Introduction
Colorectal carcinoma (CRC) is one of the most common
malignancies worldwide and is a major cause of cancer-related
morbidity and mortality (1,2). Although great advances have been
achieved in the treatment of CRC over the past few decades, the
mean 5-year survival rate is estimated to be less than 10% when
metastasis occurs (3). Currently,
the precise molecular mechanisms of its pathogenesis and
progression are still largely unknown.
Glucose-regulated protein 78 (GRP78), also known as
immunoglobulin heavy chain-binding protein (BiP) or HSPA5, is a
member of heat shock protein 70 (HSP70) family, which is localized
in endoplasmic reticulum (ER). GRP78 is a critical regulator of ER
function, due to its roles in protein folding and assembly,
targeting misfolded protein for degradation, translocation of newly
synthesized polypeptides through the ER membrane and ER
Ca2+ binding (4,5). ER stress, such as glucose deprivation,
hypoxia and oxidative stress, is known to induce glucose-regulated
stress response or unfolded protein response (UPR), which results
in an accumulation of GRP78 in the ER compartment (6–9).
Recently, several studies have demonstrated the overexpression of
GRP78 in a variety of solid tumors and GRP78 overexpression
contributes to cancer cell proliferation, migration and drug
resistance (10–17). Emerging evidence indicates that
GRP78 distributes to the cell surface in various tumors and form a
complex with specific ligands, exhibiting a wide range of
biological effects. Ligation of surface GRP78 by α2-macroglobulin
activates Akt-dependent signaling and promotes proliferation and
metastasis of prostate cancer cells (18). Isthmin targets cell-surface GRP78 to
trigger apoptosis by inducing mitochondrial dysfunction of cancer
cells (19). A recent study showed
that CRC cells can secrete GRP78, which binds to cell surface GRP78
resulting in the activation of intracellular proliferation
signaling (20).
The AKT and ERK signaling pathways regulate many
biologic events in cells. Inhibition of these two pathways has been
considered as a novel antitumor strategy (21–23).
It was suggested that GRP78 oncogenic properties may rely on the
activation of AKT and ERK signaling pathways (13,24).
Our previous study has revealed that GRP78 is overexpressed in CRC,
regulates CRC cell growth and apoptosis and may be an important
indicator for malignant transformation (15). However, the exact pathway driven by
GRP78 in carcinogenesis remains unclear. In the present study,
investigated the role of GRP78 in CRC carcinogenesis using human
CRC cells transfected with GRP78 shRNA and elucidated the possible
signaling pathway.
Materials and methods
Cell culture
The human CRC cell lines RKO and SW620 were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). The cells were cultured in RPMI-1640 medium
supplemented with 10% heat inactivated fetal bovine serum (FBS;
Gibco, Grand Island, NY, USA). All cells were maintained in a
humidified incubator at 37°C and 5% CO2.
Plasmid construction and cell
transfection
Lentivirus with the GRP78 shRNA plasmid expression
vector (pLV-GRP78 shRNA) and empty vector (pLV-control) were
purchased from Shanghai GeneChem Corporation (Shanghai, China).
Transfection of pLV-GRP78 shRNA or pLV-control into RKO and SW620
cells was performed using Lipofectamine 2000 according to the
manufacturer's instructions. The transfection efficiency was
monitored by fluorescence microscopy and the stably transfected
cells were selected by G418 at a concentration of 1 mg/ml.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and colony formation assays
After transfection, cells were plated at 4,000
cells/well into 96-well plates and cultured for 1–5 days. MTT
reagent (Amresco, Solon, OH, USA) was added into wells on the
indicated day, and the cells were incubated for another 4 h at
37°C. The supernatants were then removed, and the formazan crystals
were dissolved in dimethyl sulfoxide (DMSO) (150 µl/well).
The absorbance at 490 nm for each sample was measured using a
multilabel plate reader (PerkinElmer, Waltham, MA, USA). For the
colony formation assay, 500 cells were placed in 6-well plates and
maintained in complete medium for 2 weeks. Colonies were fixed with
methanol, stained with 0.1% crystal violet and counted.
Cell cycle analysis
RKO and SW620 cells were synchronized via serum
starvation for 12 h. Cells were harvested, washed with ice-cold
phosphate-buffered saline (PBS) and fixed in 70% ethanol at 4°C
overnight. After washed twice with ice-cold PBS, the fixed cells
were stained with 50 µg/ml propidium iodide (Sigma-Aldrich,
Saint-Quentin Fallavier, France) in PBS containing 0.1% Triton
X-100 (Sangon, Shanghai, China) and 50 µg/ml RNase A
(Beyotime, Shanghai, China) for 30 min at 37°C in the dark. The
cell cycle distributions were measured using FACSCalibur flow
cytometer (Beckman Coulter, Brea, CA, USA); 10,000 events were
collected and assayed for each determination.
Transwell migration and invasion
assays
A total of 1×105 transfected cells were
resuspended in 200 µl serum-free medium and placed in the
upper compartment of a Transwell chamber (24-well insert, pore
size, 8-µm; Corning Inc., Corning, NY, USA). The lower
chamber was filled with 15% FBS as a chemoattractant and incubated
for 24 h for the migration assay and 24 h for the invasion assay.
For the invasion assay, the inserts were pre-coated with
extracellular matrix gel (BD Biosciences, Sparks, MD, USA). After
incubation, the cells on the upper surface of the membrane were
removed, and the cells on the lower surface were fixed and stained
with 0.1% crystal violet. Five visual fields of each insert were
randomly chosen and counted under a light microscope.
Cellular chemosensitivity to
5-fluorouracil (5-FU)
RKO and SW620 cells (1×104) expressing
scramble control or GRP78 shRNA were suspended in 500 µl of
serum-free RPMI-1640 medium and were seeded into 6-well plates.
Twelve hours after seeding, cells were treated with 5-FU at the
final concentration of 20 µg/ml. The cells were harvested at
the time point of 1, 6, 12 or 24 h for the western blot assay.
Western blotting
Cells were lysed in RIPA buffer (Beyotime)
supplemented with protease inhibitor cocktail and phosphatase
inhibitors (Aidlab Biotechnologies, Beijing, China). Total protein
(40 µg) was separated on 10% SDS-PAGE gel and transferred
onto polyvinylidene fluoride (PVDF) membranes. After blocked with
Tris-buffered saline with Tween-20 (TBST) containing 5% fat-free
milk for 1 h, the membranes were incubated with the primary
antibodies against GRP78 (1:250 dilution; Abcam, Cambridge, MA,
USA), AKT (1:1,000 dilution), phospho-AKT (ser473) (1:000
dilution), ERK (1:000 dilution), phospho-ERK1/2 (1:1,000 dilution)
(all from Cell Signaling Technology, Danvers, MA, USA) and β-actin
(1:8,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C
overnight. The membranes were then incubated with secondary
incubation using a horseradish peroxidase-conjugated anti-rabbit or
anti-mouse IgG (ZSGB-BIO, Beijing, China). After washing, the blots
were developed by chemiluminescent reagent (Thermo Scientific,
Waltham, MA, USA).
Tumorigenicity assay in nude mice
Five-week-old nude BALB/c athymic strain mice were
purchased from Shanghai GeneChem Corporation. Animal operation was
carried out according to the protocols approved by the Committee on
the Use of Live Animals in Teaching and Research (CULATR). License
to conduct live animal experiments was approved by Independent
Ethics Committee of Affiliated Hospital of Qingdao University.
Animals were subcutaneously injected in the right flank with
4×106 RKO cells expressing scramble control or GRP78
shRNA. Tumor growth was examined every 3 days for 6 weeks. Tumor
volume (V) was monitored by measuring the length (L) and width (W)
of the tumors and calculated with the following equation: V = (L ×
W2) × 0.5.
Statistical analysis
All experiments were repeated at least 3 times. All
data were expressed as the mean ± SD. Statistical analyses were
performed using SPSS 17.0 and 3 group data comparison was assessed
by one-way analysis of variance (ANOVA). LSD test was performed for
two group comparison. Differences were considered significant for
P-values <0.05.
Results
Knockdown of GRP78 expression inhibits
the proliferation and colony formation of CRC cells
To determine the involvement of GRP78 in the
proliferation and colony formation of CRC cells in vitro, we
knocked down the GRP78 in RKO and SW620 cells using specific shRNA.
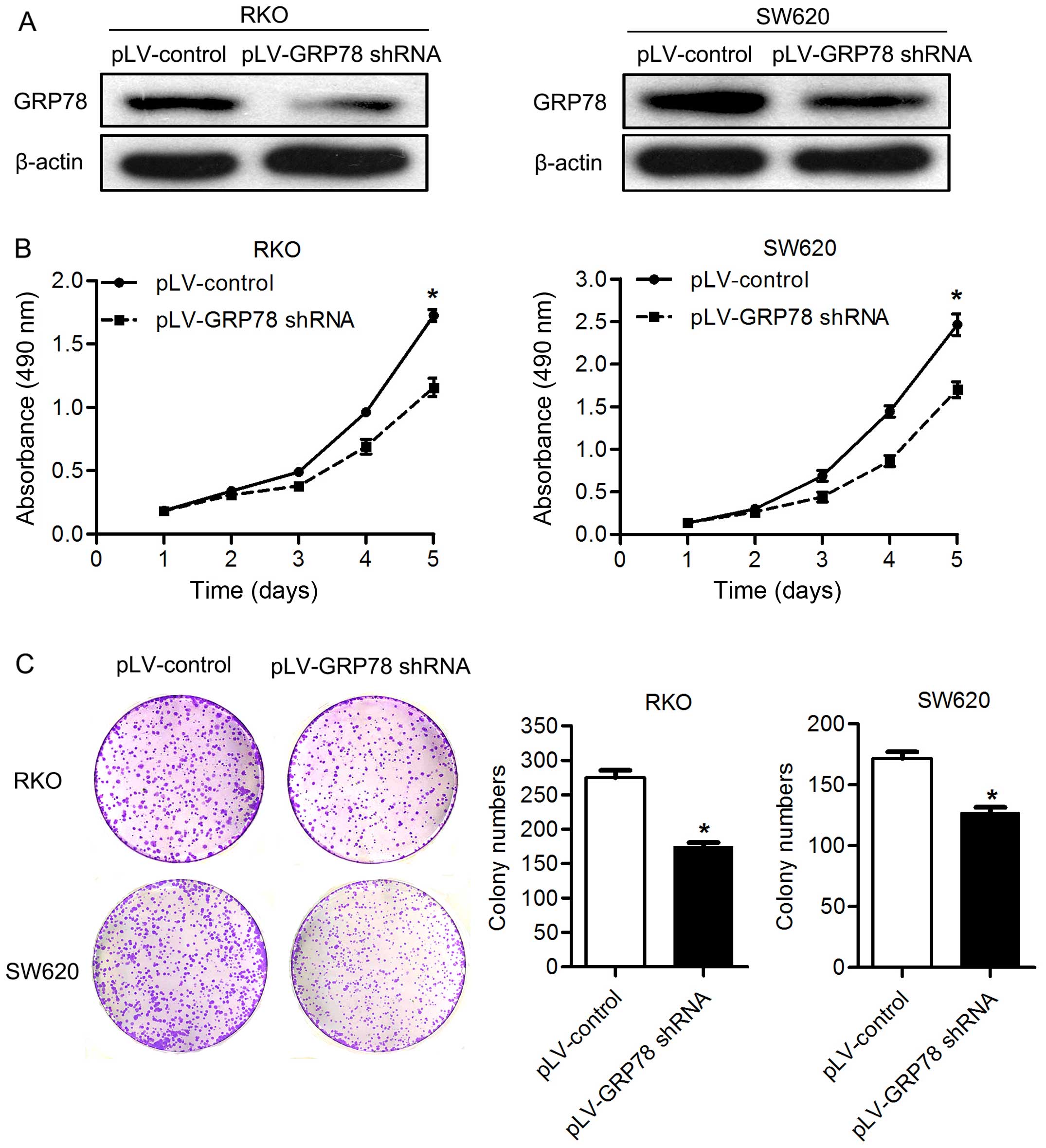
As shown in Fig. 1, GRP78 shRNA
transfection markedly decreased the expression of GRP78 in RKO and
SW620 cells (Fig. 1A). By MTT
assay, the cell proliferation was significantly lower in both RKO
and SW620 cells transfected with pLV-GRP78 shRNA than cells
transfected with pLV-control (P<0.05; Fig. 1B). Supporting this finding, we found
that knockdown of GRP78 inhibited the colony formation of both RKO
and SW620 cells in vitro. The number of colonies formed in
matrix gel was significantly lower in cells transfected with
pLV-GRP78 shRNA than cells transfected with pLV-control (P<0.05;
Fig. 1C). These results suggest
that GRP78 promotes the proliferation and colony formation of CRC
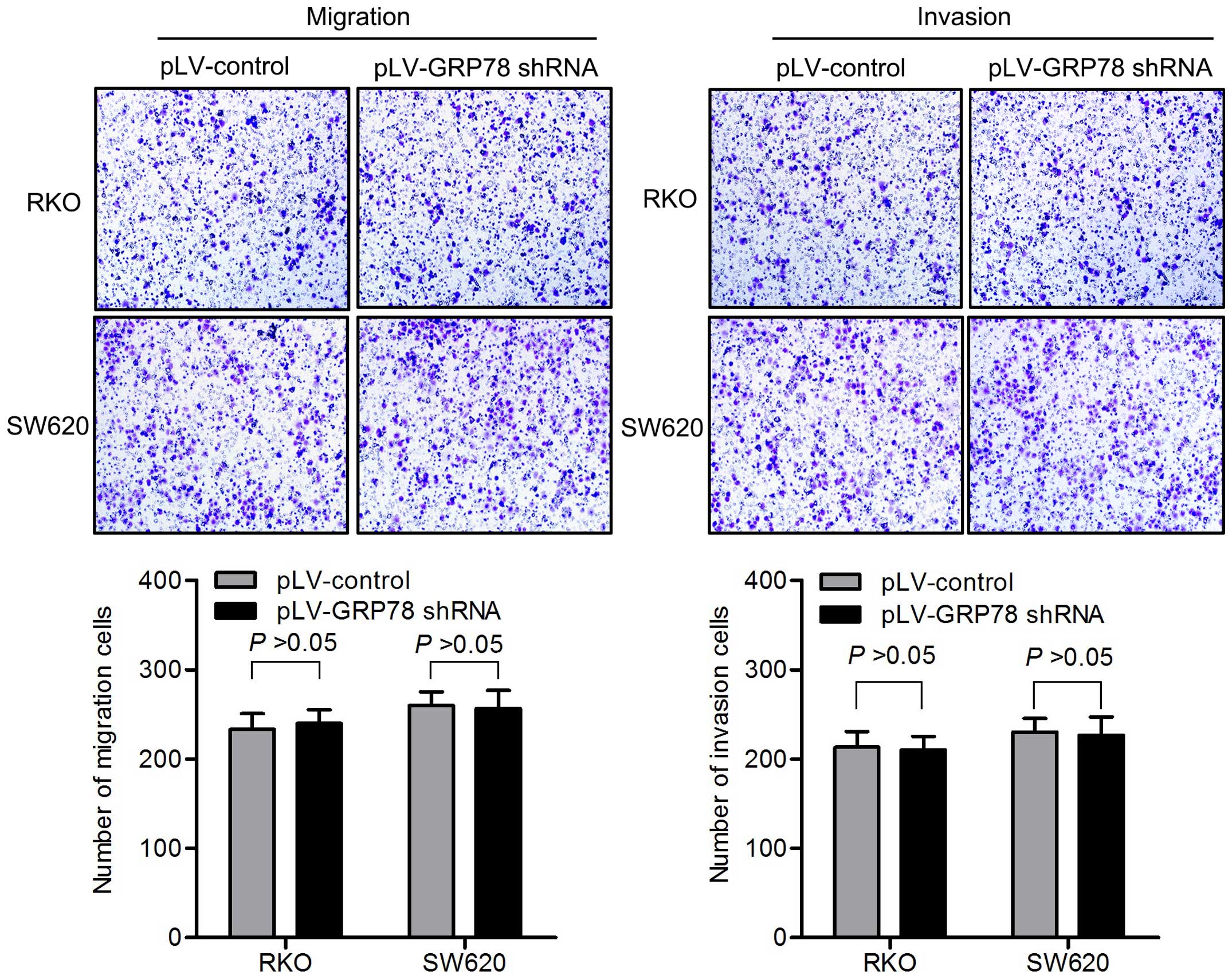
cells. The knockdown of GRP78 had no impact on migration and
invasion of CRC cells in vitro (Fig. 2).
Knockdown of GRP78 inhibits G1/S
transition and induces S phase arrest
To explore the mechanism underlying growth promotion
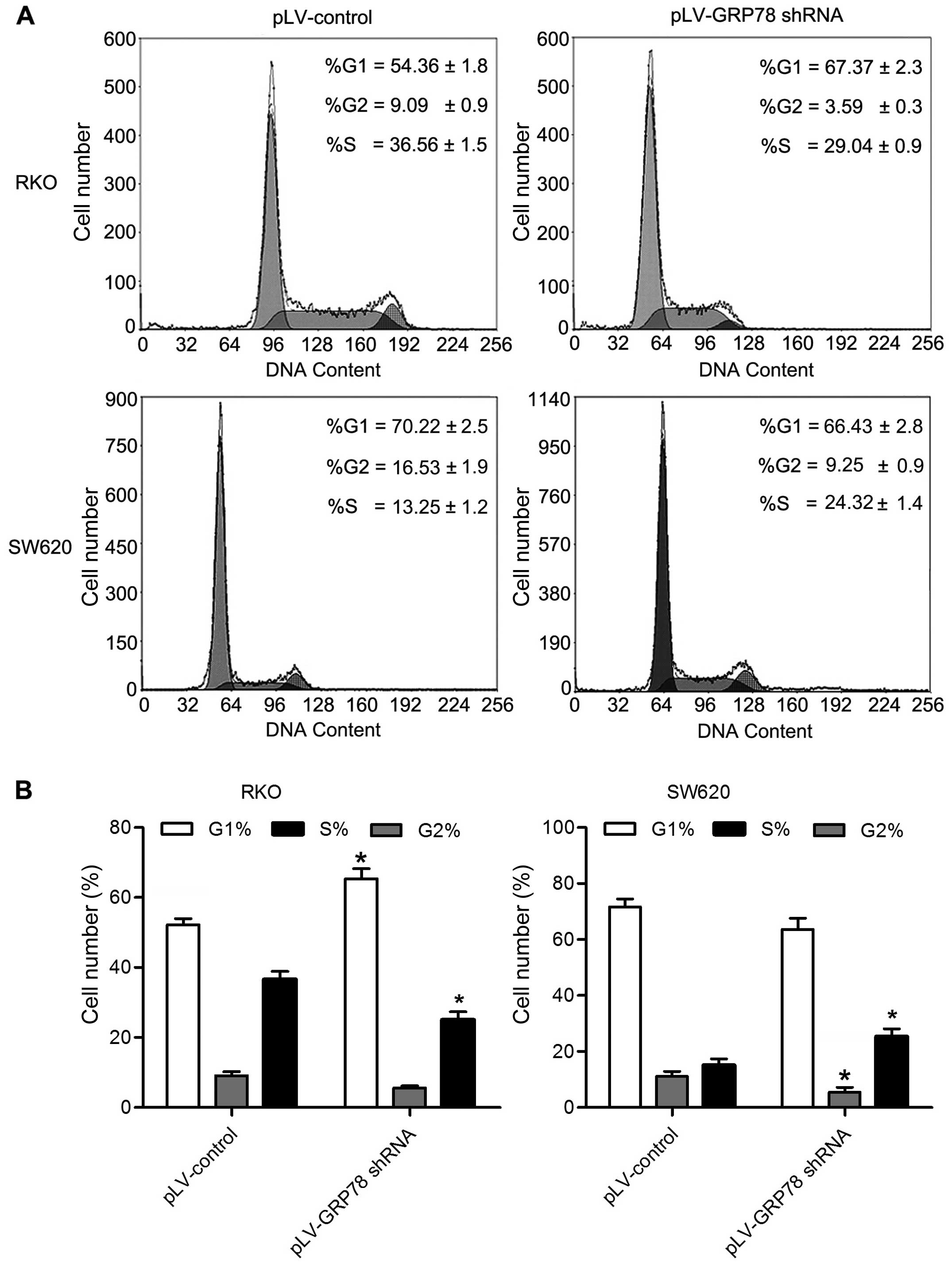
by GPR78, we determined the cell cycle of RKO and SW620 cells by
flow cytometry. As shown in Fig. 3,
the percentage of cells in G0/G1 phase was increased in RKO cells
transfected with pLV-GRP78 shRNA when compared to cells transfected
with pLV-control. In contrast, the percentage of cells in S and
G2/M phase was significantly decreased in RKO cells transfected
with pLV-GRP78 shRNA (P<0.05). Notably, the percentage of cells
in S phase was increased in SW620 cells transfected with pLV-GRP78
shRNA when compared to cells transfected with pLV-control, and G2/M
phase was significantly decreased in SW620 cells transfected with
pLV-GRP78 shRNA (P<0.05). These results suggest that knockdown
of GPR78 was able to inhibit G1/S transition and induce S phase
arrest, consistent with the finding that GRP78 promoted the
proliferation of CRC cells.
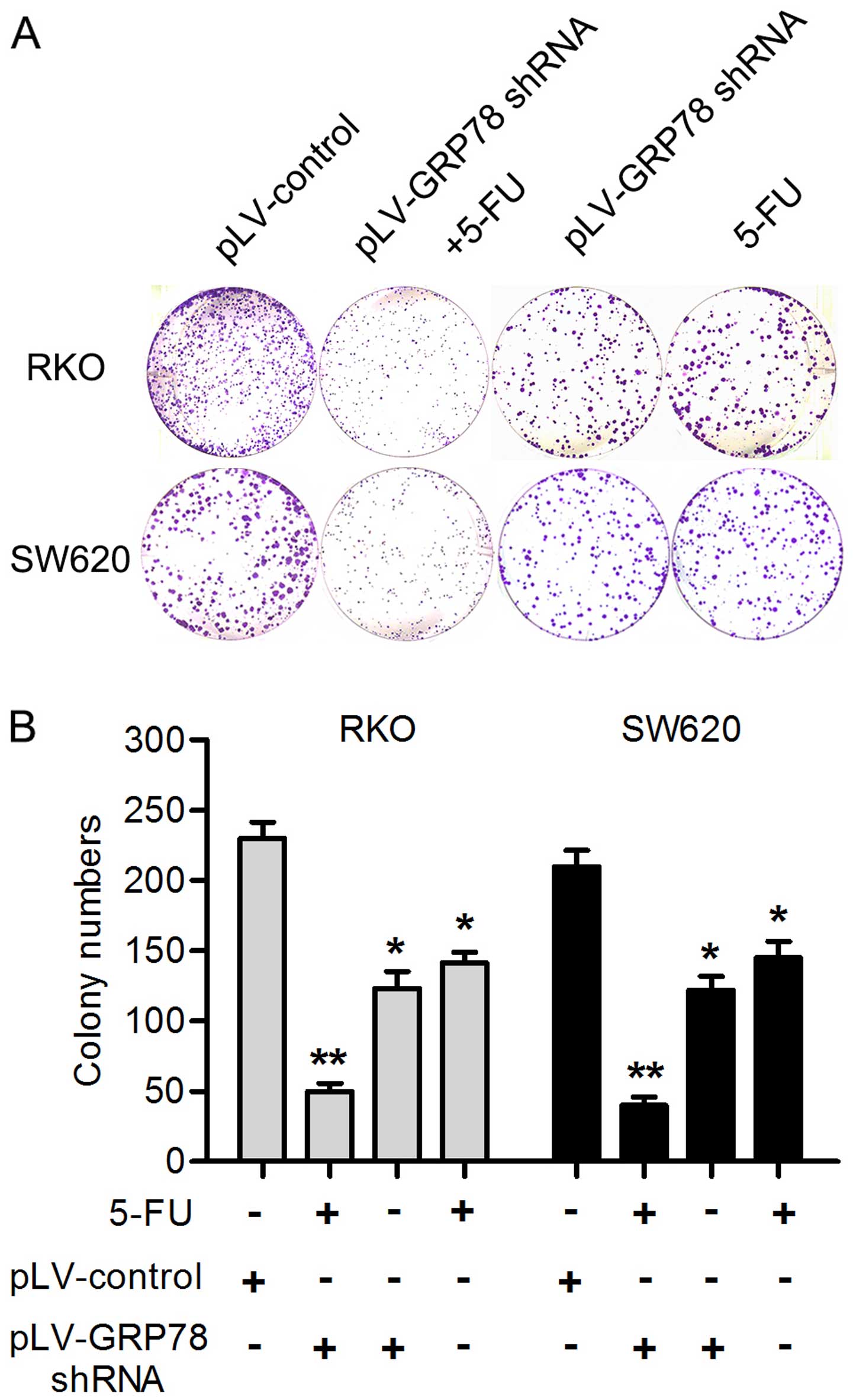
GRP78 knockdown enhances the chemotherapy
sensitivity induced by 5-FU
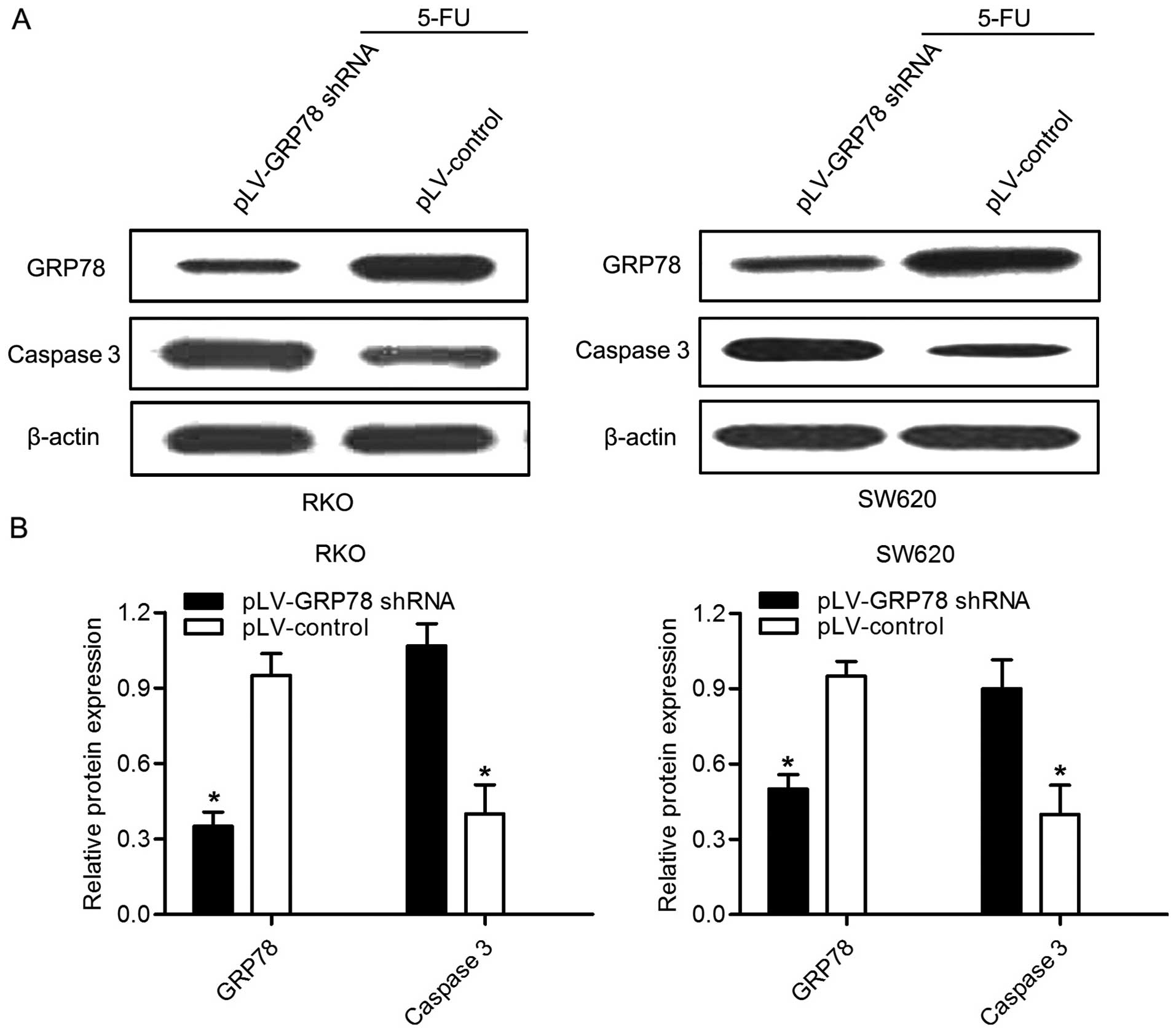
5-FU is an effective drug which is widely used in
the CRC chemotherapy. In order to evaluate whether GRP78 knockdown
had effect on the chemotherapy sensitivity of 5-FU, RKO and SW620
cells were treated with 20 µg/ml 5-FU for 24 h. Notably, as
shown in Fig. 4, the expression of
caspase 3 was significant increased in both cells after GRP78
knockdown. Given that caspase 3 plays a critical role in the
regulation of cell death, this result indicates that GRP78 may be
able to inhibit the death and apoptosis of CRC cells. In addition,
we also found that GRP78 knockdown significantly reduced the number
of RKO and SW620 cell colonies after the cells were treated with
5-FU for 24 h by colony formation assays (P<0.05; Fig. 5). This result suggest that GRP78 may
contribute to the therapeutic resistance to 5-FU in RKO and SW620
cells.
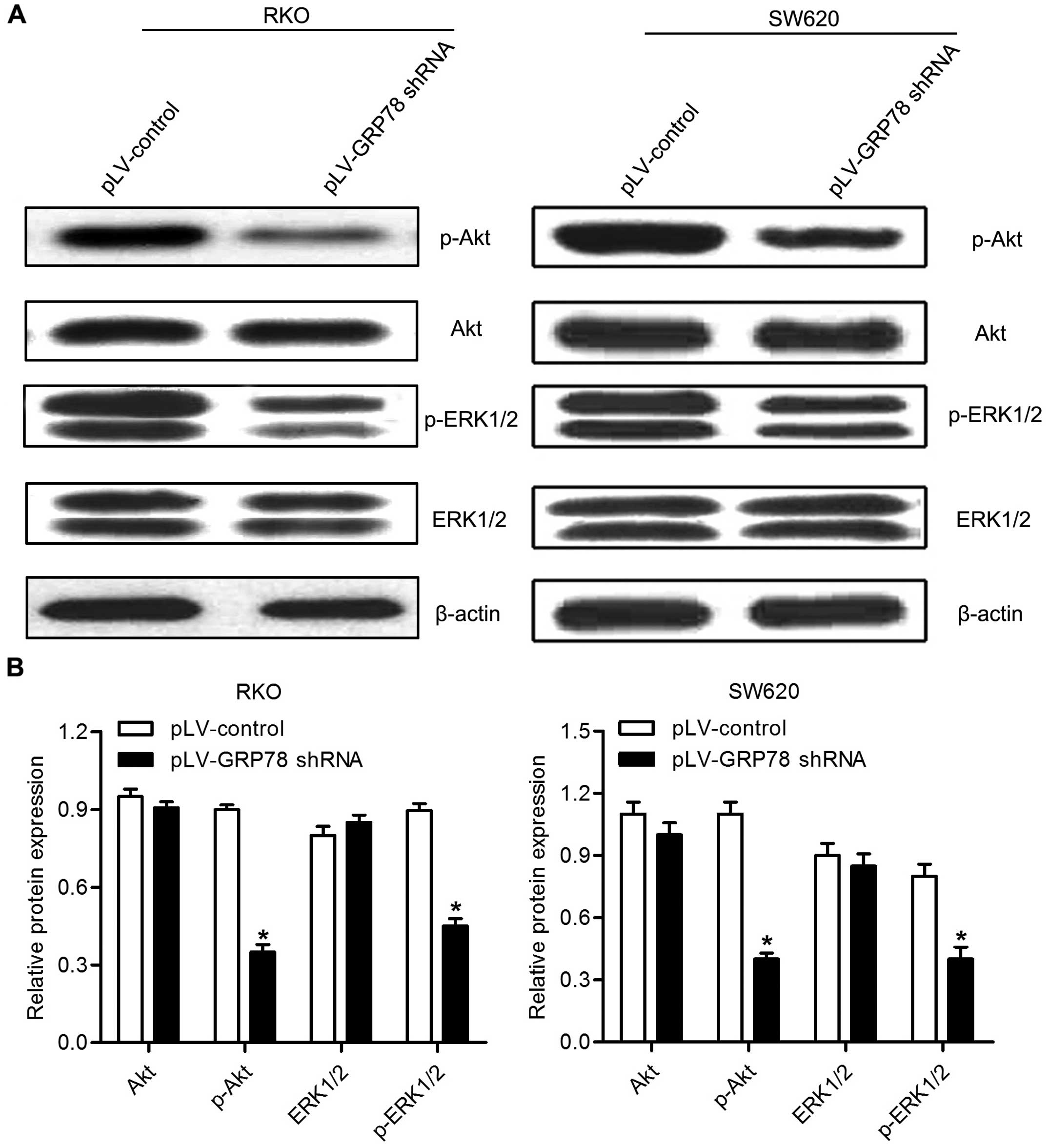
Effects of GRP78 knockdown on the
phosphorylation of AKT and ERK1/2
To reveal the potential molecular mechanism by which
GRP78 promotes the proliferation of CRC cells, we examined
phosphorylation of AKT and ERK1/2 by western blot analysis. As
shown in Fig. 6, we found that
GRP78 knockdown resulted in a downregulation of phosphorylation of
both AKT and ERK1/2 in RKO and SW620 cells. These results suggest
that GRP78 is involved in the regulation of phosphorylation of AKT
and ERK in CRC cells.
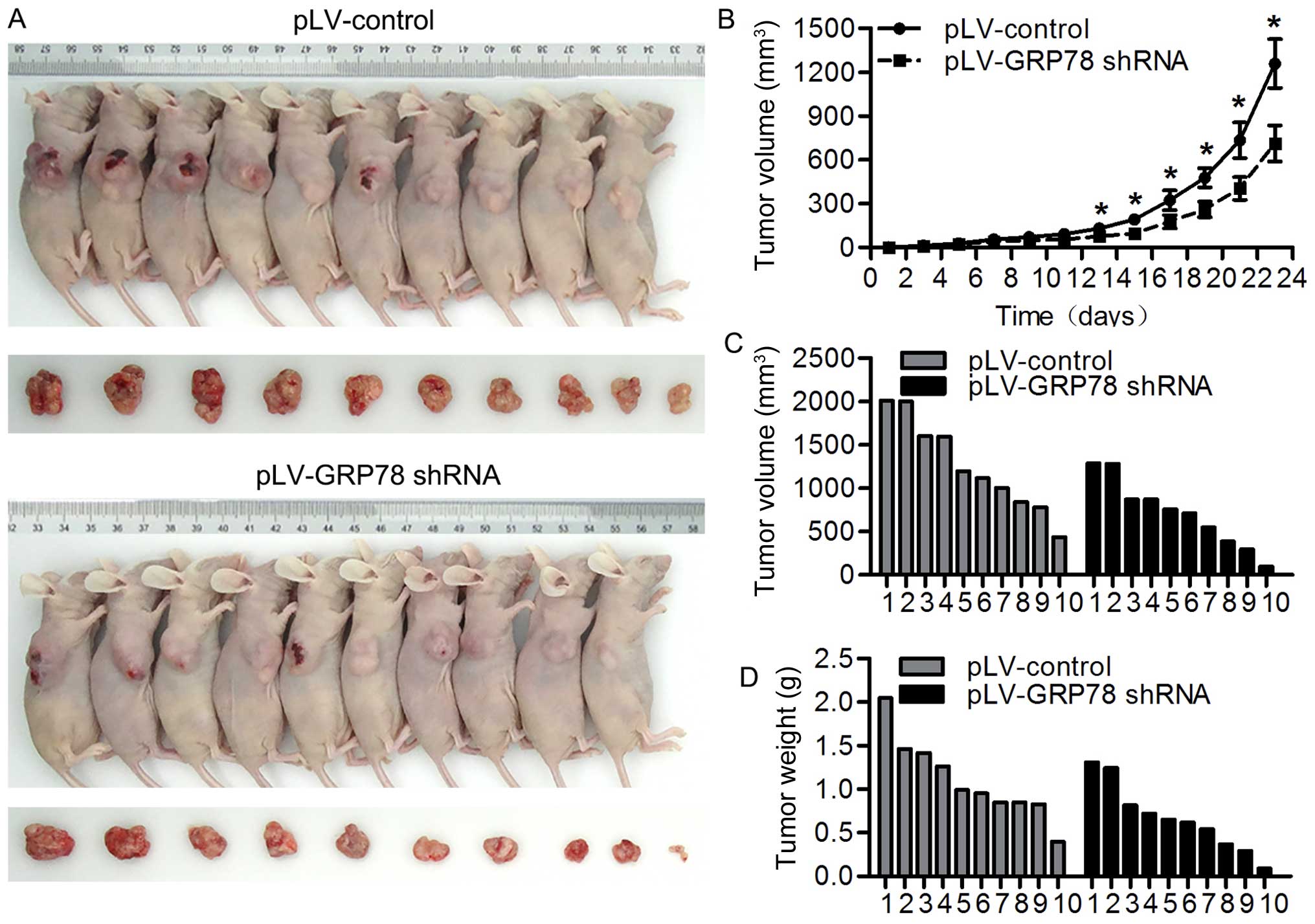
Inhibition of GRP78 suppresses tumor
growth in vivo
We observed that GRP78 inhibition suppresses the
proliferation of CRC cells in in vitro system, we wondered
whether GRP78 has a similar effect in in vivo model. To
perform this, we inoculated athymic BALB/c mice with RKO cells
stably transfected with pLV-GRP78 shRNA or pLV-control and
monitored tumor growth by measuring tumor volume every 3 days for 6
weeks. As shown in Fig. 7A–D, we
found that tumors grew at a slower rate with smaller sizes in mice
injected cells transfected with pLV-GRP78 shRNA than in those
transfected with pLV-control. These results indicate that
inhibition of GRP78 suppresses tumor growth in vivo,
confirming our finding from in vitro experiments.
Discussion
The microenvironment of a solid tumor is
characterized by glucose deprivation, acidosis and severe hypoxia,
all of which lead to the induction of GRP78 (25,26).
In the present study, we explored the potential pathways driven by
GRP78 in carcinogenesis of colorectal carcinoma (CRC).
Our previous study revealed that GRP78 regulates CRC
cell growth and apoptosis and may be an important indicator for
malignant transformation. In the present study, we found that
silencing GRP78 can suppress the proliferation of CRC cells through
inducing S phase arrest and regulating G1/S transition. These
findings indicated that GRP78 affects the proliferation of CRC
cells by regulating cell cycle progression and apoptosis. Notably,
previous study reported that downregulation of GRP78 inhibited CRC
cell growth and induced G1 cell cycle arrest by regulating G1/S
transition-related cyclins and CDK proteins (14). Therefore, the molecular mechanism
underlying the proliferation inhibition by GRP78 knockdown through
inducing S phase arrest in CRC cells need further investigation in
our further study.
Pathways which are driven by GRP78 in carcinogenesis
remain largely unclear. It has been shown that AKT-ERK signaling
pathway is critically involved in the regulation of proliferation
and survival of tumor cells in a variety of types of cancers
(21–23). Given that GRP78 regulates the
activity of AKT-ERK, we examined whether AKT and ERK signaling
pathways are involved in GRP78-mediated CRC cell proliferation. We
found that knockdown of GRP78 reduces the phosphorylation of AKT
and ERK. This result was consistent with the finding that deletion
of GRP78 can suppress the AKT activation and tumorigenesis
(24). Similar result was observed
in astrocytoma that GRP78 expression is responsible for the
promotion of cell proliferation by activating ERK1/2 and AKT
signaling pathways (13). We also
observed that GRP78 blockade differentially affected the
phosphorylation of AKT and ERK with a stronger inhibition to AKT
than ERK, which may be partially explained by the finding that cell
surface GRP78 is an upstream regulator of PI3K/AKT pathway, but not
MAPK signaling pathway, and directly forms a complex with the PI3K
subunit (27). The finding that AKT
blocks ERK signaling pathway through inhibition of Raf may also
contribute to the weak inhibition of GRP78 to phosphorylation of
ERK in RKO cells (22).
It is unclear how GRP78 regulates AKT-ERK signaling
pathway. Although primarily located in the ER lumen, GRP78 can
appear on tumor cell surface and ER stress upregulates GRP78
expression on the cell surface. It has been reported that CRC cells
(HT29, LOVO and SW480) have relatively high surface GRP78
expression (27,28). Since GRP78 shRNA can reduce both
intracellular and cell surface GRP78 expression, we speculated that
both intracellular and cell surface GRP78 are involved in
regulating AKT-ERK signaling pathway. Supporting this, studies have
found that cell surface GRP78 interacts with activated
α2 and cripto and promotes cell proliferation and
survival via ERK and AKT signaling pathways (28,29).
In addition, blockade of cell surface GRP78 using monoclonal
antibody mAb159 led to the reduction of AKT phosphorylation and the
inhibition of tumor growth both in vitro and in vivo
(27). It has been shown that AKT
and ERK signaling pathways are involved in the induction of GRP78
(30–32). However, we found that silencing
GRP78 attenuates the phosphorylation of AKT and ERK, suggesting a
positive feedback between GRP78 and AKT-ERK signaling pathway. In
addition, we also observed that GRP78 knockdown enhanced apoptosis
and reduce the number of cells colonies after the cells were
treated with 5-FU. A previous study also showed that GRP78
knockdown enhances apoptosis via the downregulation of oxidative
stress and Akt pathway after epirubicin treatment in colon cancer
DLD-1 cells (33). We speculate
that GRP78 can be considered as a novel predictor of responsiveness
to chemotherapy in CRC treatment. However, the precise molecular
mechanism remains to be investigated in our further study.
The present study findings should be elucidated with
consideration of its limitations. CRC patients usually also receive
radiotherapy if surgery is impossible. Radiotherapy can induce cell
apoptosis, helping to shrink the volume of tumors and improve the
survival rate of CRC patients. However, the present study does not
show whether GRP78 knockdown, along with radiation, would had a
better effect on inhibiting CRC cell proliferation and promoting
apoptosis. Thus, we could not precisely evaluate the potential
therapeutic role of GRP78 in the treatment of CRC patients. This
aspect should be explored in our future experiments.
In conclusion, our findings demonstrated that GRP78
contributes to the proliferation and tumorigenesis of CRC via AKT
and ERK pathways in vitro and in vivo. GRP78 plays an
important role in CRC development and progression. A better
understanding of the molecular mechanism of GPR78 in CRC
development and progression would provide novel therapeutic targets
for the treatment of CRC patients.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81201947,
81502388 and 81202142), the Natural Science Foundation of Shandong,
China (ZR2009CM014), and the Excellent Young Scientist Foundation
of Shandong Province, China (no. 2006BSB14001).
References
|
1
|
Luo YX, Chen DK, Song SX, Wang L and Wang
JP: Aberrant methylation of genes in stool samples as diagnostic
biomarkers for colorectal cancer or adenomas: A meta-analysis. Int
J Clin Pract. 65:1313–1320. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dashwood RH: Early detection and
prevention of colorectal cancer (Review). Oncol Rep. 6:277–281.
1999.PubMed/NCBI
|
|
4
|
Li J and Lee AS: Stress induction of
GRP78/BiP and its role in cancer. Curr Mol Med. 6:45–54. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Y, Liu R, Ni M, Gill P and Lee AS:
Cell surface relocalization of the endoplasmic reticulum chaperone
and unfolded protein response regulator GRP78/BiP. J Biol Chem.
285:15065–15075. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rao RV, Peel A, Logvinova A, del Rio G,
Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM and
Bredesen DE: Coupling endoplasmic reticulum stress to the cell
death program: Role of the ER chaperone GRP78. FEBS Lett.
514:122–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takada K, Hirose J, Yamabe S, Uehara Y and
Mizuta H: Endoplasmic reticulum stress mediates nitric
oxide-induced chondrocyte apoptosis. Biomed Rep. 1:315–319.
2013.
|
|
8
|
Pyrko P, Schönthal AH, Hofman FM, Chen TC
and Lee AS: The unfolded protein response regulator GRP78/BiP as a
novel target for increasing chemosensitivity in malignant gliomas.
Cancer Res. 67:9809–9816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song MS, Park YK, Lee JH and Park K:
Induction of glucose-regulated protein 78 by chronic hypoxia in
human gastric tumor cells through a protein kinase
C-epsilon/ERK/AP-1 signaling cascade. Cancer Res. 61:8322–8330.
2001.PubMed/NCBI
|
|
10
|
Daneshmand S, Quek ML, Lin E, Lee C, Cote
RJ, Hawes D, Cai J, Groshen S, Lieskovsky G, Skinner DG, et al:
Glucose-regulated protein GRP78 is up-regulated in prostate cancer
and correlates with recurrence and survival. Hum Pathol.
38:1547–1552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng HC, Takahashi H, Li XH, Hara T,
Masuda S, Guan YF and Takano Y: Overexpression of GRP78 and GRP94
are markers for aggressive behavior and poor prognosis in gastric
carcinomas. Hum Pathol. 39:1042–1049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baptista MZ, Sarian LO, Vassallo J, Pinto
GA, Soares FA and de Souza GA: Prognostic significance of GRP78
expression patterns in breast cancer patients receiving adjuvant
chemotherapy. Int J Biol Markers. 26:188–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang LH, Yang XL, Zhang X, Cheng JX and
Zhang W: Association of elevated GRP78 expression with increased
astrocytoma malignancy via Akt and ERK pathways. Brain Res.
1371:23–31. 2011. View Article : Google Scholar
|
|
14
|
Lin JA, Fang SU, Su CL, Hsiao CJ, Chang
CC, Lin YF and Cheng CW: Silencing glucose-regulated protein 78
induced renal cell carcinoma cell line G1 cell-cycle arrest and
resistance to conventional chemotherapy. Urol Oncol.
32:29.e1–29.e11. 2014. View Article : Google Scholar
|
|
15
|
Xing X, Li Y, Liu H, Wang L and Sun L:
Glucose regulated protein 78 (GRP78) is overexpressed in colorectal
carcinoma and regulates colorectal carcinoma cell growth and
apoptosis. Acta Histochem. 113:777–782. 2011. View Article : Google Scholar
|
|
16
|
Li Z, Zhang L, Zhao Y, Li H, Xiao H, Fu R,
Zhao C, Wu H and Li Z: Cell-surface GRP78 facilitates colorectal
cancer cell migration and invasion. Int J Biochem Cell Biol.
45:987–994. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roller C and Maddalo D: The molecular
chaperone GRP78/BiP in the development of chemoresistance:
Mechanism and possible treatment. Front Pharmacol. 4:102013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Misra UK, Deedwania R and Pizzo SV:
Binding of activated alpha2-macroglobulin to its cell surface
receptor GRP78 in 1-LN prostate cancer cells regulates
PAK-2-dependent activation of LIMK. J Biol Chem. 280:26278–26286.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen M, Zhang Y, Yu VC, Chong YS, Yoshioka
T and Ge R: Isthmin targets cell-surface GRP78 and triggers
apoptosis via induction of mitochondrial dysfunction. Cell Death
Differ. 21:797–810. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fu R, Yang P, Wu HL, Li ZW and Li ZY:
GRP78 secreted by colon cancer cells facilitates cell proliferation
via PI3K/Akt signaling. Asian Pac J Cancer Prev. 15:7245–7249.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar
|
|
22
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: A family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fu Y, Wey S, Wang M, Ye R, Liao CP,
Roy-Burman P and Lee AS: Pten null prostate tumorigenesis and AKT
activation are blocked by targeted knockout of ER chaperone
GRP78/BiP in prostate epithelium. Proc Natl Acad Sci USA.
105:19444–19449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Z and Li Z: Glucose regulated protein
78: A critical link between tumor microenvironment and cancer
hallmarks. Biochim Biophys Acta. 1826:13–22. 2012.PubMed/NCBI
|
|
26
|
Lee AS: GRP78 induction in cancer:
Therapeutic and prognostic implications. Cancer Res. 67:3496–3499.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra
SK, Krasnoperov V, Dong D, Liu S, Li D, et al: Monoclonal antibody
against cell surface GRP78 as a novel agent in suppressing PI3K/AKT
signaling, tumor growth, and metastasis. Clin Cancer Res.
19:6802–6811. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Misra UK, Deedwania R and Pizzo SV:
Activation and crosstalk between Akt, NF-kappaB, and unfolded
protein response signaling in 1-LN prostate cancer cells consequent
to ligation of cell surface-associated GRP78. J Biol Chem.
281:13694–13707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kelber JA, Panopoulos AD, Shani G, Booker
EC, Belmonte JC, Vale WW and Gray PC: Blockade of Cripto binding to
cell surface GRP78 inhibits oncogenic Cripto signaling via
MAPK/PI3K and Smad2/3 pathways. Oncogene. 28:2324–2336. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang LJ, Chen S, Wu P, Hu CS, Thorne RF,
Luo CM, Hersey P and Zhang XD: Inhibition of MEK blocks GRP78
up-regulation and enhances apoptosis induced by ER stress in
gastric cancer cells. Cancer Lett. 274:40–46. 2009. View Article : Google Scholar
|
|
31
|
Feng C, He K, Zhang C, Su S, Li B, Li Y,
Duan CY, Chen S, Chen R, Liu Y, et al: JNK contributes to the
tumorigenic potential of human cholangiocarcinoma cells through the
mTOR pathway regulated GRP78 induction. PLoS One. 9:e903882014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gray MJ, Mhawech-Fauceglia P, Yoo E, Yang
W, Wu E, Lee AS and Lin YG: AKT inhibition mitigates GRP78
(glucose-regulated protein) expression and contribution to
chemoresistance in endometrial cancers. Int J Cancer. 133:21–30.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chang YJ, Huang YP, Li ZL and Chen CH:
GRP78 knockdown enhances apoptosis via the down-regulation of
oxidative stress and Akt pathway after epirubicin treatment in
colon cancer DLD-1 cells. PLoS One. 7:e351232012. View Article : Google Scholar : PubMed/NCBI
|