Introduction
Helicobacter pylori is a spiral-shaped,
flagellated, micro-aerophilic Gram-negative bacillus first
described in 1982 by Marshall and Warren. H. pylori is
thought to colonize the gastric mucosa of >50% of the world's
population, with the higher prevalence in the developing countries
(1,2). However, the majority of H.
pylori-infected population is asymptomatic, but resulting in
chronic gastritis. Only 10% of infected individuals occurred
symptomatic diseases. Furthermore, experimental and epidemiological
studies have indicated that H. pylori infection indeed
increase the risk of gastric cancer. Based on this evidence, the
World Health Organization International Agency for Research on
Cancer classified H. pylori as class I carcinogen in 1994.
The estimated total of infection-attributable cancer is 1.9 million
cases every year, which is 17.8% of the global cancer burden. The
principal agent is H. pylori, ~63.4% of all stomach cancer
or 5.5% of the global cancer burden would be attributable to H.
pylori infection.
The interindividual differences in risk of H.
pylori-induced gastric diseases involve significant
heterogeneity of both host genetics and H. pylori strain
virulence factors. In the H. pylori-associated diseases
pathogenic mechanisms, several strain-specific virulence factors
were reported, such as cagA (cytotoxin-associated gene A),
vacA (vacuolating cytotoxin A), hpaA (Helicobacter
pylori adhesin A), babA (blood group antigen binding
adhesin), dupA (duodenal ulcer-promoting gene A),
iceA (induced by contact with epithelium) genes. One of the
main virulence factors is CagA, which is associated with higher
risk of gastric cancer and peptic ulcer. CagA protein can interact
with intercellular proteins and activate signaling pathways through
both tyrosine phosphorylation-dependent or independent mechanisms.
Here, we review the possible underlying pathogenic mechanisms of
the oncoprotein CagA in H. pylori-induced gastric
diseases.
Cytotoxin associated gene pathogenicity
island (cagPAI) and cytotoxin-associated gene A (cagA)
The cag PAI is an ~40-kb DNA, which likely was
acquired by horizontal gene transfer from another strain in the
course of evolution. The cag PAI contains ~31 genes including
cagA, cagB, cagC, cagL, cagM, cagI, cagY, which
encode the CagA protein and functional components of a type IV
secretion system (T4SS) (3,4). cag PAI is found in >95% East Asian
strains, whereas 60% of Western strains isolated are cag
PAI-positive (5). In some strains,
cagPAI is split into a right segment (cagI) and a left segment
(cagII) by an insertion sequence (IS605). IS605 was associated with
gastric cancer that was higher in H. pylori isolated from
patients with gastric carcinoma than in patients with duodenal
ulcer or chronic gastritis (6).
The cagA gene is ~3,500–5,000 bp located in
the beginning region of cag PAI, encoding 120–145 kDa CagA protein.
It is a recognized marker for the entire cag locus. The size of the
cagA gene and its protein varies in different strains due to
structural diversity in its C-terminal region. In Western
populations, cagA-positive strain is associated with
enhanced induction of gastritis, peptic ulcer, and higher risk of
gastric cancer. However, in East Asia cagA gene is not
associated with an increased risk of gastric diseases where almost
all strains are cagA-positive (7,8).
Franco et al constructed the Mongolian gerbils infection
model. They indicate that loss of CagA prevents the development of
cancer in this model (9). Ohnishi
et al provided first direct evidence for the role of CagA as
a bacterium-derived oncoprotein by transgenic mouse model (10).
Translocation and phosphorylation of CagA
protein
Translocation of CagA into host epithelial cells is
the first step in the processes of CagA-induced diseases. Several
different Cag proteins are involved in the translocation of CagA
(11). CagL carries a RGD
(arginine-glycine-aspartate) motif that is important for binding
and interaction with integrin α5β1 receptor on gastric epithelial
cells, and triggers CagA delivery into the target cells, as well as
downstream signaling to active tyrosine kinases including FAK, Src
and EGFR (12,13). CagM, along with CagX and CagT, forms
an outer membrane-associated T4SS subcomplex (14). CagX and CagT interact directly, the
C-terminal region of CagX is important for CagT interaction, and
CagT depends on CagX for its stabilization (15). CagE is one of the energy providing
components of CagA translocation. CagE is an inner membrane
associated active NTPase and has multiple interacting partners
including the inner membrane proteins CagV and Cagβ (16).
As reported, CagA also facilitates its translocation
into host epithelial cells by T4SS-induced externalization of
phosphatidylserine from inner leaflet of the plasma membrane. The
protein binds to phosphatidylserine via Lys-Xn-Arg-X-Arg
(K-Xn-R-X-R) motif present in the central region of CagA. The 2
arginine residues in K-Xn-R-X-R motif are highly conserved among
CagA proteins derived from H. pylori strains (13,17).
It has previously been reported that C-terminal CagA secretion
signal and N-terminal CagA domain (D1) are crucial for efficient
translocation (18). Collectively,
these findings indicate that all components of this type IV
secretion system, including the effector protein CagA, are encoded
on the cag pathogenicity island.
Once the protein has entered these target cells,
CagA localizes to the inner surface of the cellular membrane, once
again by the interaction between the K-Xn-R-X-R motif with
phosphatidylserine (13). Then
parts of CagA proteins undergo tyrosine phosphorylation at the
C-terminal Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs by Src family kinases
and Abl kinase, while other CagA molecules remain unphosphorylated
(19–21).
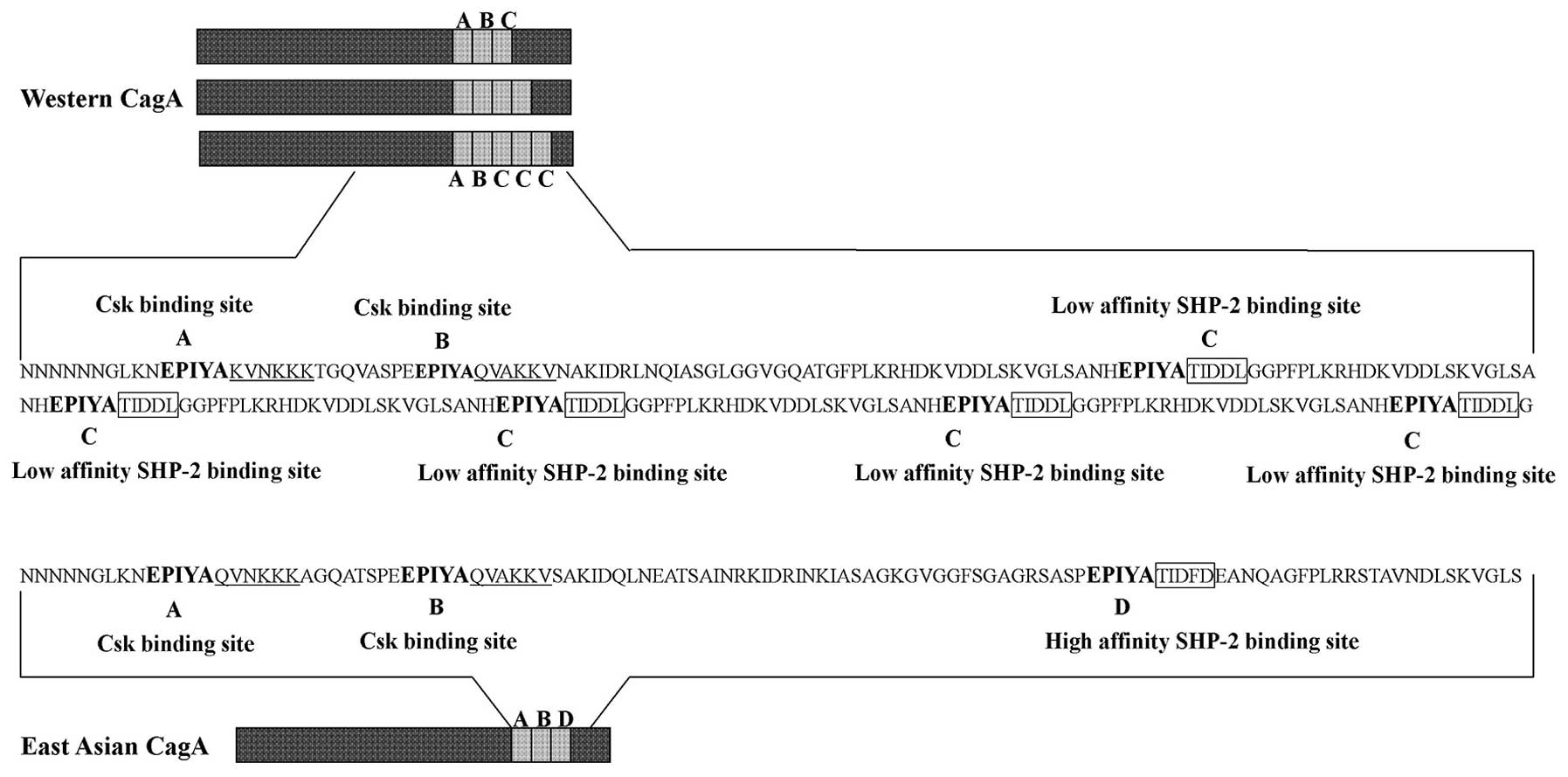
CagA can be tyrosine phosphorylated at EPIYA motifs,
which is found as part of repetitive region in the C-terminal of
CagA. Based on the amino acid sequences surrounding each of the
EPIYA motifs, four distinct EPIYA motifs (EPIYA-A, -B, -C, -D) have
been classified. EPIYA-A and EPIYA-B are present throughout the
world, EPIYA-C is found in strains isolated from Western countries.
In contrast, East Asian strains carry East Asian CagA, which
contains EPIYA-D (22,23) (Fig.
1). Through database searching and in silico analysis,
Zhang et al revealed a strong non-random distribution of the
EPIYA-B motif polymorphisms (including EPIYT and EPIYA) in Western
H. pylori isolates, and provide evidence that the EPIYT are
significantly less associated with gastric cancer than the EPIYA.
CagA B-motif phosphorylation status is essential for its
interaction with host PI3-kinase during colonization and that CagA
with an EPIYT B-motif had significantly attenuated induction of
interleukin-8 and the hummingbird phenotype, had higher affinity
with PI3-kinase, and enhanced induction of AKT compared to the
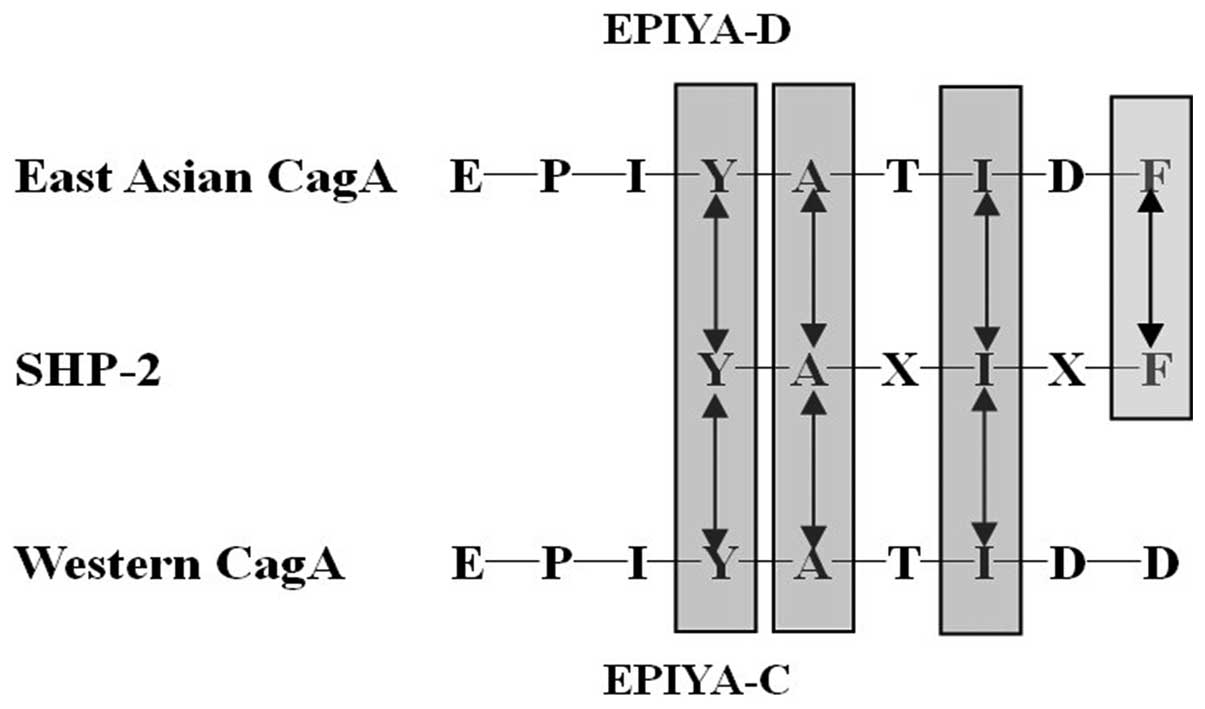
EPIYA (24). It was reported that
the two SH2 domains from SHP-2 (Src homology 2-containing protein
tyrosine phosphatase-2) bind to highly related sequences
pY-(S/T/A/V/I)-X-(V/I/L)-X-(W/F). Intriguingly, the consensus motif
perfectly matches the SHP-2-binding site of EPIYA-D (pY-A-T-I-D-F).
Furthermore, EPIYA-C (pY-A-T-I-D-D) replacement of the pY + 5
position in Western CagA, reduces the binding affinity to SHP-2.
This East Asian-specific sequence conferred stronger SHP-2 binding
and morphologically transforming activities to Western CagA. So the
East Asian CagA or larger numbers of EPIYA-C in Western CagA was
associated with atrophic gastritis and increased the risk of
gastric cancer. Several recent reports have demonstrated that the
ABCCC type can induce the intestinal metaplasia, IL-8, perturbation
of Crk adaptor proteins, anti-apoptotic effect and carcinogenic
effect more significantly than ABC type (25–28)
(Fig. 2).
CagA phosphorylation-dependent or
independent effects
Perturb host cell functions by CagA
phosphorylation-dependent effects
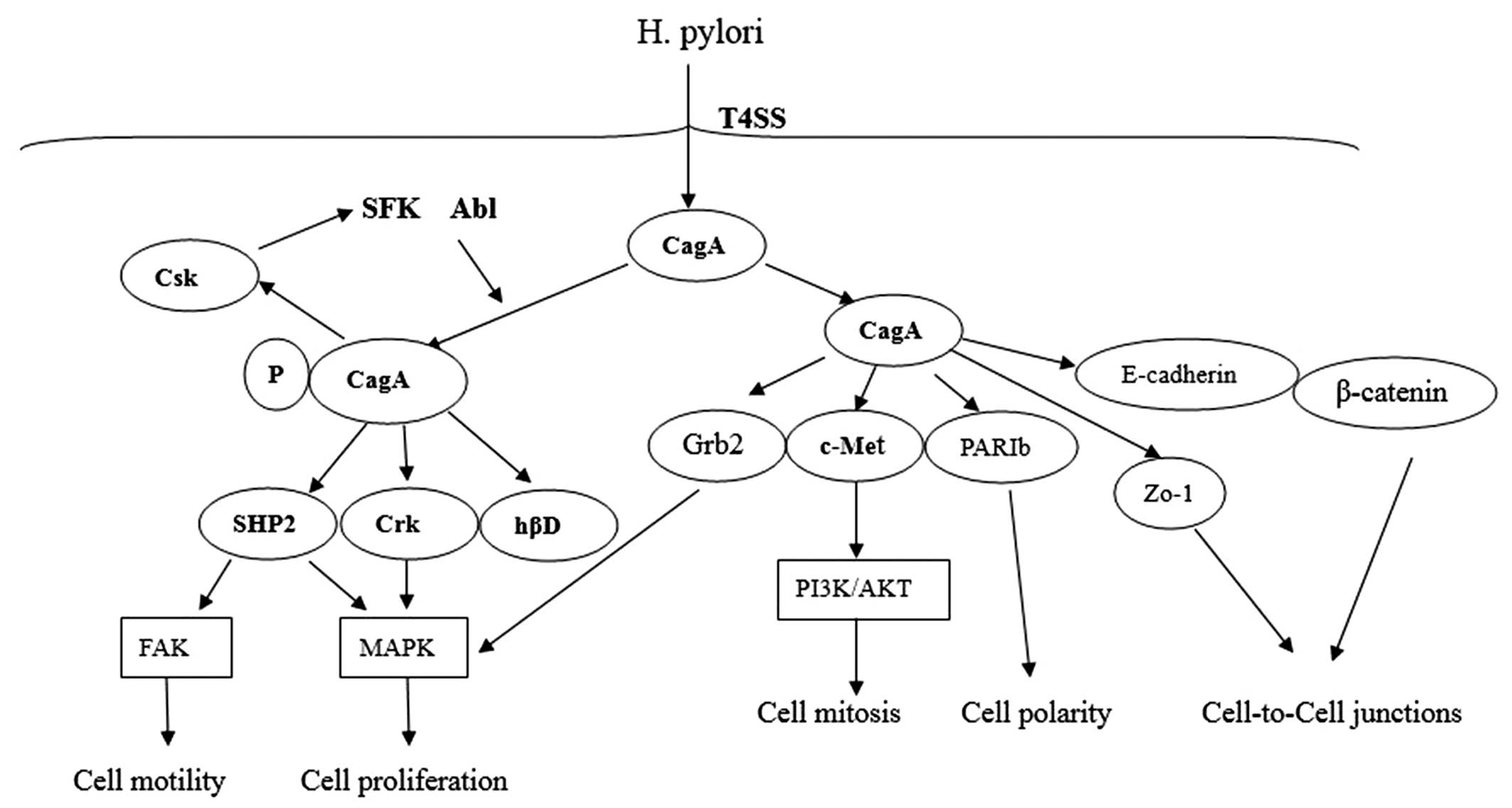
Once within the host cells, CagA undergoes tyrosine
phosphorylation at EPIYA motifs by Src family kinases (SFKs) and
Abl kinase. Along with the report that c-Src only phosphorylated
EPIYA-C or EPIYA-D, whereas c-Abl phosphorylated all EPIYA motifs.
CagA proteins were phosphorylated on 1 or 2 EPIYA motifs, but never
simultaneously on 3 motifs. Furthermore, none of the phosphorylated
EPIYA motifs alone was sufficient for deregulation of cell growth
and motility. Western CagA EPIYA-A and EPIYA-C were preferred
combination phosphorylation, either across two CagA molecules or
simultaneously on one (21,29).
CagA proteins were probably from a dimer in host
cell by a 16-amino-acid CagA multimerization (CM) sequence, which
is located downstream of the EPIYA-C motif in C-terminal region
(30). After phosphorylated CagA
(pCagA) from a homodimer through the CM sequence, the pCagA dimer
can bind with a single SHP-2 molecule via its two homologous SH2
domains (31). The pCagA-SHP-2
complex triggers the phosphatase activity of SHP-2, which in return
causes the dephosphorylation of focal adhesion kinase (FAK) and
activation of Ras/MAPK/ERK signaling pathway (32–34).
In addition to SHP-2, the phosphorylated EPIYA-A, -B
can specifically bind to the C-terminal Src kinase (Csk) and
activate Csk, and then the inhibitory tyrosine residues of SFKs are
phosphorylated by Csk. So Csk is characterized as a negative
regulator of SFKs, which results in reduced EPIYA phosphorylation
of CagA. Therefore, the CagA-Csk interaction may establish a
negative feedback mechanism that prevents damage to the host cells
from H. pylori. This could be harmful to the long-term
colonization of H. pylori in stomach (35).
CagA can associate with Crk adaptor proteins (Crk-I,
Crk-II, Crk-L) EPIYA-phosphorylation-dependently. CagA-Crk
interaction plays a critical role in promoting cell scattering by
inducing several downstream signaling pathways, such as
SoS1/H-Ras-Raf-MEK and C3G-Rap1/B-Raf-MEK (36). The human β-defensins (hβDs) are
antimicrobial peptides that are highly active against H.
pylori during early infection via EGFR-dependent activation of
MAP kinase and JAK/STAT signaling pathways. However, during
prolonged infection, hβD1 and hβD3 is subsequently downregulated by
phosphorylated CagA (37,38) (Fig.
3).
Disruption of epithelial cells by CagA
phosphorylation-independent effects
CagA also exerts numerous effects within host cells
in a tyrosine phosphorylation-independent manner. A CRPIA
(conserved repeat responsible for phosphorylation-independent
activity) motif, FPLKRHDKVDDLSKVG, in the C-terminal region of
CagA, which is distinct from the EPIYA motifs used for
phosphorylation. The CRPIA motif in non-phosphorylated CagA was
involved in interacting with the hepatocyte growth factor scatter
factor receptor c-Met, which is involved in invasive growth of
tumor cells. CagA binds c-Met and could represent an adaptor
protein, which associates with phospholipase Cγ (PLCγ) and
activates phosphatidylinositol 3-kinase/Akt signaling. This in turn
led to the activation of β-catenin and NF-κB signaling, which
promotes proliferation and inflammation (39,40).
CagA can interact with Grb2, which results in the
activation of the Ras/MEK/ERK pathway and leads to cell scattering
as well as proliferation. This ability of CagA is independent from
the tyrosine phosphorylation. However, the EPIYA sequences are
indispensable for the Grb2 binding and induction of the cellular
responses (41).
Moreover, CagA associates with the epithelial
tight-junction scaffolding protein ZO-1 and the transmembrane
protein JAM (junctional adhesion molecule), causing an ectopic
assembly of tight-junction components at sites of bacterial
attachment, and altering the composition and function of the
apical-junctional complex (42).
CagA physically interacts with E-cadherin
independently of EPIYA motif phosphorylation. The CagA/E-cadherin
interaction impairs the E-cadherin/β-catenin complex, causing
cytoplasmic and nuclear accumulation of β-catenin (43). Then it leads to nuclear
translocation of free β-catenin, where it binds to the
transcriptional cofactors of the Wnt pathway and upregulates the
transcription of targeted genes such as axin, cyclin and myc
(44). H. pylori alters the
E-cadherin/β-catenin complex, leading to formation of a
multiproteic complex composed of CagA, c-Met, E-cadherin, and
p120-catenin. This complex abrogates c-Met and p120-catenin
tyrosine phosphorylation and suppresses the cell-invasive phenotype
induced by H. pylori (45).
CagA deregulation of β-catenin requires residues 1,009–1,086 and
residues 908–1,012 of Western CagA and East Asian CagA,
respectively, and is mediated by the CM motif of CagA (46).
CagA also disrupts the tight junction and causes
loss of apical-basolateral polarity by interaction with PAR1/MARK
kinase, which is a central regulator of cell polarity. Association
of CagA inhibits PAR1 kinase activity and prevents atypical protein
kinase C (aPKC)-mediated PAR1 phosphorylation. The PAR-aPKC system
is the molecular machinery that converts initial polarity cues in
the establishment of complementary membrane domains along the
polarity axis. CagA-PAR1 complex dissociates PAR1 from the
membrane, collectively causing junctional and polarity defects. The
PAR1 also promotes CagA multimerization, which stabilizes the
CagA-SHP2 interaction (47–49). This interaction is also dependent on
the CM motif of CagA. The CM sequence of CagA isolated from East
Asian H. pylori binds PAR1b more strongly than that of CagA
isolated from Western H. pylori. Within Western CagA
species, the ability to bind PAR1b is proportional to the number of
CM sequences (50).
Lu et al found that CagA binds not only PAR1b
but also other PAR1 isoforms, with order of strength as follows:
PAR1b > PAR1d > or = PAR1a > PAR1c. They also indicate
that malfunctioning of microtubules and myosin II by CagA-mediated
PAR1 inhibition cooperates with deregulated SHP-2 in the
morphogenetic activity of CagA (51). In MDCK tissue culture model,
association of CagA with MARK2 not only causes disruption of apical
junctions, but also inhibition of tubulogenesis and cell
differentiation (52). Furthermore
inhibition of PAR1b kinase activity contributed to an increased
hummingbird phenotype. CagA-mediated inhibition of PAR1b and then
prevented PAR1b mediated phosphorylation, which inactivates a
RhoA-specific GEF, GEF-H1 and thereby strengthens the hummingbird
phenotype induced by CagA-stimulated SHP2 (53). Moreover, the interaction of CagA
with PAR1 is associated with reduction of an inhibitor NF-κB,
called IκB kinase, which regulates microtubule stability by
phosphorylating microtubule-associated proteins (MAPs). Since
microtubule destabilization leads to the activation of NF-κB by
promoting IκBα degradation, impairment of the microtubule system by
CagA-PAR1 interaction may give a cytoskeletal cue that stimulates
IκB degradation (54).
Additionally, CagA was described to affect activity of protein
kinase C-related kinase 2 (PRK2), which acts downstream of Rho
GTPases and is known to affect cytoskeletal rearrangements and cell
polarity (55) (Fig. 3).
CagA destroys the host cells via epigenetic
modifications
Epigenetics may be defined as the mechanisms that
initiate and maintain heritable patterns of gene function and
regulation in a heritable manner without affecting the sequence of
the genome. Epigenetic modifications include DNA methylation,
post-translational modifications of histone proteins, microRNA
expression, genomic imprinting and chromatin remodeling (56,57)
(Fig. 4).
DNA methylation
DNA methylation is an important epigenetic
modification involved in the regulation of numerous biological
processes. In mammals, DNA methylation mainly occurs in the context
of cytosine-phosphate-guanine (CpG) dinucleotides (58). H. pylori infection potently
induces methylation of CpG islands (CGIs) to various degrees.
Methylation levels of specific CGIs seemed to reflect gastric
cancer risk in H. pylori-negative individuals (OR 95% CI
2.2–32). The promoter CpG islands of FLNc, HAND1, THBD, p41ARC,
HRASLS and LOX gene were reportedly altered by H.
pylori infection (59–62). In vitro experiments showed
significant CagA aberrant epigenetic silencing of let-7 expression
leading to Ras upregulation by histone and DNA methylation
(63). A significantly increased
risk of RUNX3 methylation (OR, 4.28; 95% CI, 1.19–15.49) was
observed with a high consumption of nuts in patients with
CagA-positive H. pylori infection (64).
MicroRNAs
miRNAs are small, non-coding RNAs, which regulate
gene expression in a sequence-specific manner. miRNAs have been
implicated in the etiology, progression and prognosis of diseases,
and many studies have shown that profiles of miRNA expression
differ between infection and non-infection with H. pylori
(65,66). miRNAs are involved in H.
pylori-related pathology via the regulation of the
transcription and expression of various genes, playing an important
role in inflammation, cell proliferation, apoptosis and
differentiation (67,68).
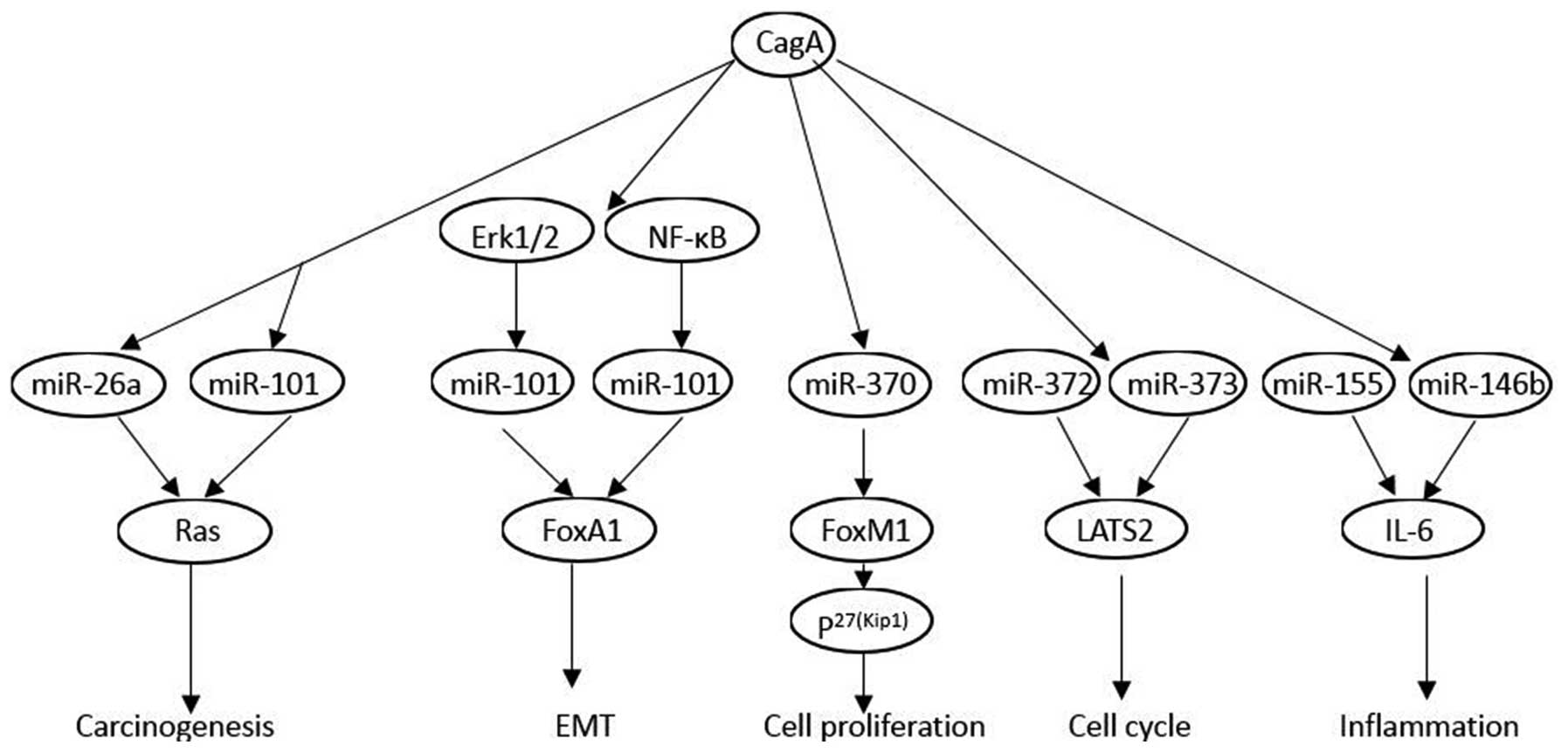
CagA may be involved in cellular regulation of
certain miRNAs in the gastric epithelium. miRNA expression patterns
in H. pylori-infected gastric mucosa are determined by
microarray. The results found that expression levels of let-7
family miRNAs are significantly altered following infection with
CagA positive strain (69). CagA
enhanced c-myc, DNA methyltransferase 3B (DNMT3B) and enhancer of
zeste homologue 2 (EZH2) expression and attenuated miR-26a and
miR-101 expression, which resulted in the attenuation of let-7
expression (63). Using mammalian
miRNA profile microarrays, miR-1290 and miR-584 expressions are
upregulated in CagA-transformed cells. miR-1290 is upregulated in
an Erk1/2-dependent manner, and miR-584 is activated by NF-κB.
Foxa1 is an important target of miR-584 and miR-1290, which promote
the epithelial-mesenchymal transition significantly (70). In vitro, CagA inhibited
miR-370 expression, which led to overexpression of FoxM1. The
upregulated FoxM1 expression altered the expression of p27 (Kip1),
and promoted proliferation in gastric cells (71). CagA may function as an initiator in
the process of carcinogenesis by upregulating miR-222, which
further participates in the progression of cancer by promoting
proliferation and inhibiting RECK (reversion-inducing cysteine-rich
protein with Kazal motifs) (72).
Shortly after H. pylori infection, miR-372 and miR-373
synthesis is highly inhibited, leading to the post-transcriptional
release of LATS2 expression and thus, to a cell cycle arrest at the
G1/S transition. This downregulation of a specific cell
cycle-regulating microRNA is dependent on the translocation of CagA
into the host cells (73). The
integrative analysis and immunohistochemistry staining validation
indicated that miR-155 and miR-146b are upregulated in H.
pylori-positive gastroduodenal ulcer. Further experiments in
gastric epithelial cells revealed that CagA mediated upregulation
of miR-155 and miR-146b, which decreases IL6 overexpression
(74).
Histone modifications
Nucleosomes are represented by DNA wrapped around
eight histone proteins, H2A, H2B, H3, and H4. Histones are primary
protein components of eukaryotic chromatin and play a role in gene
regulation. A histone modification is a covalent post-translational
modification (PTM) to histone proteins which include methylation,
phosphorylation, acetylation, ubiquitylation, and sumoylation
(75,76).
Substantial research has investigated the effects of
H. pylori infection on histone modification. Chromatin
immunoprecipitation analysis of NCI-N87 and primary gastric cells
revealed that H. pylori induce cell cycle control factor
p21WAF1 overexpression. H. pylori is associated
with hyperacetylation of histone H4 which can release HDAC-1 from
the p21WAF1 promoter (77). cagPAI-dependent decreases of H3
phosphorylation levels at serine 10 (pH3Ser10) and threonine 3
(pH3Thr3) are observed. H. pylori causes a strong decrease
of the cell division cycle 25 (CDC25C) phosphatase (78). Liang et al demonstrated that
RBP2, a newly identified H3K4 demethylase, can be induced by CagA
via PI3K/AKT-Sp1 pathway depending on AKT phosphorylation.
Furthermore, the novel CagA-PI3K/AKT-Sp1-RBP2-Cyclin D1 pathway
links chronic inflammation to tumor during GC development (79).
In conclusion, the CagA protein is encoded by
cag PAI and delivered into the host cells during the T4SS
system. Following translocation, Src and Abl kinases phosphorylate
CagA on EPIYA motifs. Phosphorylated CagA can interact with SHP2,
Csk, Crk and hβD, which trigger several intracellular signaling
pathways resulting in epithelial cell gene expression. The
unphosphorylated CagA directly interacts with certain intracellular
proteins such as PARIb, E-cadherin/β-catenin, c-Met, Grb2 and ZO-1,
then disrupts the cell-to-cell junctions and gastric epithelial
cell polarity.
References
|
1
|
Pacchiani N, Censini S, Buti L and Covacci
A: Echoes of a distant past: The cag pathogenicity island of
Helicobacter pylori. Cold Spring Harb Perspect Med. 3:a0103552013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cavaleiro-Pinto M, Peleteiro B, Lunet N
and Barros H: Helicobacter pylori infection and gastric cardia
cancer: Systematic review and meta-analysis. Cancer Causes Control.
22:375–387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Akopyants NS, Clifton SW, Kersulyte D,
Crabtree JE, Youree BE, Reece CA, Bukanov NO, Drazek ES, Roe BA and
Berg DE: Analyses of the cag pathogenicity island of Helicobacter
pylori. Mol Microbiol. 28:37–53. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Censini S, Lange C, Xiang Z, Crabtree JE,
Ghiara P, Borodovsky M, Rappuoli R and Covacci A: cag, a
pathogenicity island of Helicobacter pylori, encodes type
I-specific and disease-associated virulence factors. Proc Natl Acad
Sci USA. 93:14648–14653. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamaoka Y: Helicobacter pylori typing as a
tool for tracking human migration. Clin Microbiol Infect.
15:829–834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lai CH, Perng CL, Lan KH and Lin HJ:
Association of IS605 and cag-PAI of Helicobacter pylori isolated
from patients with gastrointestinal diseases in Taiwan.
Gastroenterol Res Pract. 2013:3562172013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Torres J, Perez-Perez GI, Leal-Herrera Y
and Munoz O: Infection with CagA+ Helicobacter pylori
strains as a possible predictor of risk in the development of
gastric adenocarcinoma in Mexico. Int J Cancer. 78:298–300. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Batista SA, Rocha GA, Rocha AM, Saraiva
IE, Cabral MM, Oliveira RC and Queiroz DM: Higher number of
Helicobacter pylori CagA EPIYA C phosphorylation sites increases
the risk of gastric cancer, but not duodenal ulcer. BMC Microbiol.
11:612011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Franco AT, Johnston E, Krishna U, Yamaoka
Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P and
Peek RM Jr: Regulation of gastric carcinogenesis by Helicobacter
pylori virulence factors. Cancer Res. 68:379–387. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohnishi N, Yuasa H, Tanaka S, Sawa H,
Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, et
al: Transgenic expression of Helicobacter pylori CagA induces
gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl
Acad Sci USA. 105:1003–1008. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Backert S, Tegtmeyer N and Fischer W:
Composition, structure and function of the Helicobacter pylori cag
pathogenicity island encoded type IV secretion system. Future
Microbiol. 10:955–965. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tegtmeyer N, Hartig R, Delahay RM, Rohde
M, Brandt S, Conradi J, Takahashi S, Smolka AJ, Sewald N and
Backert S: A small fibronectin-mimicking protein from bacteria
induces cell spreading and focal adhesion formation. J Biol Chem.
285:23515–23526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murata-Kamiya N, Kikuchi K, Hayashi T,
Higashi H and Hatakeyama M: Helicobacter pylori exploits host
membrane phosphatidylserine for delivery, localization, and
pathophysiological action of the CagA oncoprotein. Cell Host
Microbe. 7:399–411. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fischer W: Assembly and molecular mode of
action of the Helicobacter pylori Cag type IV secretion apparatus.
FEBS J. 278:1203–1212. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gopal GJ, Pal J, Kumar A and Mukhopadhyay
G: C-terminal domain of CagX is responsible for its interaction
with CagT protein of Helicobacter pylori type IV secretion system.
Biochem Biophys Res Commun. 456:98–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shariq M, Kumar N, Kumari R, Kumar A,
Subbarao N and Mukhopadhyay G: Biochemical analysis of CagE: A
VirB4 homologue of Helicobacter pylori Cag-T4SS. PLoS One.
10:e01426062015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Odenbreit S, Püls J, Sedlmaier B, Gerland
E, Fischer W and Haas R: Translocation of Helicobacter pylori CagA
into gastric epithelial cells by type IV secretion. Science.
287:1497–1500. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schindele F, Weiss E, Haas R and Fischer
W: Quantitative analysis of CagA type IV secretion by Helicobacter
pylori reveals substrate recognition and translocation
requirements. Mol Microbiol. 100:188–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stein M, Bagnoli F, Halenbeck R, Rappuoli
R, Fantl WJ and Covacci A: c-Src/Lyn kinases activate Helicobacter
pylori CagA through tyrosine phosphorylation of the EPIYA motifs.
Mol Microbiol. 43:971–980. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tegtmeyer N and Backert S: Role of Abl and
Src family kinases in actin-cytoskeletal rearrangements induced by
the Helicobacter pylori CagA protein. Eur J Cell Biol. 90:880–890.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tammer I, Brandt S, Hartig R, König W and
Backert S: Activation of Abl by Helicobacter pylori: A novel kinase
for CagA and crucial mediator of host cell scattering.
Gastroenterology. 132:1309–1319. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu Y-L, Zheng S, Du Q, Qian K-D and Fang
P-C: Characterization of CagA variable region of Helicobacter
pylori isolates from Chinese patients. World J Gastroenterol.
11:880–884. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vaziri F, Peerayeh SN, Alebouyeh M,
Maghsoudi N, Azimzadeh P, Siadat SD and Zali MR: Novel effects of
Helicobacter pylori CagA on key genes of gastric cancer signal
transduction: A comparative transfection study. Pathog Dis.
73:ftu0212015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang XS, Tegtmeyer N, Traube L, Jindal S,
Perez-Perez G, Sticht H, Backert S and Blaser MJ: A specific A/T
polymorphism in Western tyrosine phosphorylation B-motifs regulates
Helicobacter pylori CagA epithelial cell interactions. PLoS Pathog.
11:e10046212015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ferreira RM, Machado JC, Leite M, Carneiro
F and Figueiredo C: The number of Helicobacter pylori CagA EPIYA C
tyrosine phosphorylation motifs influences the pattern of gastritis
and the development of gastric carcinoma. Histopathology.
60:992–998. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Higashi H, Tsutsumi R, Fujita A, Yamazaki
S, Asaka M, Azuma T and Hatakeyama M: Biological activity of the
Helicobacter pylori virulence factor CagA is determined by
variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci
USA. 99:14428–14433. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Monstein H-J, Karlsson A, Ryberg A and
Borch K: Application of PCR amplicon sequencing using a single
primer pair in PCR amplification to assess variations in
Helicobacter pylori CagA EPIYA tyrosine phosphorylation motifs. BMC
Res Notes. 3:352010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
De Souza D, Fabri LJ, Nash A, Hilton DJ,
Nicola NA and Baca M: SH2 domains from suppressor of cytokine
signaling-3 and protein tyrosine phosphatase SHP-2 have similar
binding specificities. Biochemistry. 41:9229–9236. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mueller D, Tegtmeyer N, Brandt S, Yamaoka
Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A and Backert
S: c-Src and c-Abl kinases control hierarchic phosphorylation and
function of the CagA effector protein in Western and East Asian
Helicobacter pylori strains. J Clin Invest. 122:1553–1566. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ren S, Higashi H, Lu H, Azuma T and
Hatakeyama M: Structural basis and functional consequence of
Helicobacter pylori CagA multimerization in cells. J Biol Chem.
281:32344–32352. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamazaki S, Yamakawa A, Ito Y, Ohtani M,
Higashi H, Hatakeyama M and Azuma T: The CagA protein of
Helicobacter pylori is translocated into epithelial cells and binds
to SHP-2 in human gastric mucosa. J Infect Dis. 187:334–337. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Montagner A, Yart A, Dance M, Perret B,
Salles JP and Raynal P: A novel role for Gab1 and SHP2 in epidermal
growth factor-induced Ras activation. J Biol Chem. 280:5350–5360.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Higashi H, Nakaya A, Tsutsumi R, Yokoyama
K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y,
Teishikata Y, et al: Helicobacter pylori CagA induces
Ras-independent morphogenetic response through SHP-2 recruitment
and activation. J Biol Chem. 279:17205–17216. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsutsumi R, Takahashi A, Azuma T, Higashi
H and Hatakeyama M: Focal adhesion kinase is a substrate and
downstream effector of SHP-2 complexed with Helicobacter pylori
CagA. Mol Cell Biol. 26:261–276. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsutsumi R, Higashi H, Higuchi M, Okada M
and Hatakeyama M: Attenuation of Helicobacter pylori CagA × SHP-2
signaling by interaction between CagA and C-terminal Src kinase. J
Biol Chem. 278:3664–3670. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Suzuki M, Mimuro H, Suzuki T, Park M,
Yamamoto T and Sasakawa C: Interaction of CagA with Crk plays an
important role in Helicobacter pylori-induced loss of gastric
epithelial cell adhesion. J Exp Med. 202:1235–1247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bauer B, Pang E, Holland C, Kessler M,
Bartfeld S and Meyer TF: The Helicobacter pylori virulence effector
CagA abrogates human β-defensin 3 expression via inactivation of
EGFR signaling. Cell Host Microbe. 11:576–586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel SR, Smith K, Letley DP, Cook KW,
Memon AA, Ingram RJ, Staples E, Backert S, Zaitoun AM, Atherton JC,
et al: Helicobacter pylori downregulates expression of human
β-defensin 1 in the gastric mucosa in a type IV secretion-dependent
fashion. Cell Microbiol. 15:2080–2092. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Churin Y, Al-Ghoul L, Kepp O, Meyer TF,
Birchmeier W and Naumann M: Helicobacter pylori CagA protein
targets the c-Met receptor and enhances the motogenic response. J
Cell Biol. 161:249–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Suzuki M, Mimuro H, Kiga K, Fukumatsu M,
Ishijima N, Morikawa H, Nagai S, Koyasu S, Gilman RH, Kersulyte D,
et al: Helicobacter pylori CagA phosphorylation-independent
function in epithelial proliferation and inflammation. Cell Host
Microbe. 5:23–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mimuro H, Suzuki T, Tanaka J, Asahi M,
Haas R and Sasakawa C: Grb2 is a key mediator of Helicobacter
pylori CagA protein activities. Mol Cell. 10:745–755. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Amieva MR, Vogelmann R, Covacci A,
Tompkins LS, Nelson WJ and Falkow S: Disruption of the epithelial
apical-junctional complex by Helicobacter pylori CagA. Science.
300:1430–1434. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Murata-Kamiya N, Kurashima Y, Teishikata
Y, Yamahashi Y, Saito Y, Higashi H, Aburatani H, Akiyama T, Peek RM
Jr, Azuma T, et al: Helicobacter pylori CagA interacts with
E-cadherin and deregulates the β-catenin signal that promotes
intestinal transdifferentiation in gastric epithelial cells.
Oncogene. 26:4617–4626. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Neal JT, Peterson TS, Kent ML and
Guillemin K: H. pylori virulence factor CagA increases intestinal
cell proliferation by Wnt pathway activation in a transgenic
zebrafish model. Dis Model Mech. 6:802–810. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oliveira MJ, Costa AM, Costa AC, Ferreira
RM, Sampaio P, Machado JC, Seruca R, Mareel M and Figueiredo C:
CagA associates with c-Met, E-cadherin, and p120-catenin in a
multiproteic complex that suppresses Helicobacter pylori-induced
cell-invasive phenotype. J Infect Dis. 200:745–755. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kurashima Y, Murata-Kamiya N, Kikuchi K,
Higashi H, Azuma T, Kondo S and Hatakeyama M: Deregulation of
beta-catenin signal by Helicobacter pylori CagA requires the
CagA-multimerization sequence. Int J Cancer. 122:823–831. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Saadat I, Higashi H, Obuse C, Umeda M,
Murata-Kamiya N, Saito Y, Lu H, Ohnishi N, Azuma T, Suzuki A, et
al: Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt
epithelial cell polarity. Nature. 447:330–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Goldstein B and Macara IG: The PAR
proteins: Fundamental players in animal cell polarization. Dev
Cell. 13:609–622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Suzuki A and Ohno S: The PAR-aPKC system:
Lessons in polarity. J Cell Sci. 119:979–987. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lu HS, Saito Y, Umeda M, Murata-Kamiya N,
Zhang HM, Higashi H and Hatakeyama M: Structural and functional
diversity in the PAR1b/MARK2-binding region of Helicobacter pylori
CagA. Cancer Sci. 99:2004–2011. 2008.PubMed/NCBI
|
|
51
|
Lu H, Murata-Kamiya N, Saito Y and
Hatakeyama M: Role of partitioning-defective 1/microtubule
affinity-regulating kinases in the morphogenetic activity of
Helicobacter pylori CagA. J Biol Chem. 284:23024–23036. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zeaiter Z, Cohen D, Müsch A, Bagnoli F,
Covacci A and Stein M: Analysis of detergent-resistant membranes of
Helicobacter pylori infected gastric adenocarcinoma cells reveals a
role for MARK2/Par1b in CagA-mediated disruption of cellular
polarity. Cell Microbiol. 10:781–794. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yamahashi Y and Hatakeyama M: PAR1b takes
the stage in the morphogenetic and motogenetic activity of
Helicobacter pylori CagA oncoprotein. Cell Adhes Migr. 7:11–18.
2013. View Article : Google Scholar
|
|
54
|
Suzuki N, Murata-Kamiya N, Yanagiya K,
Suda W, Hattori M, Kanda H, Bingo A, Fujii Y, Maeda S, Koike K, et
al: Mutual reinforcement of inflammation and carcinogenesis by the
Helicobacter pylori CagA oncoprotein. Sci Rep. 5:100242015.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mishra JP, Cohen D, Zamperone A, Nesic D,
Muesch A and Stein M: CagA of Helicobacter pylori interacts with
and inhibits the serine-threonine kinase PRK2. Cell Microbiol.
17:1670–1682. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sandoval J and Esteller M: Cancer
epigenomics: Beyond genomics. Curr Opin Genet Dev. 22:50–55. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Verma M: The role of epigenomics in the
study of cancer biomarkers and in the development of diagnostic
tools. Adv Exp Med Biol. 867:59–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hernando-Herraez I, Garcia-Perez R, Sharp
AJ and Marques-Bonet T: DNA methylation: Insights into human
evolution. PLoS Genet. 11:e10056612015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Maekita T, Nakazawa K, Mihara M, Nakajima
T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M,
et al: High levels of aberrant DNA methylation in Helicobacter
pylori-infected gastric mucosae and its possible association with
gastric cancer risk. Clin Cancer Res. 12:989–995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yoshida T, Kato J, Maekita T, Yamashita S,
Enomoto S, Ando T, Niwa T, Deguchi H, Ueda K, Inoue I, et al:
Altered mucosal DNA methylation in parallel with highly active
Helicobacter pylori-related gastritis. Gastric Cancer. 16:488–497.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tomita H, Takaishi S, Menheniott TR, Yang
X, Shibata W, Jin G, Betz KS, Kawakami K, Minamoto T and Tomasetto
C: Inhibition of gastric carcinogenesis by the hormone gastrin is
mediated by suppression of TFF1 epigenetic silencing.
Gastroenterology. 140:879–891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Guo XB, Guo L, Zhi QM, Ji J, Jiang JL,
Zhang RJ, Zhang JN, Zhang J, Chen XH, Cai Q, et al: Helicobacter
pylori induces promoter hypermethylation and downregulates gene
expression of IRX1 transcription factor on human gastric mucosa. J
Gastroenterol Hepatol. 26:1685–1690. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hayashi Y, Tsujii M, Wang J, Kondo J,
Akasaka T, Jin Y, Li W, Nakamura T, Nishida T, Iijima H, et al:
CagA mediates epigenetic regulation to attenuate let-7 expression
in Helicobacter pylori-related carcinogenesis. Gut. 62:1536–1546.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang YW, Eom SY, Yim DH, Song YJ, Yun HY,
Park JS, Youn SJ, Kim BS, Kim YD and Kim H: Evaluation of the
relationship between dietary factors, CagA-positive Helicobacter
pylori infection, and RUNX3 promoter hypermethylation in gastric
cancer tissue. World J Gastroenterol. 19:1778–1787. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xiao C and Rajewsky K: MicroRNA control in
the immune system: Basic principles. Cell. 136:26–36. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Libânio D, Dinis-Ribeiro M and
Pimentel-Nunes P: Helicobacter pylori and microRNAs: Relation with
innate immunity and progression of preneoplastic conditions. World
J Clin Oncol. 6:111–132. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Noto JM and Peek RM: The role of microRNAs
in Helicobacter pylori pathogenesis and gastric carcinogenesis.
Front Cell Infect Microbiol. 1:212012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Matsushima K, Isomoto H, Inoue N, Nakayama
T, Hayashi T, Nakayama M, Nakao K, Hirayama T and Kohno S: MicroRNA
signatures in Helicobacter pylori-infected gastric mucosa. Int J
Cancer. 128:361–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhu Y, Jiang Q, Lou X, Ji X, Wen Z, Wu J,
Tao H, Jiang T, He W, Wang C, et al: MicroRNAs up-regulated by CagA
of Helicobacter pylori induce intestinal metaplasia of gastric
epithelial cells. PLoS One. 7:e351472012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Feng Y, Wang L, Zeng J, Shen L, Liang X,
Yu H, Liu S, Liu Z, Sun Y, Li W, et al: FoxM1 is overexpressed in
Helicobacter pylori-induced gastric carcinogenesis and is
negatively regulated by miR-370. Mol Cancer Res. 11:834–844. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li N, Tang B, Zhu ED, Li BS, Zhuang Y, Yu
S, Lu DS, Zou QM, Xiao B and Mao XH: Increased miR-222 in H.
pylori-associated gastric cancer correlated with tumor progression
by promoting cancer cell proliferation and targeting RECK. FEBS
Lett. 586:722–728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Belair C, Baud J, Chabas S, Sharma CM,
Vogel J, Staedel C and Darfeuille F: Helicobacter pylori interferes
with an embryonic stem cell micro RNA cluster to block cell cycle
progression. Silence. 2:72011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cheng SF, Li L and Wang LM: miR-155 and
miR-146b negatively regulates IL6 in Helicobacter pylori
(cagA+) infected gastroduodenal ulcer. Eur Rev Med
Pharmacol Sci. 19:607–613. 2015.PubMed/NCBI
|
|
75
|
Harr JC, Gonzalez-Sandoval A and Gasser
SM: Histones and histone modifications in perinuclear chromatin
anchoring: From yeast to man. EMBO Rep. 17:139–155. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Schones DE, Cui K, Cuddapah S, Roh TY,
Barski A, Wang Z, Wei G and Zhao K: Dynamic regulation of
nucleosome positioning in the human genome. Cell. 132:887–898.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Xia G, Schneider-Stock R, Diestel A,
Habold C, Krueger S, Roessner A, Naumann M and Lendeckel U:
Helicobacter pylori regulates p21 (WAF1) by histone H4 acetylation.
Biochem Biophys Res Commun. 369:526–531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Fehri LF, Rechner C, Janssen S, Mak TN,
Holland C, Bartfeld S, Brüggemann H and Meyer TF: Helicobacter
pylori-induced modification of the histone H3 phosphorylation
status in gastric epithelial cells reflects its impact on cell
cycle regulation. Epigenetics. 4:577–586. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liang X, Zeng J, Wang L, Shen L, Li S, Ma
L, Ci X, Yu J, Jia M, Sun Y, et al: Histone demethylase RBP2
induced by Helicobactor pylori CagA participates in the malignant
transformation of gastric epithelial cells. Oncotarget.
5:5798–5807. 2014. View Article : Google Scholar : PubMed/NCBI
|