Introduction
Gastric cancer (GC), the fourth common malignant
tumor type, ranks as the second leading cause of cancer-related
mortality worldwide (1). Currently,
the main therapies for GC treatment include surgery, radiation
therapy and chemotherapy. However even following maximal
trimodality therapies, the prognosis of advanced GC patients
remains poor with a median overall survival (OS) of less than 12
months and a 5-year survival rate of less than 5% (2). Elucidating the molecular mechanisms
underlying GC progression is essential for developing novel
diagnostic and therapeutic strategies against this disease.
Cap-dependent translation plays a crucial role in
the initiation and progression of many types of cancers by
enhancing the translation initiation of many oncogenic mRNAs such
as cyclin D1, Bcl-2, MMP9 and VEGF that regulate cell
proliferation, survival, metastasis and angiogenesis, respectively
(3). Cap-dependent translation is
controlled by the eukaryotic translation initiation factor 4F
(eIF4F) complex, which comprises eIF4E, eIF4G and eIF4A and is
essential for translation initiation by recruiting the 40S ribosome
subunit to the 5′ cap mRNA (4). The
availability of eIF4E is the rate-limiting factor for the assembly
and activity of the eIF4F complex and is controlled by the eIF4E
expression level and the phosphorylation status of eIF4E-binding
protein 1 (4E-BP1) (5).
Unphosphorylated 4E-BP1 negatively regulates the formation of the
eIF4F complex by competing with eIF4G for binding to eIF4E.
Phosphorylation of 4E-BP1 releases its binding to eIF4E, allowing
the binding between eIF4G and eIF4E and translation initiation
(6). The activity of 4E-BP1 is
mainly regulated by the mammalian target of rapamycin complex 1
(mTORC1), an important downstream target of AKT signaling. mTORC1
phosphorylates 4E-BP1 on Thr37 and Thr46, which facilitates
subsequent phosphorylation of Ser65 and Thr70, leading to the
dissociation of 4E-BP1 and eIF4E and the assembly of the eIF4F
complex (7). eIF4E was reported to
be overexpressed in gastric tumor tissues compared with that in
adjacent normal tissues and silencing of eIF4E was found to inhibit
the proliferation of GC cells (8).
The AKT/mTORC1 pathway is frequently activated in GC and is
directly associated with its progression (9). Several groups have reported higher
expression of p-4E-BP1 in GC tissue samples than that noted in
normal tissue samples (10–12). All of these studies indicate that
cap-dependent translation contributes to gastric tumorigenesis and
may serve as a potential therapeutic target for GC.
Hematopoietic pre-B-cell leukemia transcription
factor interacting protein (HPIP) was initially identified as a
co-repressor for PBX-HOX transcriptional complex through a yeast
two-hybrid screening of a hematopoietic cDNA library (13). Subsequent studies have demonstrated
that HPIP plays a critical role in erythroid differentiation by
serving as a target of GATA1, an erythroid lineage-specific
transcription factor (14).
Recently, increasing studies indicate that HPIP is implicated in
carcinogenesis. HPIP has been reported to be overexpressed in many
types of cancers, including colorectal cancer (15), astrocytoma (16), thyroid carcinoma (17), breast cancer (18), non-small cell lung cancer (19) and hepatocellular carcinoma (20), and plays an important role in the
development and progression of these tumors. In GC, the expression
of HPIP was found to be significantly upregulated in tumors
compared with that noted in adjacent normal tissues and was found
to be positively correlated with tumor size and metastasis
(21). HPIP overexpression was
found to increase GC cell proliferation by promoting cell cycle
progression and to enhance GC cell migration by modulating
epithelial-mesenchymal transition (EMT) (21). Although the role of HPIP in
promoting GC cell proliferation and activating G1/S transition has
been characterized, how HPIP enhances these processes remains
unclear. In colorectal cancer and hepatocellular carcinoma
(15,20), HPIP has been shown to promote cell
proliferation via enhancing AKT/mTORC1 signaling. Due to the
important role of the AKT/mTORC1 pathway in the regulation of
4E-BP1 phosphorylation, we hypothesized that HPIP may increase GC
cell proliferation by activating cap-dependent translation.
In the present study, we found that HPIP activated
cap-dependent translation in an AKT/mTORC1 signaling-dependent
manner. Furthermore, we showed that inhibition of the activity of
the eIF4F complex abolished the ability of HPIP to enhance GC cell
proliferation both in vitro and in a xenograft mouse model
in vivo, while enhancing activity of the eIF4F complex by
silencing 4E-BP1 expression reversed the inhibitory effect of HPIP
knockdown on GC cell proliferation. Our data indicate that HPIP
promotes GC cell proliferation by enhancing cap-dependent
translation.
Materials and methods
Antibodies and reagents
Antibodies against AKT, pAKT, 4EBP-1, p4EBP-1 (S65),
P70S6K, pP70S6K (T389), eIF4E, eIF4G and cyclin D1 were purchased
from Cell Signaling Technology, Inc. (Beverly, MA, USA). The
anti-tubulin antibody was purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Antibody against HPIP was obtained from
Proteintech Group, Inc. (Chicago, IL, USA). MK2206 and PP242 were
obtained from Selleckchem (Houston, TX, USA). 4EGI-1 was provided
by Calbiochem (Darmstadt, Germany).
Cells and cell culture
SGC-7901 and BGC-823 cells were maintained in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS; Hyclone, Logan, UT, USA) and pen/strep (100 U/ml
penicillin and 100 mg/l streptomycin; Cellgro Mediatech, Inc.,
Manassas, VA, USA). Cells were cultured in a 37°C incubator at 5%
CO2.
To generate stable cell lines with overexpression of
HPIP, SGC-7901 and BGC-823 cells were transfected with the
pCMV-2B-HPIP vector or pCMV-2B backbone with Lipofectamine 3000
(Promega, Madison, WI, USA) according to the manufacturer's
instructions, followed by selection with 600 µg/ml G418
(Calbiochem) for 2 weeks.
For establishing stable HPIP-knockdown cell lines,
SGC-7901 and BGC-823 cells were infected with appropriate amounts
of lentiviral particles harboring HPIP shRNA or control shRNA
(GeneChem, Co., Ltd., Shanghai, China). Infected cells were
selected with 1 µg/ml puromycin (Sigma-Aldrich, St. Louis, MO, USA)
for 1 week.
Western blotting
Cells were lysed in RIPA buffer (50 mM Tris-HCl, pH
7.5, 150 mM NaCl, 0.1% SDS, 1% NP-40, 1% sodium deoxycholate, 1 mM
EDTA, 1 mM EGTA, 1X protease inhibitor cocktail) on ice. Equal
amounts of total protein were resolved by SDS-PAGE and transferred
to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ,
USA). After being blocked with 5% nonfat milk for 1 h at room
temperature, the membranes were probed with primary antibodies
overnight at 4°C, followed by incubation with horseradish
peroxidase-conjugated secondary antibodies for 2 h at room
temperature. Detection was performed using ECL™ Advance western
blotting detection kit (GE Healthcare, Buckinghamshire, UK).
siRNA and transient transfections
siRNAs against AKT, Raptor and 4E-BP1 were obtained
from Invitrogen Life Technologies. SGC-7901 and BGC-823 cells were
transfected with control siRNAs or siRNAs against AKT, Raptor or
4EBP1 using Lipofectamine RNAiMAX (Invitrogen Life Technologies)
according to the manufacturer's instructions. For siRNAs against
AKT or Raptor, the cells were lysed and subjected to western
blotting at 48 h post-transfection. For siRNAs against 4E-BP1, the
cells were lysed and subjected to assays for cap-dependent
translation, cell cycle and western blotting at 48 h
post-transfection. For Cell Counting Kit-8 (CCK-8) assay, the cells
were seeded into 96-well plates 24 h after transfection.
Bicistronic luciferase assays
SGC-7901 and BGC-823 cells were transiently
transfected with a bicistronic luciferase reporter plasmid,
pcDNA3-rLuc-PolioIRES-fLuc, which directs cap-dependent translation
of the Renilla luciferase (RL) gene and cap-independent
Polio IRES-mediated translation of the firefly (FL) gene. Cells
were harvested and analyzed for luciferase activity using
Dual-Luciferase reporter assay kit (Promega) at 48 h
post-transfection. Cap-dependent translational activity was
determined by calculating the ratio of Renilla/firefly
luciferase activity. Assays were performed in triplicate and
results are presented as means ± standard deviation (SD).
m7GTP pull-down assay
Cells were harvested in m7GTP lysis
buffer (20 mM Tris, 100 mM KCl, 1 mM dithiothreitol, 20 mM
β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA,
1 mM EGTA, 0.25 mM Na3VO4, 10 mM NaF and 1X
protease inhibitor cocktail) on ice. Thirty microliters of
m7GTP-Sepharose beads (GE Healthcare, Chalfont St.
Giles, UK) was added to 500 µl cell lysate and gently rocked at 4°C
for 3 h. Precipitates were collected, washed 3 times with 500 µ
lysis buffer at 4°C, and then eluted by boiling in SDS sample
buffer for western blotting. The original lysate was performed as a
loading control.
Cell cycle analysis
Cells were fixed with 70% ethanol overnight at
−20°C, washed twice with ice-cold PBS and then treated with RNase A
(0.1 mg/ml) in PBS for 30 min at 37°C. Propidium iodide (50 µg/ml)
was added to the cellular suspension. DNA contents were determined
by a FACSCanto flow cytometer (BD Biosciences, Mississauga, ON,
Canada).
Cell proliferation assays
Cells were seeded in 96-well plates at 1,500
cells/well. Cell proliferation was assessed with CCK-8 (Dojindo
Laboratories, Tokyo, Japan) assay according to the manufacturer's
instructions and the absorbance was determined at a wavelength of
450 nm using a microplate reader. The optical density (OD) value of
the treatment group was normalized to the values from the untreated
control group.
Soft agar assays
For anchorage-independent growth assay, the cells
were seeded in 6-well plates at 5,000 cells/well, with a bottom
layer of 0.35% low melting point agarose (BD Biosciences) in
DMEM/10% FBS and a top layer of 0.5% agarose in the same medium.
Three weeks later, the number of colonies with diameters >100 µm
were counted.
GC xenografts and treatments
Five six-week-old female BALB/c nude mice were
obtained from the Animal Center of Sun Yat-Sen University
(Guangzhou, China). BGC-823 cells (5×106) stably
overexpressing HPIP or control cells in serum-free medium were
injected into the flanks of the nude mice, respectively (n=12).
After 10 days, the mice injected with BGC-823 cells stably
overexpressing HPIP or control cells were randomly allocated to 2
groups (n=6/group) according to body weight and tumor volume and
treated with 4EGI-1 (50 mg/kg/day, intraperitoneal injection) or
vehicle control. 4EGI-1 was dissolved at 90 mg/ml in DMSO and
stored at −80°C. Immediately prior to injection, 4EGI-1 was thawed
and diluted 1:9 in saline (0.9% sodium chloride). The tumor volume
and the body weight of the mice were monitored at the indicated
times. One month after treatment, all mice were sacrificed.
Xenograft primary tumors were harvested for western blotting.
Statistical analysis
All data were analyzed using the unpaired Student's
t-test with the SPSS statistical software program (version 13.0;
SPSS, Inc., Chicago, IL, USA). The data are presented as means ±
SD. P-values <0.05 were considered statistically
significant.
Results
HPIP activates cap-dependent
translation
Given that HPIP was reported to promote GC cell
proliferation and cap-dependent translation promotes cell cycle
progression via enhancing the expression of cyclin D1 (21), we hypothesized that HPIP may
activate cap-dependent translation in GC cells. To test this
hypothesis, the cap-dependent translation rate in the control and
the HPIP-overexpressing cells was examined using a dual-luciferase
reporter system which can monitor the ratio between cap-dependent
(Renilla luciferase) and cap-independent IRES-mediated
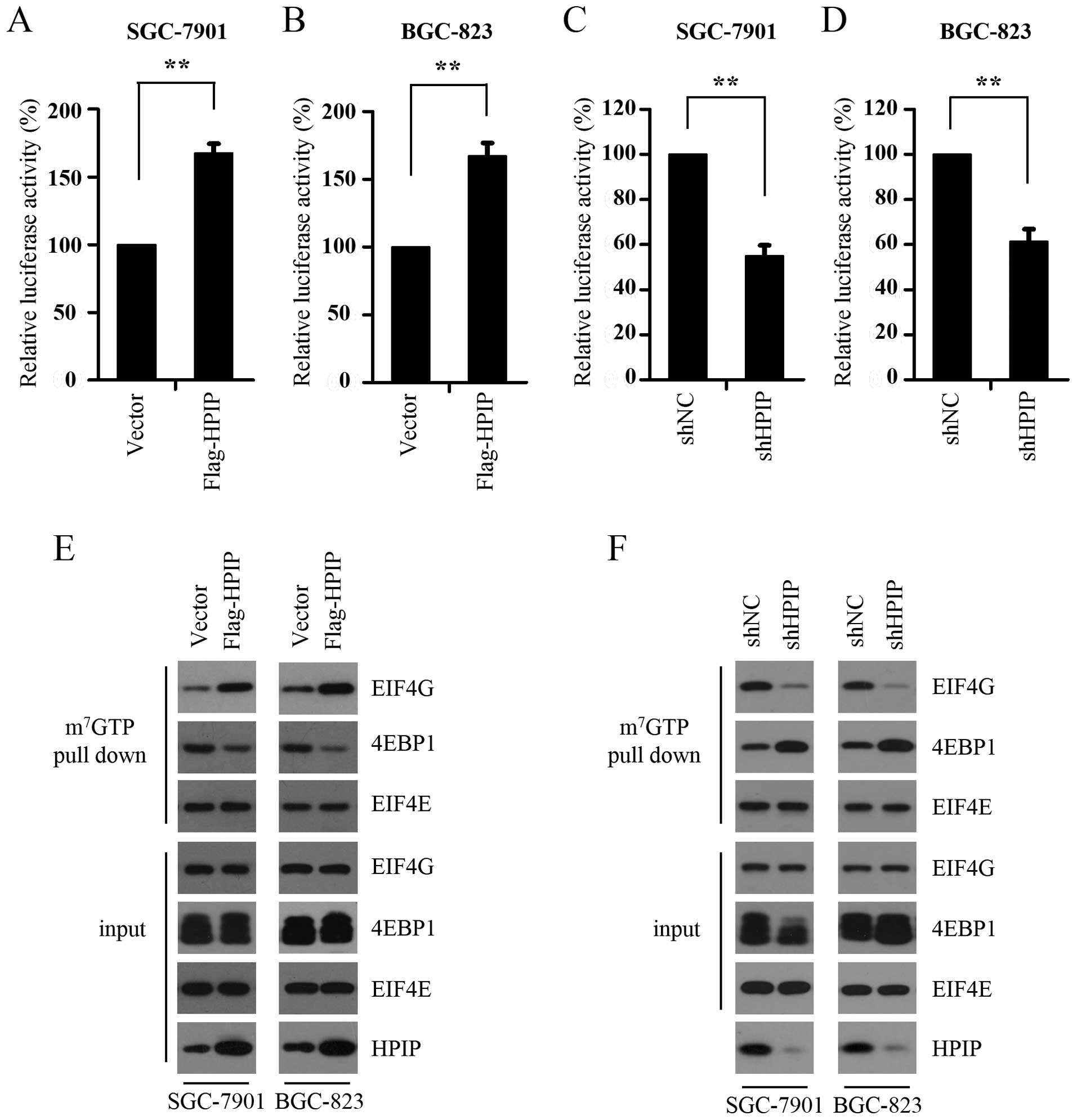
(firefly luciferase) translation initiation. As shown in Fig. 1A and B, HPIP overexpression promoted
cap-dependent translation in both the SGC-7901 and BGC-823 cells.
To confirm this result, we further determined the cap-dependent
translation rate in the control and HPIP-silenced SGC-7901 and
BGC-823 cells. We found that HPIP depletion profoundly suppressed
cap-dependent translation (Fig. 1C and
D). It is known that cap-dependent translation is regulated by
the assembly of the eIF4F complex, which can be monitored with
7-methyl GTP Sepharose bead assay. We subsequently detected the
effect of HPIP overexpression on the assembly of the eIF4F complex
and found that HPIP overexpression markedly increased the
interaction between eIF4E and eIF4G, while suppressed the binding
of eIF4E and 4E-BP1 (Fig. 1E),
indicating the increased assembly of the eIF4F complex in the
HPIP-overexpressing GC cells. Consistent with this result, HPIP
knockdown decreased the assembly of the eIF4F complex in both
SGC-7901 and BGC-823 cell lines (Fig.
1F). Taken together, these findings suggest that HPIP activates
cap-dependent translation by promoting the assembly of the eIF4F
complex in GC cells.
HPIP increases cap-dependent
translation in an AKT/mTORC1-dependent manner
Cap-dependent translation is mainly regulated by
AKT/mTORC1 signaling, which phosphorylates 4E-BP1 and relieves its
binding to eIF4E, allowing the formation of the eIF4F complex to
initiate translation. As previous studies have demonstrated that
HPIP enhances AKT/mTORC1 signaling in thyroid cancer cells and
breast cancer cells, we first detected whether HPIP regulates
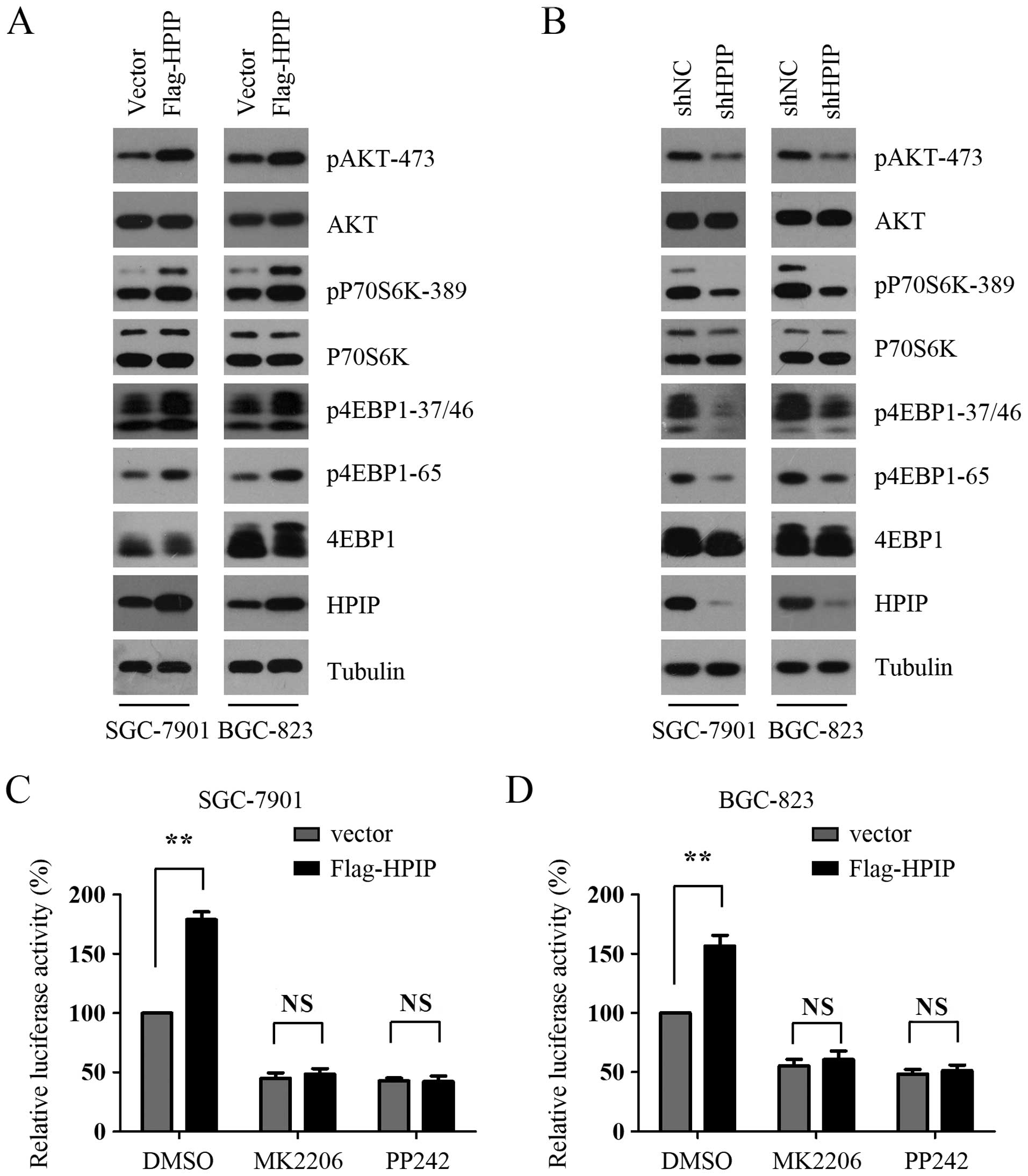
AKT/mTORC1 signaling in GC cells. As shown in Fig. 2A, HPIP overexpression enhanced the
phosphorylation of AKT in both the SGC-7901 and BGC-823 cells.
Furthermore, the phosphorylation levels of p70S6K and 4E-BP1,
downstream substrates of mTORC1, were also significantly increased
in the HPIP-overexpressing SGC-7901 and BGC-823 cells (Fig. 2A), indicating that HPIP enhances
AKT/mTORC1 signaling in GC cells. We next examined the effect of
HPIP knockdown on AKT/mTORC1 signaling and found that HPIP
silencing decreased the phosphorylation of AKT, p70S6K and 4E-BP1
(Fig. 2B). To investigate whether
AKT/mTORC1 signaling activation is essential for HPIP-mediated
cap-dependent translation, control and HPIP-overexpressing GC cells
were treated with MK2206 and PP242, which are AKT and mTORC1
pathway inhibitors, respectively. The results showed that MK2206
and PP242 antagonized the HPIP-mediated cap-dependent translation
in both the SGC-7901 and BGC-823 cells (Fig. 2C and D). To further confirm the role
of AKT/mTORC1 signaling activation in this process, we analyzed
HPIP-mediated cap-dependent translation after silencing the
expression of AKT or Raptor, a specific and essential component of
mTORC1. We found that AKT or Raptor depletion abrogated
HPIP-mediated cap-dependent translation in both the SGC-7901 and
BGC-823 cells (Fig. 2E and F).
Collectively, these data suggest that AKT/mTORC1 signaling plays an
indispensable role in the regulation of HPIP-mediated cap-dependent
translation.
HPIP promotes GC cell proliferation by
activating cap-dependent translation
Previous studies have reported that HPIP promotes GC
cell proliferation by enhancing the expression of cyclin D1 and
G1/S transition. Since cap-dependent translation promotes cancer
cell cycle progression by initiating the translation of cyclin D1,
we ascertained whether cap-dependent translation is required for
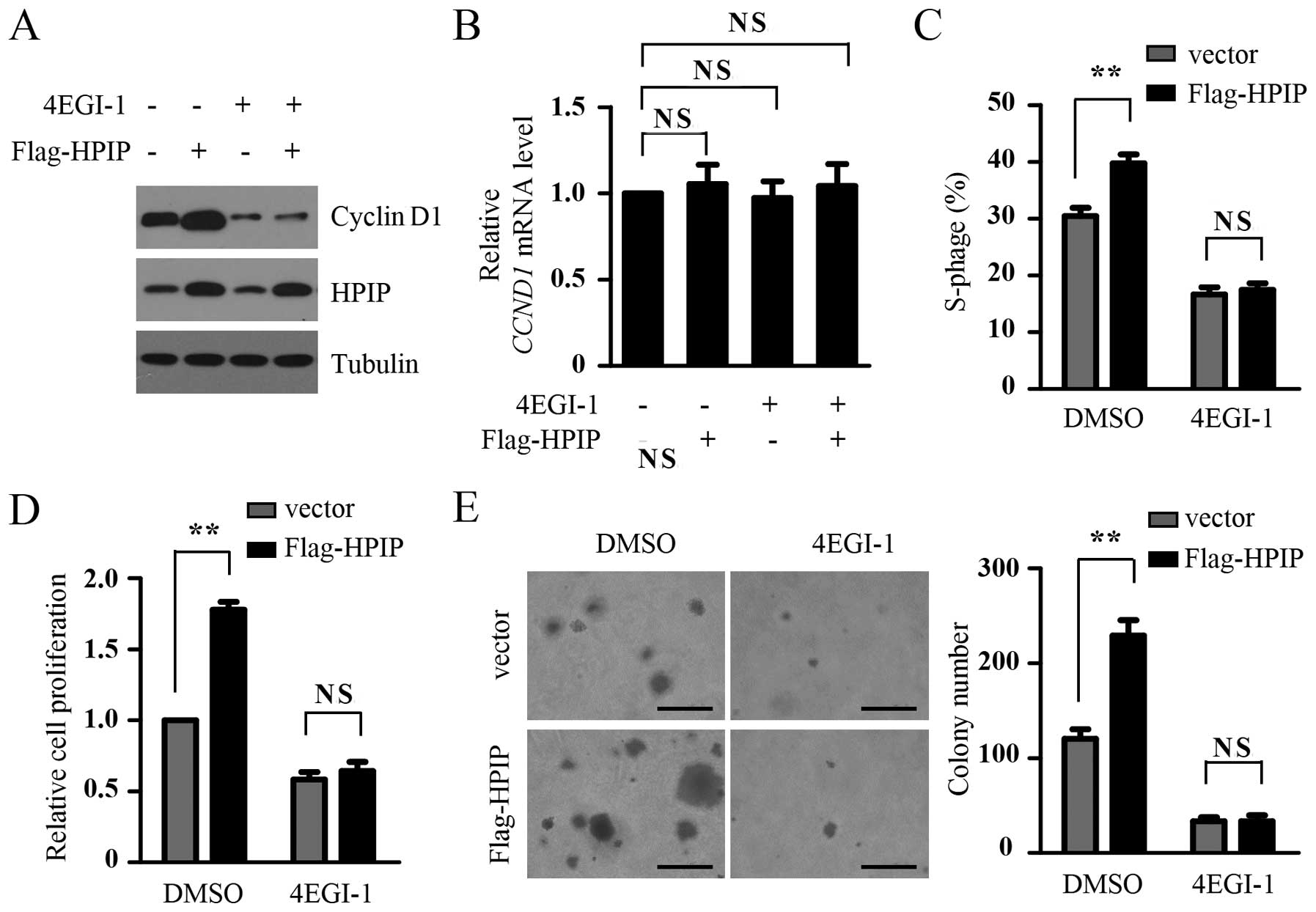
HPIP-mediated GC cell proliferation. Consistent with the previous
results, in the present study, cyclin D1 expression, the proportion
of cells in the S phase and cell proliferation were increased in
the HPIP-overexpressing BGC-823 cells. However, the treatment of
4EGI-1, a selective eIF4E/eIF4G interaction inhibitor, abolished
the ability of HPIP to enhance the expression of cyclin D1, the
proportion of cells in the S phase and cell proliferation in the
BGC-823 cells (Fig. 3A, C and D).
Contrary to the increase in cyclin D1 at the protein level by HPIP
overexpression and the downregulation by 4EGI-1 treatment, HPIP
overexpression and 4EGI-1 treatment had little effect on the mRNA
levels of cyclin D1, suggesting that the downregulation of cyclin
D1 by 4EGI-1 (Fig. 3A) resulted
from the suppression of cyclin D1 translational inhibition rather
than its gene transcription (Fig.
3B). Furthermore, soft agar assay showed that HPIP promoted
anchorage-independent proliferation of the BGC-823 cells, while
4EGI-1 treatment attenuated the effect of HPIP (Fig. 3E). Taken together, our data suggest
that HPIP enhanced GC cell proliferation by activating
cap-dependent translation.
HPIP regulates cap-dependent
translation and cell proliferation by inactivation of 4E-BP1
Hypo-phosphorylated 4E-BP1 suppresses the assembly
of the eIF4F complex by binding competitively to EIF4E, resulting
in the inhibition of cap-dependent translation. The above studies
demonstrated that HPIP activated cap-dependent translation in an
AKT/mTORC1 signaling-dependent manner. Since AKT/mTORC1 signaling
enhances cap-dependent translation by phosphorylating 4E-BP1, which
leads to the disassociation of 4E-BP1 and EIF4E and contributes to
the formation of eIF4F complex, we determined the importance of
4E-BP1 in mediating the effects of HPIP on cap-dependent
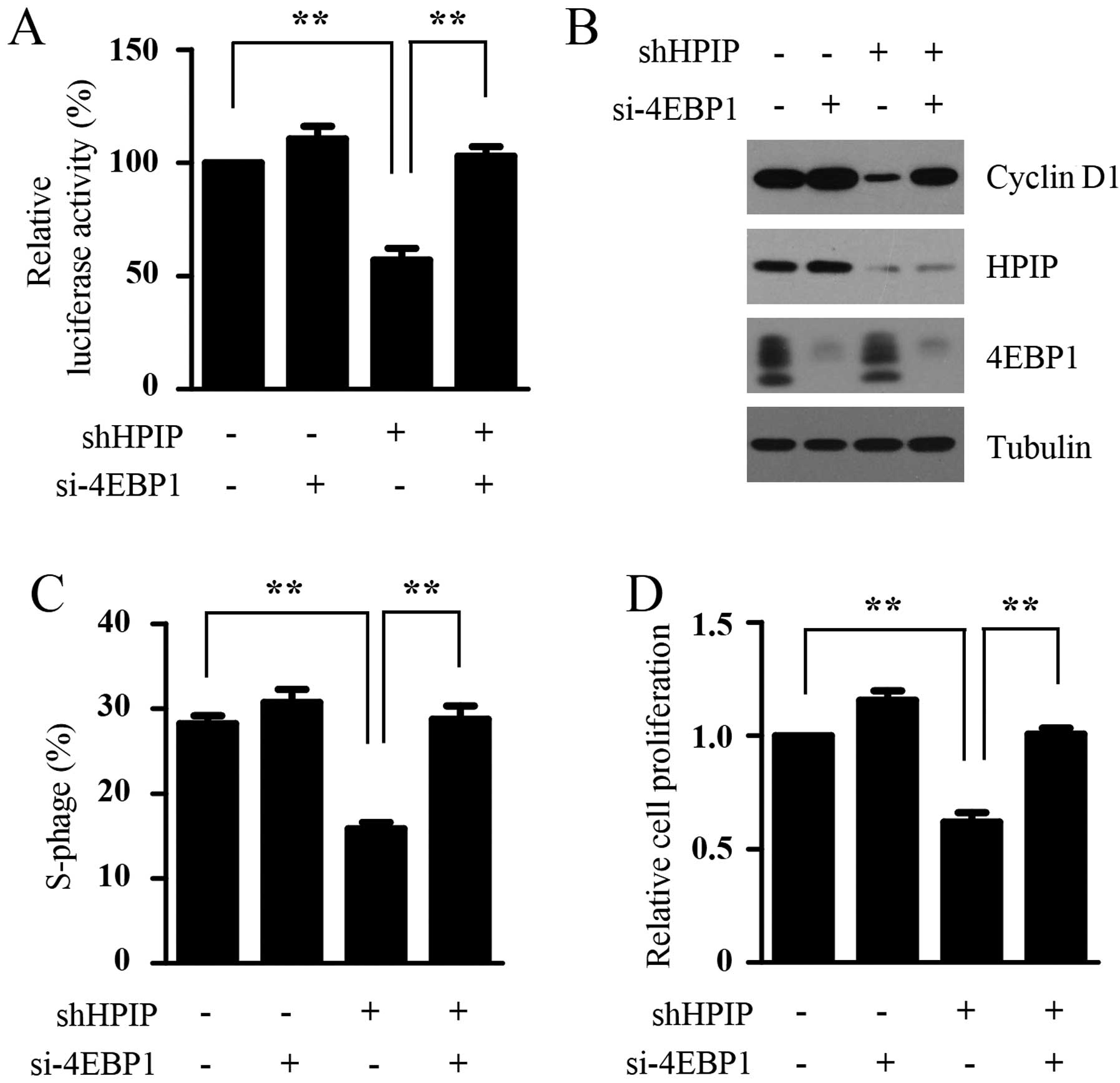
translation and cell proliferation. To test this idea, we silenced
the expression of 4E-BP1 in the control and HPIP-depleted BGC-823
cells and detected whether 4E-BP1 depletion attenuated the
inhibitory effect of HPIP knockdown on cap-dependent translation,
cyclin D1 expression, the proportion of S phase cells and cell
proliferation in the GC cells. As shown in Fig. 4A-D, HPIP knockdown suppressed
cap-dependent translation, cyclin D1 expression, the proportion of
S phase cells and cell proliferation in the BGC-823 cells, while
4E-BP1 depletion markedly rescued the inhibitory effect. Taken
together, our results further confirmed that HPIP promotes cell
proliferation by activating cap-dependent translation and suggest
that inactivation of 4E-BP1 is essential for HPIP-mediated
cap-dependent translation and cell proliferation.
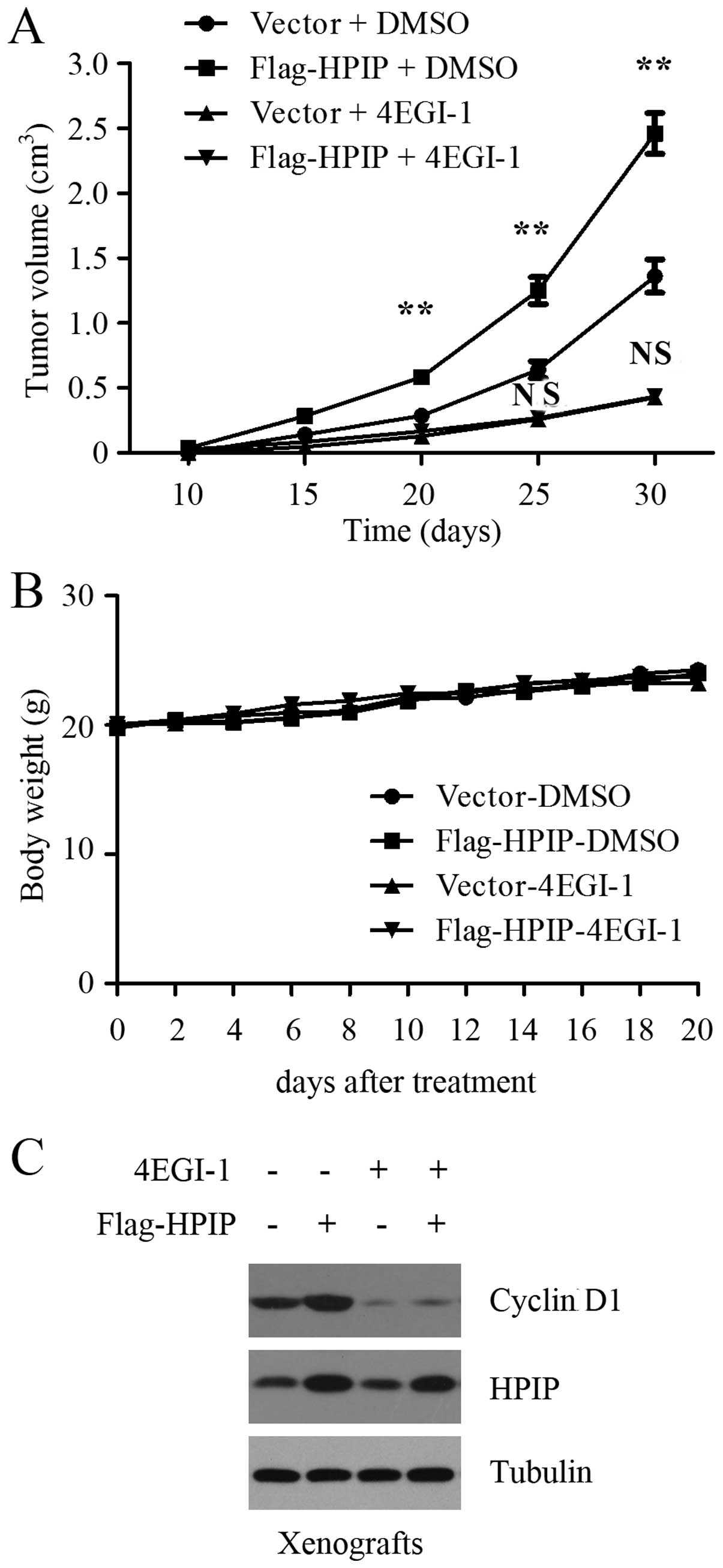
Targeting translation initiation with
4EGI-1 suppresses the ability of HPIP to promote gastric tumor
growth in vivo
Having established that HPIP promotes GC cell
proliferation by activating cap-dependent translation in
vitro, we next examined whether blockage of translation
initiation attenuated the ability of HPIP to promote tumor growth
in vivo. After the generation of GC xenografts using control
and HPIP-overexpressing BGC-823 cells, xenograft mice were randomly
grouped and treated with vehicle control or 4EGI-1 for weeks. As
expected, HPIP overexpression markedly promoted gastric tumor
growth in nude mice, as shown by a representative tumor growth
curve (Fig. 5A). Consistent with
the results from cell cultures, 4EGI-1 treatment suppressed the
ability of HPIP to promote gastric tumor growth in vivo
(Fig. 5A). There was no obvious
difference in mouse body weights among the groups during the 20 day
treatment (Fig. 5B), indicating
that the treatment was well tolerated. In addition, the BGC-823
tumors with HPIP overexpression showed increased expression of
cyclin D1, but this increase was profoundly reversed by 4EGI-1
treatment (Fig. 5C). These results
clearly indicate that HPIP promotes GC cell proliferation by
activating cap-dependent translation in vivo.
Discussion
Aberrantly expressed HPIP plays a critical role in
tumorigenesis. Overexpression of HPIP has been found in patients
with various types of cancers and is associated with a poor
clinical outcome. HPIP promotes cancer cell proliferation by
regulating cell cycle progression and cell migration via enhancing
EMT. HPIP was reported to increase cell proliferation and migration
by regulation of AKT/mTORC1 signaling in colorectal cancer and
hepatocellular carcinoma (15,20).
In breast cancer, HPIP was found to enhance estrogen receptor (ER)
signaling and promote tumor growth by activating AKT signaling
(18). A recent study demonstrated
that the expression of HPIP is higher in GC tissues than that noted
in adjacent normal gastric tissues and is significantly correlated
with several important clinicopathologic factors. HPIP knockdown
profoundly suppressed tumor growth in vitro and in an
allograft murine model (21).
However, the molecular mechanism by which HPIP promotes GC cell
proliferation remains unknown. Given that HPIP activates the
AKT/mTORC1 pathway in other cancers and cap-dependent translation
is controlled by this pathway, in the present study, we first
detected the effect of HPIP on cap-dependent translation. Using
bicistronic luciferase reporter assays, we found that HPIP enhanced
cap-dependent translation in GC cells. The results of 7-methyl GTP
Sepharose bead assays confirmed this conclusion.
Cap-dependent translation is negatively regulated by
4E-BP1, which inhibits the formation of the eIF4F complex by
competing with eIF4G for binding to eIF4E. The phosphorylation of
4E-BP1 controls its activity and is regulated by AKT/mTORC1
signaling, one of the most frequently activated pathways in GC
patients (9). mTORC1 promotes
cap-dependent translation initiation through relieving
4E-BP1-mediated inhibition of eIF4E. Since we found that HPIP
enhanced cap-dependent translation in GC cells, we next determined
whether this effect depends on AKT/mTORC1 signaling. Our results
indicated that HPIP also increased AKT/mTORC1 signaling in GC cells
and suppressing AKT activity with MK2006 or mTORC1 activity with
PP242 antagonized HPIP-mediated cap-dependent translation.
Inhibition of AKT/mTORC1 signaling by silencing the expression of
AKT or Raptor further confirmed this result. These data suggest
that activation of AKT/mTORC1 signaling is essential for
HPIP-mediated cap-dependent translation.
Cap-dependent translation contributes to cancer cell
proliferation by initiating the translation of oncogenes associated
with the regulation of cell cycle progression, such as cyclin D1
and c-Myc. cyclin D1 is commonly overexpressed in GC tissues and
promotes cell proliferation by driving G1/S phase transition
(21). Since previous studies
demonstrated that HPIP promotes GC cell proliferation and we found
that HPIP enhanced cap-dependent translation in GC cells, we next
determined whether HPIP promotes GC cell proliferation by
activating cap-dependent translation. We found that the blocking of
cap-dependent translation with 4EGI-1, a selective eIF4E/eIF4G
interaction inhibitor, abolished the ability of HPIP to promote the
expression of cyclin D1 and cell proliferation of GC cells. HPIP
depletion suppressed the phosphorylation of 4E-BP1, leading to the
disruption of the eIF4F complex and inhibition of cap-dependent
translation. We released the sequestered eIF4E and rescued the
formation of eIF4F by silencing the expression of 4E-BP1 and found
that 4E-BP1 knockdown effectively reversed the inhibitory effect of
HPIP depletion on cap-dependent translation and cell proliferation
in GC, suggesting the importance of 4E-BP1 phosphorylation and
cap-dependent translation in mediating the oncogenic role of HPIP.
Overexpression of p-4E-BP1 has been shown in 68.4% of GC tissue
samples tested, profoundly higher than in 29% of normal tissue
samples (12). Whether the
expression level of HPIP is positively associated with that of
phosphorylated 4E-BP1 in clinical tissues remains to be addressed
and is a research direction we are currently pursuing. Recently,
mTORC1/4E-BP1 signaling has been shown to play an important role in
cancer cell migration by translational regulation of Snail, a
fundamental regulator in EMT and cancer metastasis (22). Therefore, it will also be
interesting to examine if HPIP promotes GC cell migration by
enhancing the translational initiation of Snail in the future.
Our results also suggest that direct targeting of
cap-dependent translation with 4EGI-1 abrogates the ability of HPIP
to promote gastric tumor growth in vivo, indicating that
targeting the eIF4F complex may provide a promising treatment
strategy for GC patients with elevated HPIP expression. Indeed,
several translation initiation inhibitors, including eIF4E-specific
antisense oligonucleotides (ASOs) and silvestrol that suppresses
cap-dependent translation by inhibiting the activity of eIF4A, have
recently exerted effective antitumor effects with limited toxicity
in mice (23–26). In addition, targeting mTORC1
presents an alternative strategy for blocking cap-dependent
translation. However, everolimus (RAD001), an oral inhibitor of
mTORC1, failed to show improved survival in the phase III GRANITE-1
study for advanced GC patients (2).
Weak or only transient inhibition of mTORC1-mediated 4E-BP1
phosphorylation and cap-dependent translation by everolimus may
explain the limited anticancer efficacy in clinical trials
(27,28). In the present study, we also found
that 10 nM RAD001 treatment had little effect on cap-dependent
translation in GC cells and failed to inhibit HPIP-mediated
cap-dependent translation (data not shown). ATP-competitive mTOR
kinase inhibitors, including OSI-027, PP242, AZD8055 and Torin 1,
all strongly inhibit mTORC1-mediated 4E-BP1 phosphorylation and
exhibit significant antitumor activity in many cancer xenograft
models (29–32). Several of these have entered
clinical trials and hold potential as a future strategy for cancer
therapeutics (33–35).
Acknowledgements
This study was supported by the Research Program of
Science and Technology at Universities of Inner Mongolia Autonomous
Region (NJZY11127 to Y.Z.).
Glossary
Abbreviations
Abbreviations:
|
HPIP
|
hematopoietic pre-B-cell leukemia
transcription factor interacting protein
|
|
GC
|
gastric cancer
|
|
eIF4E
|
eukaryotic translation initiation
factor 4E
|
|
4E-BP1
|
eIF4E-binding protein 1
|
|
mTORC1
|
mammalian target of rapamycin complex
1
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohtsu A, Ajani JA, Bai YX, Bang YJ, Chung
HC, Pan HM, Sahmoud T, Shen L, Yeh KH, Chin K, et al: Everolimus
for previously treated advanced gastric cancer: results of the
randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol.
31:3935–3943. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kong J and Lasko P: Translational control
in cellular and developmental processes. Nat Rev Genet. 13:383–394.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spilka R, Ernst C, Mehta AK and Haybaeck
J: Eukaryotic translation initiation factors in cancer development
and progression. Cancer Lett. 340:9–21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pettersson F, Del Rincon SV and Miller WH
Jr: Eukaryotic translation initiation factor 4E as a novel
therapeutic target in hematological malignancies and beyond. Expert
Opin Ther Targets. 18:1035–1048. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pelletier J, Graff J, Ruggero D and
Sonenberg N: Targeting the eIF4F translation initiation complex: a
critical nexus for cancer development. Cancer Res. 75:250–263.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chiarini F, Evangelisti C, McCubrey JA and
Martelli AM: Current treatment strategies for inhibiting mTOR in
cancer. Trends Pharmacol Sci. 36:124–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang S, Guo R, Zhang Z, Liu D, Xu H, Xu
Z, Wang X and Yang L: Upregulation of the eIF4E signaling pathway
contributes to the progression of gastric cancer, and targeting
eIF4E by perifosine inhibits cell growth. Oncol Rep. 29:2422–2430.
2013.PubMed/NCBI
|
|
9
|
Yang W, Raufi A and Klempner SJ: Targeted
therapy for gastric cancer: molecular pathways and ongoing
investigations. Biochim Biophys Acta. 1846:232–237. 2014.PubMed/NCBI
|
|
10
|
Yang HY, Xue LY, Xing LX, Wang J, Wang JL,
Yan X and Zhang XH: Putative role of the mTOR/4E-BP1 signaling
pathway in the carcinogenesis and progression of gastric cardiac
adenocarcinoma. Mol Med Rep. 7:537–542. 2013.PubMed/NCBI
|
|
11
|
Tapia O, Riquelme I, Leal P, Sandoval A,
Aedo S, Weber H, Letelier P, Bellolio E, Villaseca M, Garcia P, et
al: The PI3K/AKT/mTOR pathway is activated in gastric cancer with
potential prognostic and predictive significance. Virchows Arch.
465:25–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun DF, Zhang YJ, Tian XQ, Chen YX and
Fang JY: Inhibition of mTOR signalling potentiates the effects of
trichostatin A in human gastric cancer cell lines by promoting
histone acetylation. Cell Biol Int. 38:50–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abramovich C, Shen WF, Pineault N, Imren
S, Montpetit B, Largman C and Humphries RK: Functional cloning and
characterization of a novel nonhomeodomain protein that inhibits
the binding of PBX1-HOX complexes to DNA. J Biol Chem.
275:26172–26177. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Manavathi B, Lo D, Bugide S, Dey O, Imren
S, Weiss MJ and Humphries RK: Functional regulation of pre-B-cell
leukemia homeobox interacting protein 1 (PBXIP1/HPIP) in erythroid
differentiation. J Biol Chem. 287:5600–5614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng Y, Xu X, Zhang Y, Ding J, Wang Y,
Zhang X, Wu Z, Kang L, Liang Y, Zhou L, et al: HPIP is upregulated
in colorectal cancer and regulates colorectal cancer cell
proliferation, apoptosis and invasion. Sci Rep. 5:94292015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Vuurden DG, Aronica E, Hulleman E,
Wedekind LE, Biesmans D, Malekzadeh A, Bugiani M, Geerts D, Noske
DP, Vandertop WP, et al: Pre-B-cell leukemia homeobox interacting
protein 1 is overexpressed in astrocytoma and promotes tumor cell
growth and migration. Neuro-oncol. 16:946–959. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang SC, Chai DS, Chen CB, Wang ZY and
Wang L: HPIP promotes thyroid cancer cell growth, migration and EMT
through activating PI3K/AKT signaling pathway. Biomed Pharmacother.
75:33–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X, Yang Z, Zhang H, Ding L, Li X, Zhu
C, Zheng Y and Ye Q: The estrogen receptor-interacting protein HPIP
increases estrogen-responsive gene expression through activation of
MAPK and AKT. Biochim Biophys Acta. 1783:1220–1228. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pan J, Qin Y and Zhang M: HPIP promotes
non-small cell lung cancer cell proliferation, migration and
invasion through regulation of the Sonic hedgehog signaling
pathway. Biomed Pharmacother. 77:176–181. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng
X, Zhu Z, Jiao H, Lin J, Jiang K, et al: Hepatitis B virus X
protein represses miRNA-148a to enhance tumorigenesis. J Clin
Invest. 123:630–645. 2013.PubMed/NCBI
|
|
21
|
Feng Y, Li L, Zhang X, Zhang Y, Liang Y,
Lv J, Fan Z, Guo J, Hong T, Ji B, et al: Hematopoietic pre-B cell
leukemia transcription factor interacting protein is overexpressed
in gastric cancer and promotes gastric cancer cell proliferation,
migration, and invasion. Cancer Sci. 106:1313–1322. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cai W, Ye Q and She QB: Loss of 4E-BP1
function induces EMT and promotes cancer cell migration and
invasion via cap-dependent translational activation of snail.
Oncotarget. 5:6015–6027. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Graff JR, Konicek BW, Vincent TM, Lynch
RL, Monteith D, Weir SN, Schwier P, Capen A, Goode RL, Dowless MS,
et al: Therapeutic suppression of translation initiation factor
eIF4E expression reduces tumor growth without toxicity. J Clin
Invest. 117:2638–2648. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cencic R, Carrier M, Galicia-Vázquez G,
Bordeleau ME, Sukarieh R, Bourdeau A, Brem B, Teodoro JG, Greger H,
Tremblay ML, et al: Antitumor activity and mechanism of action of
the cyclopenta[b]benzofuran, silvestrol. PLoS One. 4:e52232009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blagden SP and Willis AE: The biological
and therapeutic relevance of mRNA translation in cancer. Nat Rev
Clin Oncol. 8:280–291. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang H, Huang F, Wang J, Wang P, Lv W,
Hong L, Li S and Zhou J: The synergistic inhibition of breast
cancer proliferation by combined treatment with 4EGI-1 and MK2206.
Cell Cycle. 14:232–242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choo AY, Yoon SO, Kim SG, Roux PP and
Blenis J: Rapamycin differentially inhibits S6Ks and 4E-BP1 to
mediate cell-type-specific repression of mRNA translation. Proc
Natl Acad Sci USA. 105:17414–17419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choo AY and Blenis J: Not all substrates
are treated equally: implications for mTOR, rapamycin-resistance
and cancer therapy. Cell Cycle. 8:567–572. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhagwat SV, Gokhale PC, Crew AP, Cooke A,
Yao Y, Mantis C, Kahler J, Workman J, Bittner M, Dudkin L, et al:
Preclinical characterization of OSI-027, a potent and selective
inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer
Ther. 10:1394–1406. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feldman ME, Apsel B, Uotila A, Loewith R,
Knight ZA, Ruggero D and Shokat KM: Active-site inhibitors of mTOR
target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol.
7:e382009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chresta CM, Davies BR, Hickson I, Harding
T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini
P, et al: AZD8055 is a potent, selective, and orally bioavailable
ATP-competitive mammalian target of rapamycin kinase inhibitor with
in vitro and in vivo antitumor activity. Cancer Res. 70:288–298.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thoreen CC, Kang SA, Chang JW, Liu Q,
Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM and Gray NS: An
ATP-competitive mammalian target of rapamycin inhibitor reveals
rapamycin-resistant functions of mTORC1. J Biol Chem.
284:8023–8032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Benjamin D, Colombi M, Moroni C and Hall
MN: Rapamycin passes the torch: a new generation of mTOR
inhibitors. Nat Rev Drug Discov. 10:868–880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Naing A, Aghajanian C, Raymond E, Olmos D,
Schwartz G, Oelmann E, Grinsted L, Burke W, Taylor R, Kaye S, et
al: Safety, tolerability, pharmacokinetics and pharmacodynamics of
AZD8055 in advanced solid tumours and lymphoma. Br J Cancer.
107:1093–1099. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Asahina H, Nokihara H, Yamamoto N, Yamada
Y, Tamura Y, Honda K, Seki Y, Tanabe Y, Shimada H, Shi X, et al:
Safety and tolerability of AZD8055 in Japanese patients with
advanced solid tumors; a dose-finding phase I study. Invest New
Drugs. 31:677–684. 2013. View Article : Google Scholar : PubMed/NCBI
|