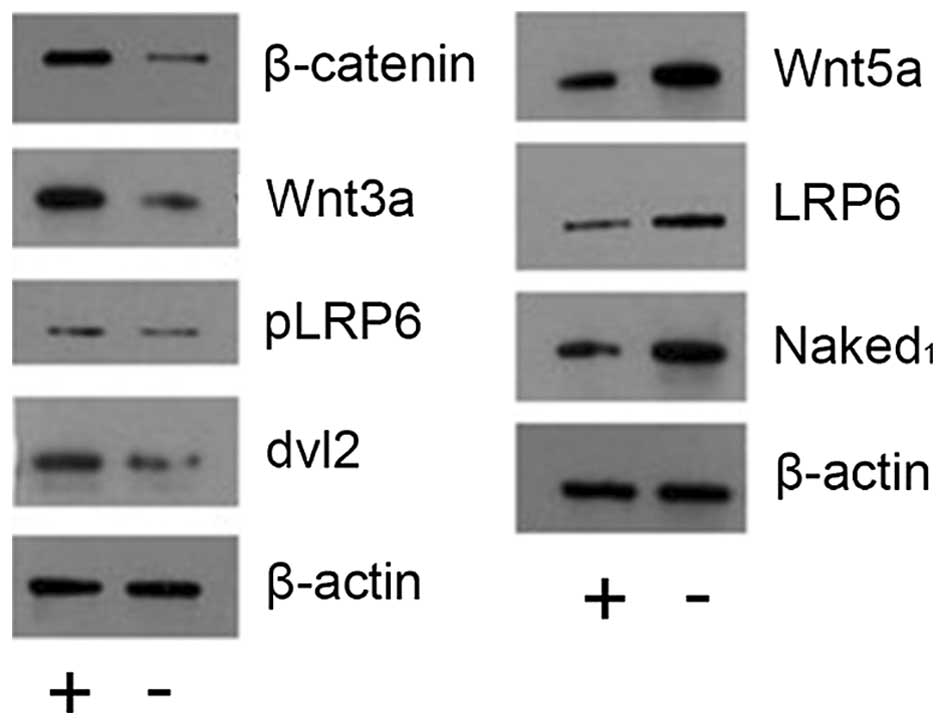
CD200 activates the Wnt/β-catenin
signaling pathway
We identified 12 differentially expressed genes
associated with the Wnt signaling pathway, by pathway analysis.
Thus, we hypothesized that CD200 regulation of colorectal cancer
occurrence and development may be closely associated with the Wnt
signaling pathway. Western blot analyses confirmed that β-catenin,
Wnt3a, Plrp6 and DVL2 were upregulated in the CD200+
population, while Wnt5a, LRP6 and Naked1 were downregulated
(Fig. 8). These results suggest
that CD200 likely activates the Wnt/β-catenin signaling
pathway.
Discussion
Colorectal cancer is one of the most common
digestive carcinomas in China, and its morbidity rate has increased
in recent years. The CCSC model has the potential to revolutionize
the colorectal cancer field from both a pathogenetic and
therapeutic point of view. However, the identity of CCSCs has
proved to be a difficult task. CD200 upregulation in several human
cancers and its tumor immune suppression properties suggest that it
is a potential CSC marker. In our previous study, we found that
CD200+ cells have stronger tumor formation abilities
than CD200− cells in vivo, which preliminarily
confirmed the reliability of CD200 as a CCSC marker. In the present
study, we compared the CD200+ population to the
CD200− population in in vitro colonosphere
formation ability. We demonstrated that CD200+ COLO 205
cells showed stronger self-renewal ability. In addition,
high-passage CD200+ colonosphere cells were highly
invasive in vitro. We also screened CD200+ cell
and CD200− cell gene expression profiles by gene chip,
and identified a set of upregulated genes in the CD200+
fraction. We selected several genes associated with proliferation,
invasion, stemness and immune processes for further study. We also
examined the correlation between CD200 expression and
CD44+CD133+ CSCs. By comparing the gene
expression profiles of CD200+ and
CD44+CD133+ COLO 205 cells, we found that
many genes were upregulated in both populations, thus further
supporting a role for CD200 molecule in the CSC signature.
Overall, the present study further confirmed the
reliability of CD200 as a CCSC marker by in vitro
experiments. We also showed a relationship between CD200 and FAS
expressions, which may help in understanding the CD200 role in the
colorectal cancers immune tolerance process. Indeed, Fas (CD95)
belongs to the tumor necrosis factor (TNF) and nerve growth factor
(NGF) receptor superfamilies, and it is expressed on the cell
surface. Upon binding to its receptor FasR, Fas triggers the death
signal cascade and induces apoptosis of sensitized cells (31). Fas-mediated apoptosis is involved in
functions such as tolerance acquisition and thymocyte clonal
deletion, immune response termination, T cell-mediated cytotoxicity
and T cell activation-induced cell death. Loss of Fas function and
expression of functional FasL by tumor cells may contribute to the
evasion of host immune surveillance by triggering apoptosis of
tumor-specific T lymphocytes (32,33).
Decreased FAS and/or increased FASL expression promotes malignant
transformation and progression of pancreatic, breast, colorectal
and gastric cancer, and many other tumors (34–37).
In summary, FAS is downregulated in CD200+ COLO 205
cells. Decreased Fas expression leads to tumor escape from immune
surveillance. Thus, we hypothesize that FAS is associated with the
immune inhibitory effect of CD200.
Another gene of interest is PPARD. This gene encodes
a peroxisome proliferator-activated receptor (PPAR) family member.
PPARs mediate several biological processes and are closely related
to several chronic diseases, including obesity, diabetes,
atherosclerosis and cancer (38–40).
PPAR expression is increased in a wide variety of cancers, such as
colorectal, lung, prostate and bladder cancer (41–44).
Additionally, PPARD is involved in the inhibition of inflammation.
PPARγ inhibits inflammatory gene expression activation and can
negatively interfere with pro-inflammatory transcription factor
signaling in inflammatory cells (39). PPARD agonists suppress monocyte
elaboration of inflammatory cytokines at agonist concentrations
(24). Moreover, PPARD plays a
central role in stem cell maintenance in multiple systems (45). Therefore, PPARD may play an
important role in carcinogenesis and immune process regulation. In
the present study, we found that PPARD expression was much higher
in CD200+ cells compared to CD200− cells,
suggesting that PPARD functions in the CD200-regulated tumor
immunosuppression process.
Notably, pathway analysis revealed that CD200 is
associated with Wnt/β-catenin signaling pathway activation. Wnt
signaling plays important roles in embryonic development and
tumorigenesis. Aberrant signaling leads to a variety of
developmental defects and some common human malignancies (46). Despite the well-known function of
Wnt signaling in cell growth, apoptosis, differentiation and cell
cycle, it also plays a role in immune regulation. It participates
in immune cell differentiation and development and controls
peripheral immune cell function (47,48).
As previously suggested (49),
constitutively activate Wnt/β-catenin signaling may contribute to
anti-melanoma T cell response suppression in both the induction and
effector phases by impairing DC and T cell function, partially
through IL-10 and the generation of the immunosuppressive
microenvironment. In the present study, we found that β-catenin,
Wnt3a, pLRP6 and DVL2 were upregulated in CD200+ COLO
205 cells compared to CD200− cells, while Wnt5a, LRP6
and Naked1 were downregulated.
Wnt3a is a canonical Wnt signaling pathway
activator, and treatment with Wnt3a results in an increase in
β-catenin targets (50,51). Upon Wnt stimulation, LRP6 is
phosphorylated by kinases at multiple sites. Phosphorylated LRP6
recruits axin to the membrane and presumably activates β-catenin
signaling (52). DVL2 associates
with actin filaments and cytoplasmic vesicular membranes (53) to mediate Fzd receptor endocytosis
after Wnt stimulation (54). Dvl
overexpression has been observed in certain cancers (55). High expression of β-catenin, Wnt3a,
pLRP6 and DVL2 suggest that the Wnt/β-catenin signaling pathway is
activated. Moreover, Mikels and Nusse (56) suggested that Wnt5a signals through
both the canonical and non-canonical Wnt pathways. Purified Wnt5a
inhibits Wnt3a-induced canonical Wnt signaling by downregulating
β-catenin-induced reporter gene expression. However, Wnt5a can also
activate β-catenin signaling in the presence of Frizzled-4. As
shown by Zhang et al (57),
Wnt5a inhibits melanoma proliferation and melanogenesis via
non-canonical Wnt/Ror2 pathway activation and inhibition of the
canonical Wnt pathway. Naked1 is a negative Wnt signaling
regulator, and it inhibits the canonical Wnt/β-catenin pathway by
binding to dishevelled proteins and directs dishevelled activity
towards the planar cell polarity pathway (58,59).
Taken together, these data suggest that the Wnt/β-catenin signaling
pathway is activated in CD200+ COLO 205 colorectal
cancer cells. Wnt/β-catenin signaling is a target of CD200-mediated
immunosuppression. However, our studies only examined expression of
several common proteins related to Wnt signaling pathway. Future
studies may focus on dual-luciferase assays to determine whether
CD200 activates the Wnt/β-catenin signaling pathway.
In summary, in vitro experiments revealed
that CD200+ COLO 205 cells have greater colony formation
ability, higher invasion and CD200 expression is enriched in
CD133+CD44+ cells. These findings further
confirm CD200 as a CSC surface marker. By analyzing gene expression
profiles, we examined genes of particular interest that are likely
involved in CSC properties. Numerous genes identified in the
present study, may also be good CCSC marker candidates, and their
evaluation could increase specificity in CCSC identification.
Although further studies are required to identify the functional
roles of these genes in the CSC phenotype and their relationship
with the CD200 immunosuppression mechanism, these findings hold
promise for potential future approaches in elucidating the
mechanism of colorectal cancer and developing new tumor targeting
therapies.
Acknowledgments
The present study was supported by the Scientific
and Technical Project of Guangdong Province (2010B031600097).
References
|
1
|
Choi D, Lee HW, Hur KY, Kim JJ, Park GS,
Jang SH, Song YS, Jang KS and Paik SS: Cancer stem cell markers
CD133 and CD24 correlate with invasiveness and differentiation in
colorectal adenocarcinoma. World J Gastroenterol. 15:2258–2264.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Su YJ, Lai HM, Chang YW, Chen GY and Lee
JL: Direct reprogramming of stem cell properties in colon cancer
cells by CD44. EMBO J. 30:3186–3199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ke J, Wu X, Wu X, He X, Lian L, Zou Y, He
X, Wang H, Luo Y, Wang L, et al: A subpopulation of
CD24+ cells in colon cancer cell lines possess stem cell
characteristics. Neoplasma. 59:282–288. 2012. View Article : Google Scholar
|
|
4
|
Chen Y, Yu D, Zhang H, He H, Zhang C, Zhao
W and Shao RG: CD133+EpCAM+ phenotype
possesses more characteristics of tumor initiating cells in
hepatocellular carcinoma Huh7 cells. Int J Biol Sci. 8:992–1004.
2012. View Article : Google Scholar :
|
|
5
|
Barker N, Huch M, Kujala P, van de
Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H,
van den Born M, et al: Lgr5+ve stem cells drive
self-renewal in the stomach and build long-lived gastric units in
vitro. Cell Stem Cell. 6:25–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levin TG, Powell AE, Davies PS, Silk AD,
Dismuke AD, Anderson EC, Swain JR and Wong MH: Characterization of
the intestinal cancer stem cell marker CD166 in the human and mouse
gastrointestinal tract. Gastroenterology. 139:2072–2082. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haraguchi N, Ohkuma M, Sakashita H,
Matsuzaki S, Tanaka F, Mimori K, Kamohara Y, Inoue H and Mori M:
CD133+CD44+ population efficiently enriches
colon cancer initiating cells. Ann Surg Oncol. 15:2927–2933. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hou NY, Yang K, Chen T, Chen XZ, Zhang B,
Mo XM and Hu JK: CD133+CD44+ subgroups may be
human small intestinal stem cells. Mol Biol Rep. 38:997–1004. 2011.
View Article : Google Scholar
|
|
9
|
Liao KL, Bai XF and Friedman A: The role
of CD200-CD200R in tumor immune evasion. J Theor Biol. 328:65–76.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coles SJ, Hills RK, Wang EC, Burnett AK,
Man S, Darley RL and Tonks A: Expression of CD200 on AML blasts
directly suppresses memory T-cell function. Leukemia. 26:2148–2151.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Coles SJ, Wang EC, Man S, Hills RK,
Burnett AK, Tonks A and Darley RL: CD200 expression suppresses
natural killer cell function and directly inhibits patient
anti-tumor response in acute myeloid leukemia. Leukemia.
25:792–799. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Costello DA, Lyons A, Denieffe S, Browne
TC, Cox FF and Lynch MA: Long term potentiation is impaired in
membrane glycoprotein CD200-deficient mice: A role for Toll-like
receptor activation. J Biol Chem. 286:34722–34732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moreaux J, Veyrune JL, Reme T, De Vos J
and Klein B: CD200: A putative therapeutic target in cancer.
Biochem Biophys Res Commun. 366:117–122. 2008. View Article : Google Scholar
|
|
14
|
Kretz-Rommel A, Qin F, Dakappagari N,
Ravey EP, McWhirter J, Oltean D, Frederickson S, Maruyama T, Wild
MA, Nolan MJ, et al: CD200 expression on tumor cells suppresses
antitumor immunity: New approaches to cancer immunotherapy. J
Immunol. 178:5595–5605. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Talebian F and Bai XF: The role of tumor
expression of CD200 in tumor formation, metastasis and
susceptibility to T lymphocyte adoptive transfer therapy.
OncoImmunology. 1:971–973. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kawasaki BT, Mistree T, Hurt EM, Kalathur
M and Farrar WL: Co-expression of the toleragenic glycoprotein,
CD200, with markers for cancer stem cells. Biochem Biophys Res
Commun. 364:778–782. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X, Li M, Wang J, Yeung CM, Zhang H,
Kung HF, Jiang B and Lin MC: The BH3-only protein, PUMA, is
involved in oxaliplatin-induced apoptosis in colon cancer cells.
Biochem Pharmacol. 71:1540–1550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li YF, Xiao B, Lai ZS, Tu SF, Wang YY and
Zhang XL: Spheres isolated from Colo205 cell line possess cancer
stem-like cells under serum-free culture condition. Nan Fang Yi Ke
Da Xue Xue Bao. 28:236–240. 2008.In Chinese. PubMed/NCBI
|
|
19
|
Alexaki VI, Javelaud D, Van Kempen LC,
Mohammad KS, Dennler S, Luciani F, Hoek KS, Juàrez P, Goydos JS,
Fournier PJ, et al: GLI2-mediated melanoma invasion and metastasis.
J Natl Cancer Inst. 102:1148–1159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gest C, Joimel U, Huang L, Pritchard LL,
Petit A, Dulong C, Buquet C, Hu CQ, Mirshahi P, Laurent M, et al:
Rac3 induces a molecular pathway triggering breast cancer cell
aggressiveness: Differences in MDA-MB-231 and MCF-7 breast cancer
cell lines. BMC Cancer. 13:632013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Spindler KL, Lindebjerg J, Lahn M,
Kjaer-Frifeldt S and Jakobsen A: Protein kinase C-beta II (PKC-beta
II) expression in patients with colorectal cancer. Int J Colorectal
Dis. 24:641–645. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sakai J: Activation of 'fat burning
sensor' peroxisome proliferator-activated receptor delta induces
fatty acid beta-oxidation in skeletal muscle and attenuates
metabolic syndrome. Seikagaku. 76:517–524. 2004.In Japanese.
PubMed/NCBI
|
|
23
|
Peters JM, Lee SS, Li W, Ward JM,
Gavrilova O, Everett C, Reitman ML, Hudson LD and Gonzalez FJ:
Growth, adipose, brain, and skin alterations resulting from
targeted disruption of the mouse peroxisome proliferator-activated
receptor beta (delta). Mol Cell Biol. 20:5119–5128. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang C, Ting AT and Seed B: PPAR-gamma
agonists inhibit production of monocyte inflammatory cytokines.
Nature. 391:82–86. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li HJ, Wang CY, Mi Y, Du CG, Cao GF, Sun
XC, Liu DJ and Shorgan B: FasL-induced apoptosis in bovine oocytes
via the Bax signal. Theriogenology. 80:248–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohkubo T and Yamazaki J: T-type
voltage-activated calcium channel Cav3.1, but not
Cav3.2, is involved in the inhibition of proliferation
and apoptosis in MCF-7 human breast cancer cells. Int J Oncol.
41:267–275. 2012.PubMed/NCBI
|
|
27
|
Toyota M, Ho C, Ohe-Toyota M, Baylin SB
and Issa JP: Inactivation of CACNA1G, a T-type calcium channel
gene, by aberrant methylation of its 5′ CpG island in human tumors.
Cancer Res. 59:4535–4541. 1999.PubMed/NCBI
|
|
28
|
López-Vicente L, Armengol G, Pons B, Coch
L, Argelaguet E, Lleonart M, Hernández-Losa J, de Torres I and
Ramon y Cajal S: Regulation of replicative and stress-induced
senescence by RSK4, which is down-regulated in human tumors. Clin
Cancer Res. 15:4546–4553. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lévy P, Robin H, Bertrand F, Kornprobst M
and Capeau J: Butyrate-treated colonic Caco-2 cells exhibit
defective integrin-mediated signaling together with increased
apoptosis and differentiation. J Cell Physiol. 197:336–347. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Turecková J, Vojtechová M, Kucerová D,
Velek J and Tuhácková Z: Sodium butyrate-mediated differentiation
of colorectal cancer cells: Regulation of PKCβII by PI 3-kinase.
Int J Mol Med. 15:329–335. 2005.
|
|
31
|
Cullen SP, Henry CM, Kearney CJ, Logue SE,
Feoktistova M, Tynan GA, Lavelle EC, Leverkus M and Martin SJ:
Fas/CD95-induced chemokines can serve as 'find-me' signals for
apoptotic cells. Mol Cell. 49:1034–1048. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Walker PR, Saas P and Dietrich PY: Role of
Fas ligand (CD95L) in immune escape: The tumor cell strikes back. J
Immunol. 158:4521–4524. 1997.PubMed/NCBI
|
|
33
|
O'Connell J, O'Sullivan GC, Collins JK and
Shanahan F: The Fas counterattack: Fas-mediated T cell killing by
colon cancer cells expressing Fas ligand. J Exp Med. 184:1075–1082.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bernstorff WV, Glickman JN, Odze RD,
Farraye FA, Joo HG, Goedegebuure PS and Eberlein TJ: Fas
(CD95/APO-1) and Fas ligand expression in normal pancreas and
pancreatic tumors. Implications for immune privilege and immune
escape. Cancer. 94:2552–2560. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gryko M, Guzińska-Ustymowicz K, Pryczynicz
A, Cepowicz D, Kukliński A, Czyżewska J, Kemona A and Kędra B:
Correlation between Fas and FasL proteins expression in normal
gastric mucosa and gastric cancer. Folia Histochem Cytobiol.
49:142–147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang W, Zheng Z, Yu W, Lin H, Cui B and
Cao F: Polymorphisms of the FAS and FASL genes and risk of breast
cancer. Oncol Lett. 3:625–628. 2012.PubMed/NCBI
|
|
37
|
Hoogwater FJ, Steller EJ, Westendorp BF,
Borel Rinkes IH and Kranenburg O: CD95 signaling in colorectal
cancer. Biochim Biophys Acta. 1826:189–198. 2012.PubMed/NCBI
|
|
38
|
Wu HT, Chen W, Cheng KC, Ku PM, Yeh CH and
Cheng JT: Oleic acid activates peroxisome proliferator-activated
receptor δ to compensate insulin resistance in steatotic cells. J
Nutr Biochem. 23:1264–1270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fan Y, Wang Y, Tang Z, Zhang H, Qin X, Zhu
Y, Guan Y, Wang X, Staels B, Chien S, et al: Suppression of
pro-inflammatory adhesion molecules by PPAR-delta in human vascular
endothelial cells. Arterioscler Thromb Vasc Biol. 28:315–321. 2008.
View Article : Google Scholar
|
|
40
|
Cohen G, Riahi Y, Shamni O, Guichardant M,
Chatgilialoglu C, Ferreri C, Kaiser N and Sasson S: Role of lipid
peroxidation and PPAR-δ in amplifying glucose-stimulated insulin
secretion. Diabetes. 60:2830–2842. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mansure JJ, Nassim R and Kassouf W:
Peroxisome proliferator-activated receptor gamma in bladder cancer:
A promising therapeutic target. Cancer Biol Ther. 8:6–15. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tsukahara T, Hanazawa S, Kobayashi T,
Iwamoto Y and Murakami-Murofushi K: Cyclic phosphatidic acid
decreases proliferation and survival of colon cancer cells by
inhibiting peroxisome proliferator-activated receptor γ.
Prostaglandins Other Lipid Mediat. 93:126–133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gupta RA, Wang D, Katkuri S, Wang H, Dey
SK and DuBois RN: Activation of nuclear hormone receptor peroxisome
proliferator-activated receptor-delta accelerates intestinal
adenoma growth. Nat Med. 10:245–247. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Genini D, Garcia-Escudero R, Carbone GM
and Catapano CV: Transcriptional and non-transcriptional functions
of PPARβ/δ in non-small cell lung cancer. PLoS One. 7:e460092012.
View Article : Google Scholar
|
|
45
|
Morales-Garcia JA, Luna-Medina R,
Alfaro-Cervello C, Cortes-Canteli M, Santos A, Garcia-Verdugo JM
and Perez-Castillo A: Peroxisome proliferator-activated receptor γ
ligands regulate neural stem cell proliferation and differentiation
in vitro and in vivo. Glia. 59:293–307. 2011. View Article : Google Scholar
|
|
46
|
Giles RH, van Es JH and Clevers H: Caught
up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta.
1653:1–24. 2003.PubMed/NCBI
|
|
47
|
Buttler K, Becker J, Pukrop T and Wilting
J: Maldevelopment of dermal lymphatics in Wnt5a-knockout-mice. Dev
Biol. 381:365–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gattinoni L, Zhong XS, Palmer DC, Ji Y,
Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et
al: Wnt signaling arrests effector T cell differentiation and
generates CD8+ memory stem cells. Nat Med. 15:808–813.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yaguchi T, Goto Y, Kido K, Mochimaru H,
Sakurai T, Tsukamoto N, Kudo-Saito C, Fujita T, Sumimoto H and
Kawakami Y: Immune suppression and resistance mediated by
constitutive activation of Wnt/β-catenin signaling in human
melanoma cells. J Immunol. 189:2110–2117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yeh JR, Zhang X and Nagano MC: Indirect
effects of Wnt3a/β-catenin signalling support mouse spermatogonial
stem cells in vitro. PLoS One. 7:e400022012. View Article : Google Scholar
|
|
51
|
Shin H, Kwack MH, Shin SH, Oh JW, Kang BM,
Kim AA, Kim J, Kim MK, Kim JC and Sung YK: Identification of
transcriptional targets of Wnt/beta-catenin signaling in dermal
papilla cells of human scalp hair follicles: EP2 is a novel
transcriptional target of Wnt3a. J Dermatol Sci. 58:91–96. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tamai K, Zeng X, Liu C, Zhang X, Harada Y,
Chang Z and He X: A mechanism for Wnt coreceptor activation. Mol
Cell. 13:149–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Capelluto DG, Kutateladze TG, Habas R,
Finkielstein CV, He X and Overduin M: The DIX domain targets
dishevelled to actin stress fibres and vesicular membranes. Nature.
419:726–729. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen W, ten Berge D, Brown J, Ahn S, Hu
LA, Miller WE, Caron MG, Barak LS, Nusse R and Lefkowitz RJ:
Dishevelled 2 recruits beta-arrestin 2 to mediate Wnt5A-stimulated
endocytosis of Frizzled 4. Science. 301:1391–1394. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pulvirenti T, Van Der Heijden M, Droms LA,
Huse JT, Tabar V and Hall A: Dishevelled 2 signaling promotes
self-renewal and tumorigenicity in human gliomas. Cancer Res.
71:7280–7290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mikels AJ and Nusse R: Purified Wnt5a
protein activates or inhibits beta-catenin-TCF signaling depending
on receptor context. PLoS Biol. 4:e1152006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang J, Li Y, Wu Y, Yang T, Yang K, Wang
R, Yang J and Guo H: Wnt5a inhibits the proliferation and
melanogenesis of melanocytes. Int J Med Sci. 10:699–706. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wharton KA Jr, Zimmermann G, Rousset R and
Scott MP: Vertebrate proteins related to Drosophila naked cuticle
bind dishevelled and antagonize Wnt signaling. Dev Biol.
234:93–106. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zeng W, Wharton KA Jr, Mack JA, Wang K,
Gadbaw M, Suyama K, Klein PS and Scott MP: nakedcuticle encodes an
inducible antagonist of Wnt signalling. Nature. 403:789–795. 2000.
View Article : Google Scholar : PubMed/NCBI
|