Introduction
Hepatocellular carcinoma (HCC) ranks the fifth most
common malignant tumor in the world. Although the outcome of HCC
patients appear to be improving due to a benefit from early
diagnosis and more effective treatment, the majority of HCC cases
are not capable of totally surgical resection and remain
insensitive to radiotherapy and chemotherapy. The overall 5-year
survival rate of HCC patients has shown no improvement over the
last several decades (1). Thus,
more effective strategies against HCC need to be developed.
Dysregulation of apoptosis is a common feature of
malignant cells and represents a significant obstacle to the
treatment of human cancer. Among the regulators of apoptosis,
survivin, a member of the inhibitor of apoptosis protein family,
plays an important role not only in inhibiting apoptosis but also
in regulating mitosis (2). Due to
its function in regulation of the balance between programmed cell
death and cell proliferation, survivin is vital for cancer cell
survival, and have been verified to be a prognostic indicator for
poor survival in several malignancies (3,4). The
most prominent feature of the survivin expression profile is that
it is rarely detected in normal tissues, whereas it is selectively
overexpressed in most tumors (5),
indicating that survivin may be a rational gene-directed target for
cancer therapy. Studies exploiting different strategies including
antisense (6), ribozymes (7), RNAi-mediation (8), survivin-directed vaccines (9), or dominant negative mutants (10) to interfere with survivin expression
and function provided direct and convincing results. The study of
molecular structure illustrated that survivin not only consists of
homodimer, which was crucial to exerting its biological function,
but also comprises some key functional sites, such as Cys-84,
Asp-71 and Thr-34 (11,12). In particular, the Thr-34 has been
identified as an important phosphorylation site by the mitotic
kinase p34cdc2-cyclin B1, and the phosphorylation at Thr-34 has a
direct correlation with anti-apoptotic function of survivin
(13). However, Thr34→Ala in
wild-type survivin would make it a surivinT34A dominant negative
mutant that lost the function being phosphorylated. This mutant is
capable of interfering with the phosphorylation of endogenous
survivin, reducing the proliferative potential of tumour cells and
enhancing tumor cell response to apoptosis inducing anticancer
agents (10).
Arsenic and its derivatives have been applied in
traditional Chinese medicine for thousands of years. Arsenic
trioxide (ATO) is being selected as a first and second line therapy
for the treatment of both newly diagnosed and all-trans-retinoic
acid (ATRA)-refractory acute promyelocytic leukemia (APL) patients
(14). Besides, an overwhelming
number of preclinical studies have demonstrated that ATO has the
ability to induce apoptosis and inhibit tumor cell growth in a wide
variety of solid tumors, including acute myeloid leukemia (15,16),
multiple myeloma (17), head and
neck (18) and glioblastoma
(19). However, preliminary reports
from phase II clinical trials on patients with hepatocellular
carcinoma (20), metastatic renal
cell carcinoma (21), and
metastatic melanoma (22) indicate
that the inconstant susceptibility of various tumor cells to this
drug limits its clinical application as a single chemotherapeutic
agent in a wider spectrum of solid malignancies. A substantial
number of preclinical studies have reported that co-treatment of
ATO with other chemotherapeutic agents has a synergistic effect on
tumor cells both in vitro and in xenograft mouse models
(23–25). To illustrate the apoptotic
activation signals and/or survival inhibition signals in
combination therapy of ATO with other drugs may provide a rational
molecular basis for novel chemotherapeutic strategies. Thus, we
tested whether the combination therapy with mouse survivinT34A and
arsenic trioxide would suppress Hepa1-6 tumor growth in a
synergistic manner. Our data demonstrated that the combination
therapy exhibited an enhanced antitumor activity in mouse HCC
models.
Materials and methods
Reagents
Arsenic trioxide and monoclonal anti-actin were
obtained from Sigma-Aldrich (St. Louis, MO, USA). Arsenic trioxide
was dissolved in 1.65 M NaOH at 5×10−2 M as a stock
solution. Monoclonal anti-caspase-9 and anti-caspase-3 were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Polyclonal anti-PARP were obtained from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). The protein assay kit was purchased from
Bio-Rad Laboratories (Hercules, CA, USA). In situ Cell Death
Detection kit was purchased from Roche (Promega, Madison, WI, USA).
All the chemicals employed in the study were of analytically pure
and of culture grade.
Cell culture
The murine HCC cell line Hepa1-6 obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA) was
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine,
100 U/ml penicillin and 100 µg/ml streptomycin. The cells were
propagated at 37°C in humidified 5% CO2 conditions. The
culture medium was replaced with fresh medium every 2 days.
Plasmid and liposome preparation
The recombinant plasmids carrying PORF-9-Msurvivin
T34A and PORF-9 null were purchased from Invivogen Corp.
Restriction endonuclease analysis PCR and DNA sequence analysis
were performed to confirm the recombinant plasmid (data not shown).
The plasmid was prepared by EndoFree Plasmid Giga kit (Qiagen,
Inc., Chatsworth, CA, USA). Genomic DNA, small DNA fragments, or
RNA were excluded in the plasmid DNA preparation as the OD260/280
ratios were between 1.8–2.0. The DNA was eventually dissolved in
sterile endotoxin free water and stored at −20°C before use. The
cationic lipids DOTAP (dioleyl-trimethylammonium propane) and
cholesterol at equimolar concentrations were dissolved in
chloroform supplemented with methanol (3:1 volume ratio) in a
rotary 100-ml round-bottomed flask, then the organic solvent was
rotated and removed under vacuum for 2 h. The lipid film was
hydrated in 5% dextrose in sterile water to yield a final
concentration of 2.5 mg/ml. Finally, the resulting mixture was
vortexed for 1 min, and then sonicated for 10 min to form small
unilamellar vesicles. The liposome was stored at 4°C. The final
null liposome was small multilamellar liposome in a size range of
80–100 nm, with a zeta potential from +50 to +60 mV. When mixed
with plasmid at a weight ration of 3:1, the size of CLDC vary from
150 to 170 nm, with zeta potential close to neutral as measured by
Malvern Mastersizer.
Apoptosis analysis by flow
cytometry
To investigate the apoptosis inducing effect of
combination treatment, we analyzed the percentage of the early
apoptotic cells by flow cytometry using the Annexin V-FITC/PI
dual-labeling technique. Aliquots of 1×105 Hepa1-6 cells
were plated into 6-well plates in 1 ml culture medium. When
incubated to 70% confluence, cells were treated with ATO (2 µM),
vector+ATO, survivinT34A, survivinT34A+ATO, or untreated control,
respectively. The cells were transfected with 2 µg plasmid/6 µg
liposome. Forty-eight hours after transfection, cells were
harvested to be stained with Annexin V-FITC kit (Beckman Coulter,
Inc., Brea, CA, USA) and to be assessed by flow cytometer.
Determination of reactive oxygen
species (ROS) production
Intracellular ROS production was detected using the
cell-permeable indicator 2,7-dichlorodihydrofluorescein diacetate
(DCFH-DA; Sigma-Aldrich), DCFH-DA was deacetylated intracellularly
by non-specific esterase, which converts into fluorescent compound
2,7-dichlorofluorescein (DCF) upon reaction with hydroxyl radical,
hydrogen peroxide or peroxynitrite. Briefly, cells were harvested
and resuspended in DCFH-DA dye reagent (20 µM) at
1×106/ml, and then were incubated at 37°C for 20 min in
the black box. After dye incubation, the cells were washed and
resuspended in cold PBS. The fluorescence was examined by flow
cytometry.
Mitochondrial transmembrane potential
measurement
Mitochondrial membrane potential (ΔΨm)
was measured by flow cytometry with the mitochondrial tracking
fluorescent dye Rhodamine 123 (Ex/Em=507 nm/529 nm), a
cell-permeable cationic dye, which preferentially enters
mitochondria due to the highly negative potential of mitochondrial
membrane. Uptake and accumulation of Rhodamine 123 in the
mitochondrion are driven by ΔΨm, therefore,
depolarization of mitochondrial membrane potential (ΔΨm)
is reflected by reduction of Rhodamine 123 staining. In brief,
cells were washed twice with PBS and incubated with Rhodamine 123
(100 mg/ml; Sigma-Aldrich) at 37°C for 30 min and then monitored by
flow cytometry.
Western blot analysis
Briefly, 1×105 Hepa1-6 cells were lysed
in lysis buffer. Cells were removed by scraping, and centrifuged at
12,500 rpm for 30 min. The protein concentration of the supernatant
was determined by the Bio-Rad protein assay kit, and whole-cell
lysates after denaturing were sepatated by 10% SDS-PAGE. Gels were
electroblotted onto a poly(vinylidene difluoride) membrane. The
membrane blots were blocked at 4°C in 5% non-fat dry milk overnight
and incubated with each antibody at a recommended dilution for 8 h
at 37°C. Followed by rinsing in solution with 10 mM Tris-HCl pH
7.5, 100 mM NaCl, and 0.1% Tween-20 (TBS-T), the gels were
incubated in horseradish peroxidase-conjugated secondary antibodies
at a dilution of 1:10,000. The immunoreactive bands were detected
by enhanced chemiluminescence (Amersham Corp., Arlington Heights,
IL, USA) followed by autoradiography. Equal loading was confirmed
by detection of β-actin.
Animal study
C57BL/6 female mice 6–8 weeks old, weighing 20–22 g,
were obtained from Beijing WeitongLihua Biological Technology Co.,
Ltd. (Beijing, China). Mice were maintained in a specific
pathogen-free facility with 12-h light-dark cycles. They were
observed for signs of tumor growth, activity, feeding, under the
guidelines of the Institutional Animal Care and Use Committee.
Hepa1-6 cell suspension at concentration of 2.5×106
cells in 100 µl PBS was injected s.c. into the backs of the C57BL/6
mice. Eight days after s.c. tumor inoculation all tumors were
palpable, 40 of these mice were randomly assigned into the
following 5 groups (n=8): i) mice received 5 µg survivinT34A/15 µg
liposome complexes+ATO; ii) mice received 5 µg survivinT34A/15 µg
liposome complexes; iii) mice received ATO (3 mg/kg); iv) mice
received vector+ATO; and v) mice received 100 µl of 0.9% NaCl
solution (NS). They received 15 i.v. administrations via tail vein
on a every 2-day basis and were monitored on a daily basis for
tumor burden, belly and other abnormalities. Before each treatment,
DNA solution was added to liposome solution at a ratio of 1 µg DNA
to 3 µg liposome to form DNA/liposome complex mixture. Then, the
mixture was incubated at room temperature for 30 min. The tumors
were measured in two dimensions by caliper and the mice were
weighed on a every 3-day basis. Tumor volume was calculated as
width2 × length × 0.52. Three days after the last
administration, all mice were sacrificed and then subcutaneous
tumors were extracted and weighed. Primary tumors were fixed in 4%
paraformaldehyde in PBS, embedded in paraffin, and cut into 3–5 µm
sections. Apoptotic index in tumor tissues were detected by
Terminal deoxynucleotidyl transferase mediated dUTPnick-end
labeling (TUNEL) staining according to the manufacturers protocol
(Promega). Images of the representative sections were taken using
the Zeiss Axiovert 400 microscope and AxioCam MRm camera. Five
equal-sized fields were randomly chosen and analyzed. Density was
visualized in each field, yielding the apoptosis index.
Statistical analysis
All statistical tests were performed using the SPSS
13.0 software. Statistical comparisons concerning tumor volume,
tumor weight were performed using one-way analysis of variance
(ANOVA); data as means ± SD were analyzed using unpaired Students
t-test. All P-values were two sides and P-values <0.05 were
defined as statistical significance. Experiments were performed at
least in duplicate.
Results
Effects of survivinT34A combined with
arsenic trioxide on tumor cells in vitro
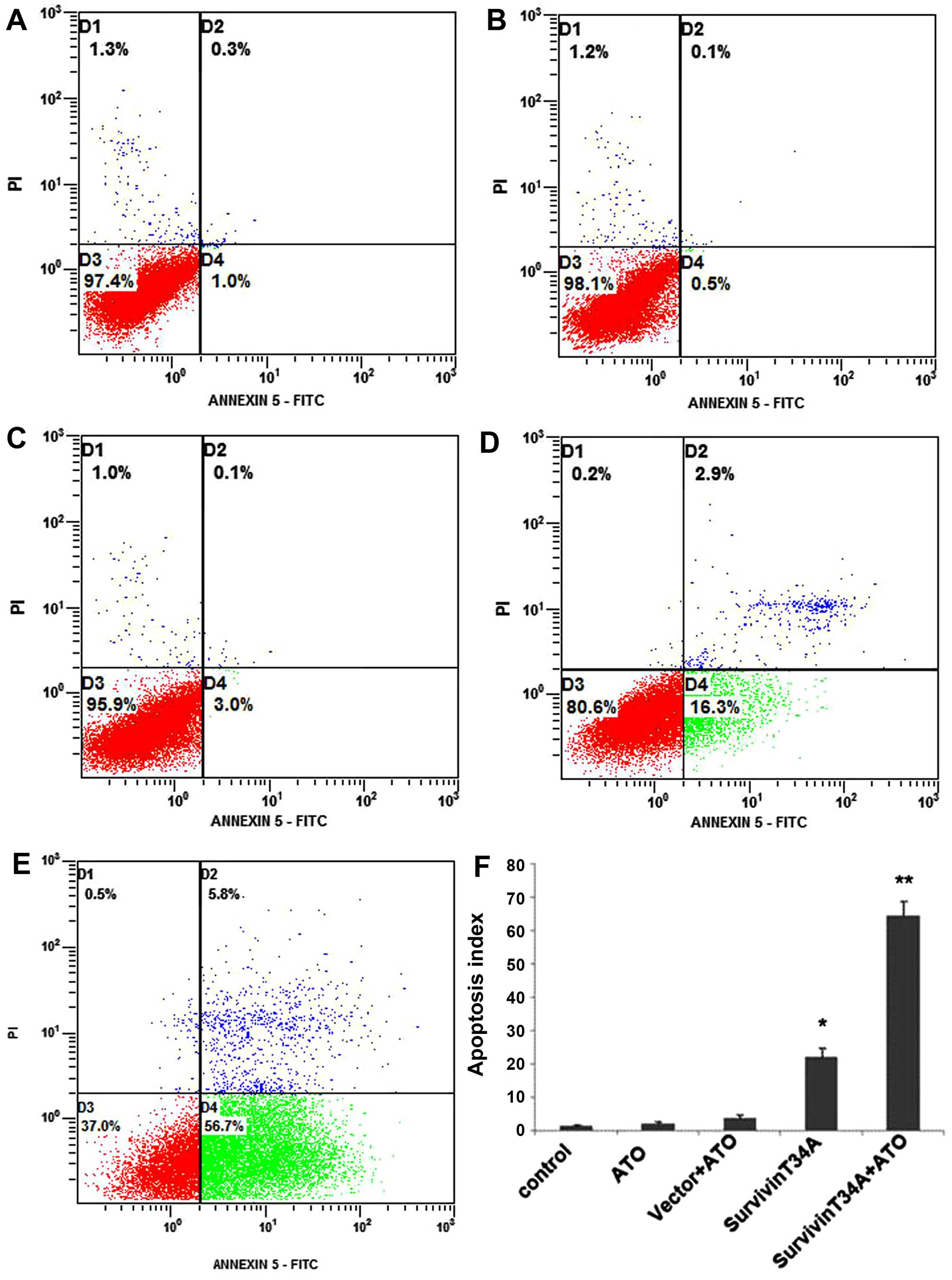
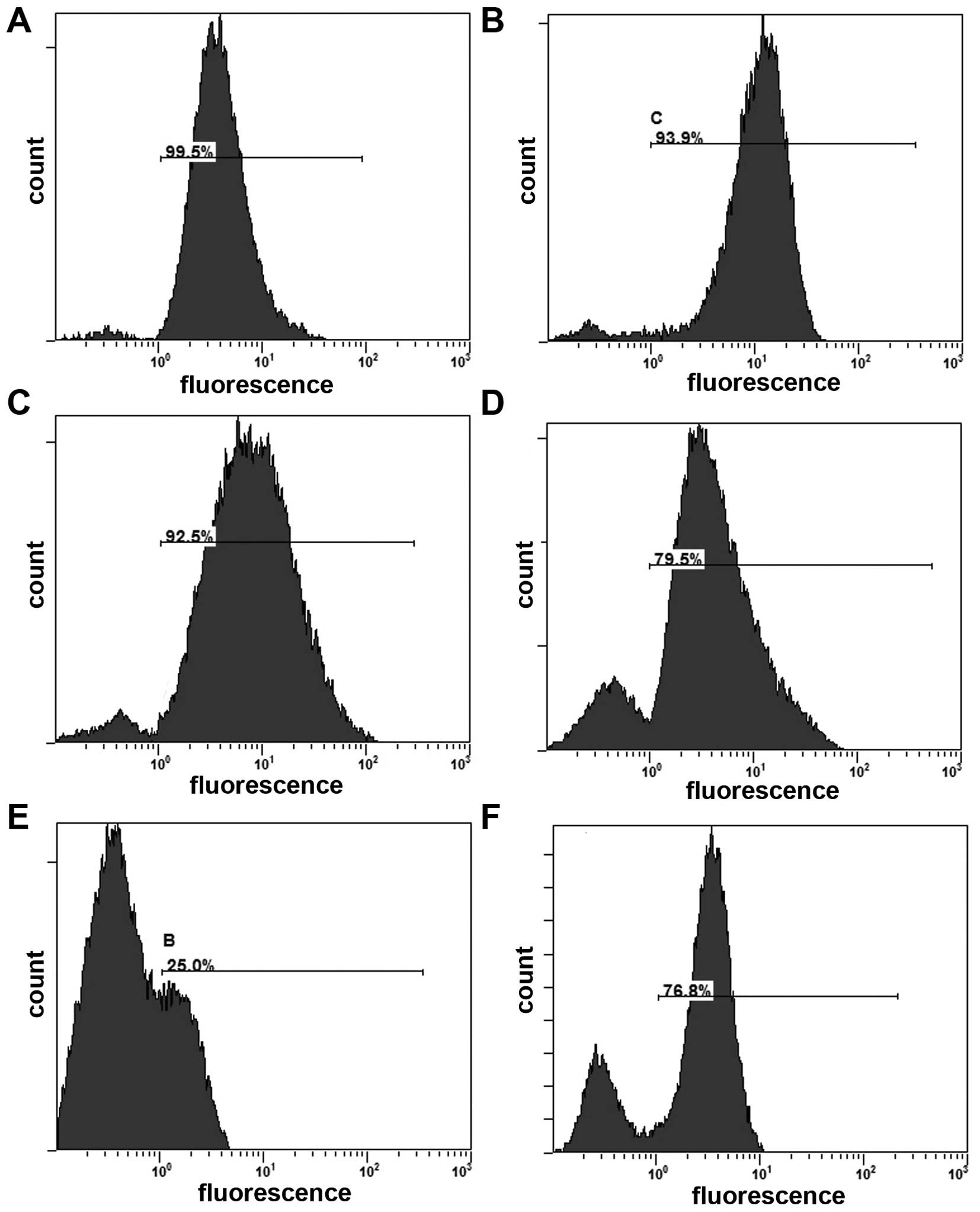
The apoptosis of Hepa1-6 cells in each treatment
group were detected by flow cytometric analysis using Annexin
V-FITC/PI staining. Both early and late apoptotic cells were
represented by Annexin V positive and PI-negative or positive cells
(right quadrants). The results revealed that treatment with
survivinT34A+arsenic trioxide induced more apoptotic cells than any
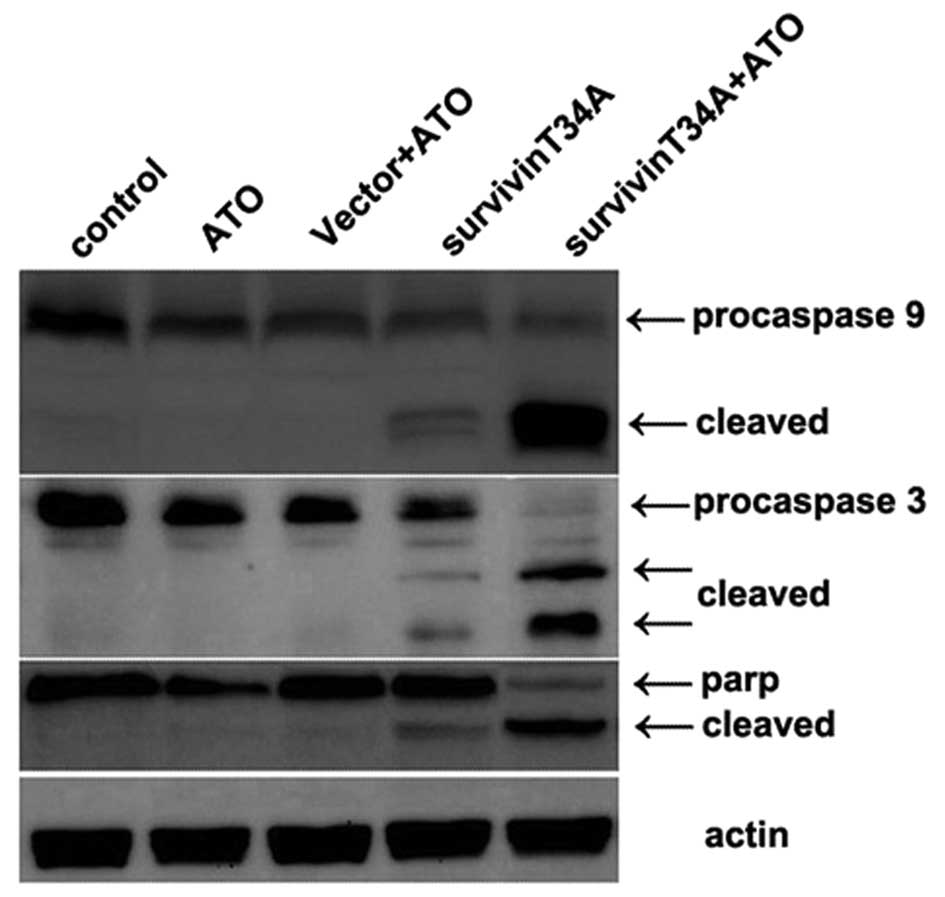
controls (Fig. 1). Caspases play an
essential role as an executor in apoptosis pathway, their
activation level directly reflects the extent of apoptosis. In
intrinsic apoptosis pathway, procaspase-9 is cleaved into an active
caspase, which in turn activates the effector pro-caspases,
including procaspase-3 and −7, to execute the process of apoptosis.
Active caspase-3 results in cleavage of poly(ADP)-ribose polymerase
(PARP). As shown in Fig. 2,
procaspase-9, procaspase-3 and PARP were significantly cleaved by
the co-treatment of survivinT34A+As2O3, but
cleavage of procaspase-3, −9 and PARP were hardly detected in the
other 3 control treatment group, except that there was slight
cleavage in single survivinT34A treatment group, which was clearly
weaker than that of co-treatment group.
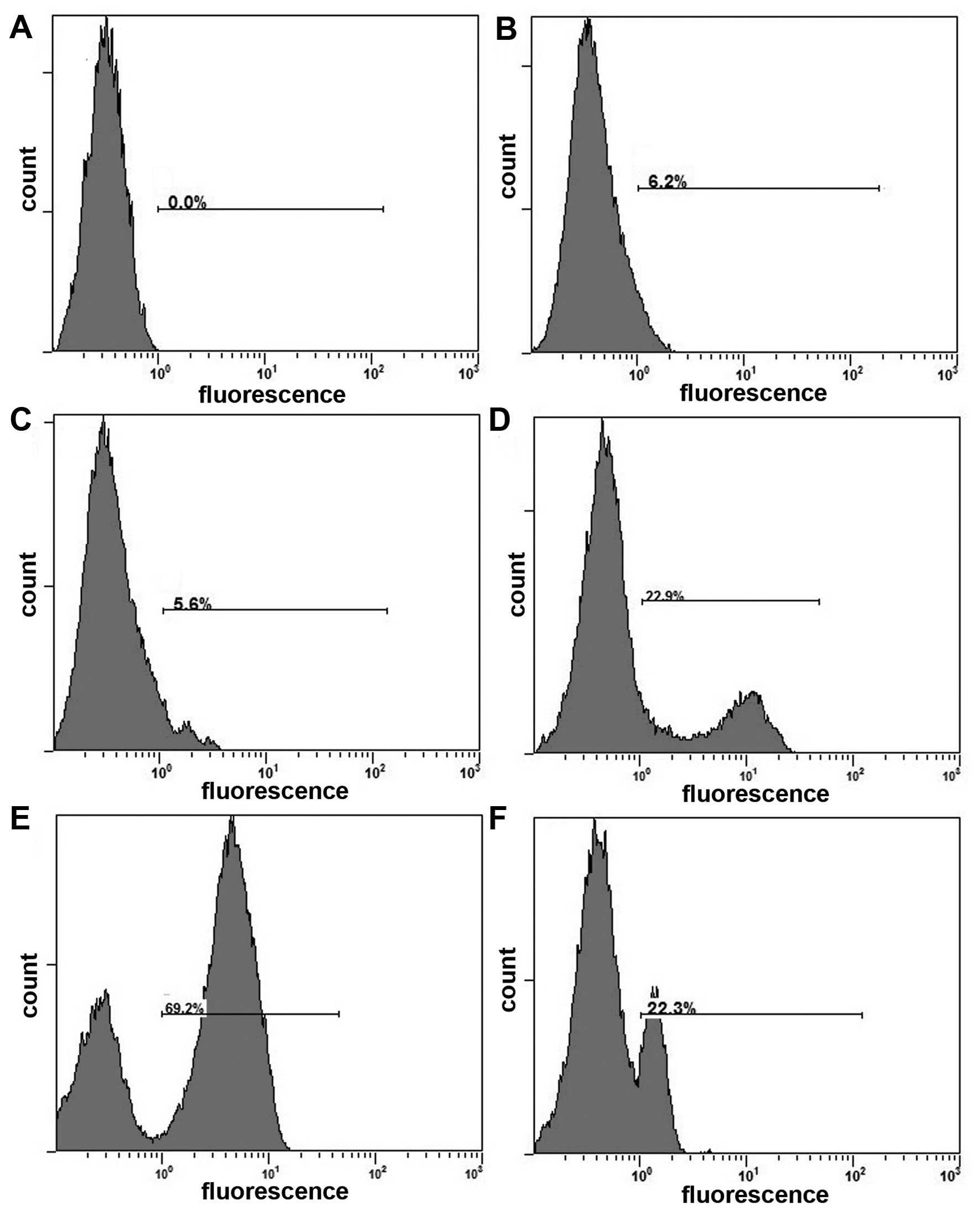
Role of ROS in the combined effect of
survivinT34A and arsenic trioxide
It has been reported that ATO induced cell death in
cancer cells by promoting the production of ROS (26). To explore the molecular mechanism
for the synergistic efficacy of combinational treatment with
survivinT34A+ATO, ROS production in response to survivinT34A alone
and combination with ATO were examined by flow cytometry using the
DHR123 fluorescence dye. As shown in Fig. 3, exposure of cells to survivinT34A
alone increases cellular ROS level, but co-treatment with
survivinT34A and As2O3 remarkably augmented
this ROS level elevation. Next, we determined whether ROS elevation
is crucial for the synergistic effects of survivinT34A and ATO on
cell apoptosis. We found that N-acetyl-l-cysteine, a free radical
scavenger, abrogated the ROS generation enhancement of the combined
treatment (Fig. 3) and
significantly attenuated the efficacy of combined treatment
(Fig. 4). Our results indicate that
ROS elevation is necessary for the induction of apoptosis by
combined treatment with survivinT34A and ATO.
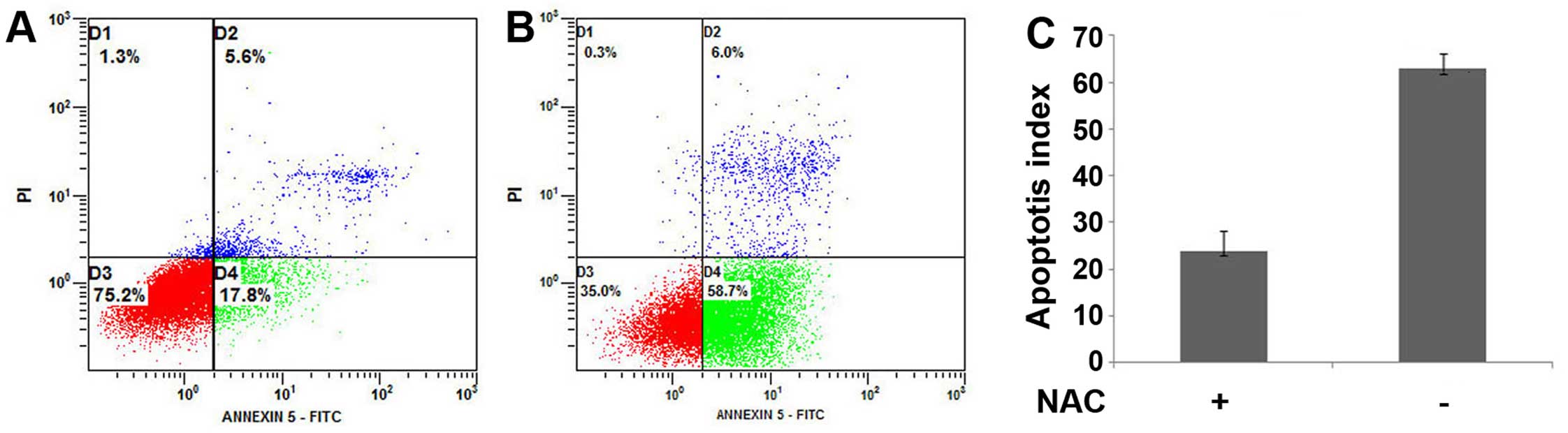
SurvivinT34A combined with arsenic
trioxide induces a loss of mitochondria membrane potential
Excessive ROS production may result in decrease in
the mitochondrial membrane potential and subsequant acceleration of
apoptosis (27). Therefore, we
assessed the changes in mitochondria membrane potential using the
Rho123 fluorescence dye. As shown in Fig. 5, the combinational treatment results
in significant reduction of Rho123 fluorescence compared to that
observed in other treatment groups. In addition, this relocation
was inhibited by N-acetyl-l-cysteine (NAC). This indicates that the
effect of survivinT34A in enhancing As2O3
induced-apoptosis involves mitochondrial damage.
Enhanced antitumor effect by
combination treatment in vivo
On the basis of the in vitro pro-apoptotic
effects of survivinT34A and ATO, we further examined the combined
anti-neoplastic effect of survivinT34A and ATO on Hepa1-6 tumors
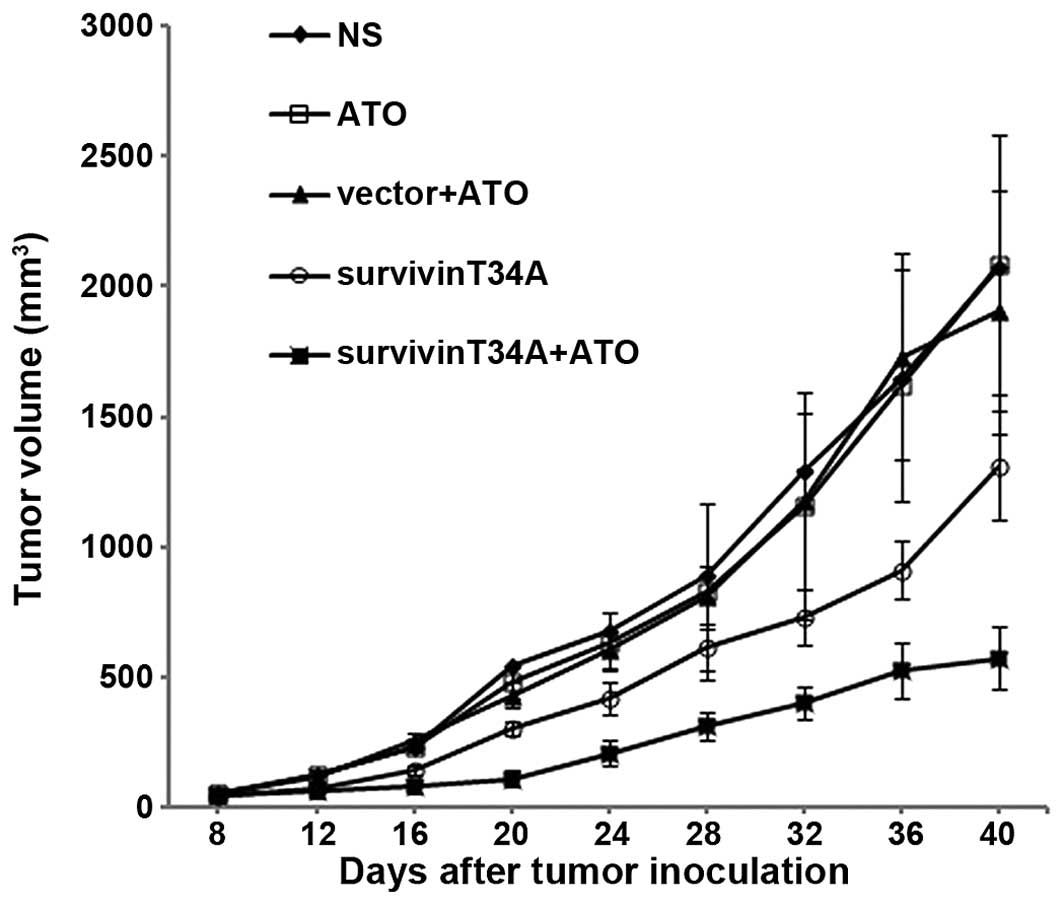
in vivo. Tumor volume of mouse assay monitored every 3 days
showed that the tumor growth was significantly inhibited by
treatment with survivinT34A+ATO while either survivinT34A or ATO
exhibits moderate antitumor efficiency. After 30 days, the average
tumor volumes in mice treated with NS, vector+ATO, ATO,
survivinT34A, and combined therapy were 2074.56±474.532,
2084.86±495.79, 1901.03±463.81, 1313.88±212.87 and 573.82±121.29
mm3, respectively (Fig.
6). Similar results were also observed in the tumor weight. The
average weight of tumors in the combination treatment group
obviously declined compared with survivinT34A alone or other
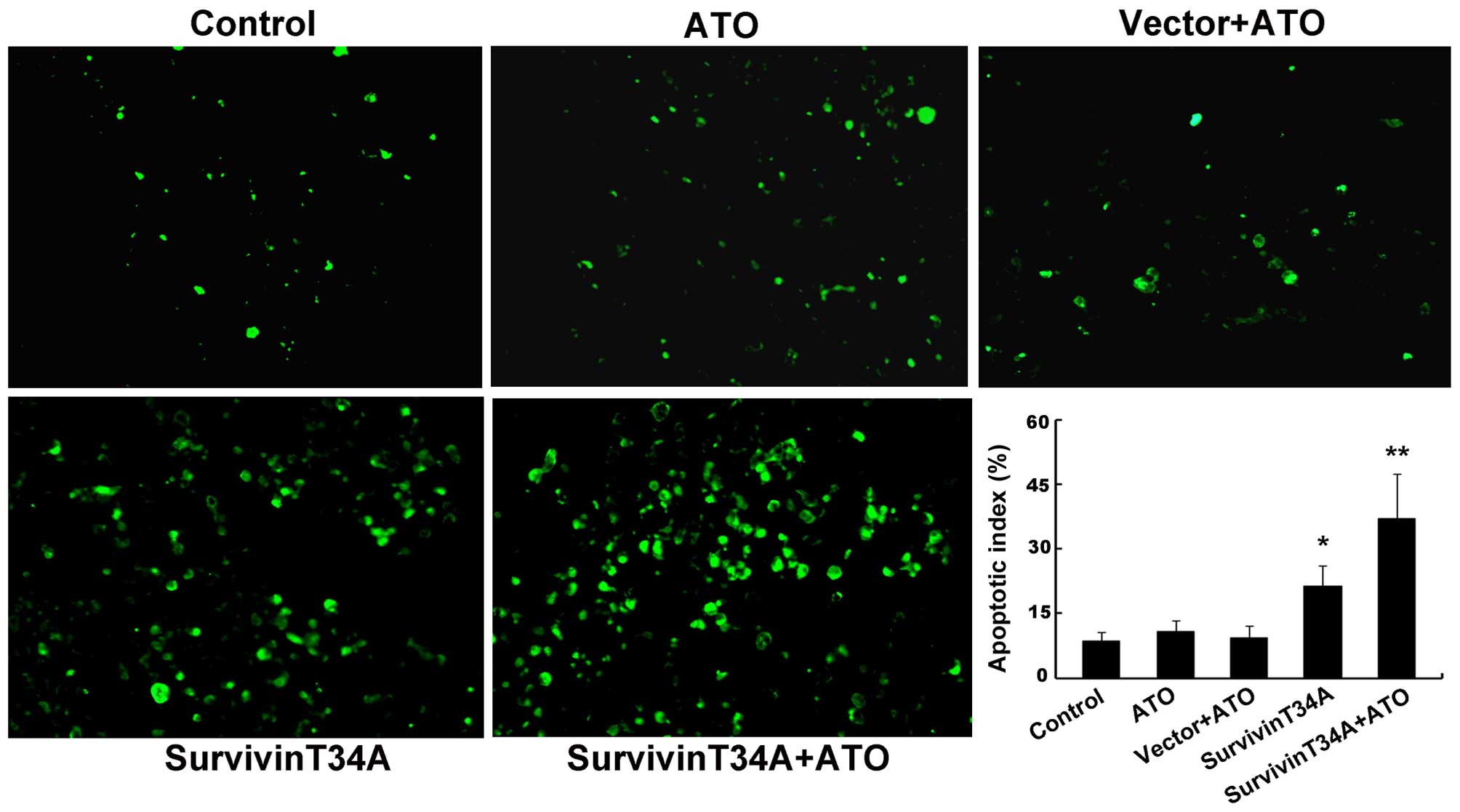
controls (data not shown). To validate whether the cell death came
from apoptosis, apoptotic cells in tumor tissue were detected by
TUNEL analysis. The results revealed that the apoptotic cells in
combination treatment group increased significantly compared to any
of the controls (Fig. 7). The
apoptotic index in the tumor tissues from the combination treatment
group was 40.6±5.8% when compared with all controls (Fig. 7). These results indicated that the
co-treatment with survivinT34A and arsenic trioxide also enhanced
cell apoptosis in Hepa1-6 tumors grafted in C57BL/6 mice.
Discussion
It is well known that treatment with survivinT34A or
ATO alone induces apoptosis via different mechanism.
Phosphorylation at Thr34 of survivin is critical for its regulation
by cyclin-dependent kinase, p34cdc2-cyclin B1, which increased
survivin expression. Moreover, the Thr34 phosphorylation site is
responsible for stabilizing anti-apoptotic protein-protein
interactions through the BIR domain of survivin (11). However, the mutant survivin of
Thr34-Ala (T34A) cannot be phosphorylated by p34cdc2-cyclin B1,
leading to the dissociation of a survivin-caspase-9 complex and
subsequent mitochondrial-dependent apoptosis with release of
cytochrome c and loss of mitochondrial transmembrane
potential during cell division (13). Whereas, ATO biochemically binds to
vicinal thiol groups of proteins with high affinity and
biologically exerts its pleiotropic toxic effects via several types
of reaction including modulation of the intracellular glutathione
redox system and oxidative injury (28), induction of mitotic arrest (29), DNA damage due to the inhibition of
DNA repair (30). As increasingly
evident, therapies based on the combination of anticancer agents
with non-overlapping mechanisms of action can result in enhanced
anticancer efficacy and reduced adverse side-effects. The targeted
disruption of survivin has been proved effective in improving the
efficacy of chemotherapy- and/or radiotherapy-induced apoptosis in
resistant tumor cells (31,32). In this regard, we examined whether
the survivinT34A, which is capable of directly inducing cell death,
could enhance the efficacy of ATO.
In the present study, several observations were made
concerning the mechanism of synergistic antitumor effect by
treatment that combines survivinT34A with Arsenic trioxide. To the
best of our knowledge, the present study has, for the first time,
demonstrated that disruption of survivin by survivinT34A augments
ATO-induced antitumor activity in Hepa1-6 both in vitro and
in vivo. Two major implications can be obtained from the
increased antitumor effects of survivinT34A and ATO. First, an
interruption of survivin levels plays an important role in reducing
apoptosis of HCC. In agreement with this, several studies have
concluded that survivin expression was correlated with poor
prognosis in patients with HCC, and upregulation of survivin is an
independent risk factor in hepatocellular carcinoma cell survival
as well as in resistance to apoptosis (33). Second, and most importantly,
survivinT34A increases arsenic trioxide-induced apoptosis. This
observation is supported by the present findings. The treatment
that combines survivinT34A with ATO significantly increased the
apoptosis of cells when compared with the survivinT34A or ATO
alone. The level of cheavage of procaspase-3 and −9 into the active
form remarkably increased in the combination group compared with
the survivinT34A or ATO groups, suggesting that the initiation of
apoptosis is through the intrinsic pathway and the process of
apoptosis has bypassed the point-of-no-return in the apoptotic
cascade. Thus, it is indicated that increased induction of
apoptosis is crucial for survivinT34A-induced enhancement of the
antitumor effects of ATO in HCC cells.
Cellular reactive oxygen species (ROS) are the major
components of the endogenous oxidants, which is essential to cell
survival, but the effect of ROS on cells is complex. At high level,
oxidative stress may stimulate cells to undergo apoptosis, whereas
in low concentration, it may initiate proliferative or survival
signaling to antagonize apoptosis (34). This implies that two antagonizing
signaling pathways coexist in the process in which cancer cells are
induced to undergo apoptosis by cytotoxic drugs. Therefore, the
effects of ROS depend on their levels. ROS accumulation has been
demonstrated to play a pivotal role in the process in which ATO
triggers tumor cell death (26).
Our results suggest that the elevation of cellular ROS level
elicited by survivinT34A plus ATO may surpass a certain threshold
that finally overrides anti-apoptotic forces, shifting the cell
survival/death balance towards cell death. NAC, as an antioxidant
and ROS scavenger, could inhibit the two-drug combination
treatment-induced apoptotic death. This strongly indicates that the
synergistic efficacy that survivinT34A and arsenic trioxide exert
on Hepa1-6 cancer cells is ROS dependent. It has been proven that
excessive ROS production may result in reduction of the
mitochondrial membrane potential and impair the mitochondrial
respiratory chain (27). In the
present study, we also suggest that the elevating intracellular ROS
level induced by the survivinT34A-As2O3
combined therapy is essential for the loss of mitochondrial
membrane potential.
In our laboratory, Peng et al (35) utilized survivinT34A plasmid (25
µg/one) complexed with cationic liposome (DOTAP:chol) by
intravenous administration to inhibit tumor growth in female BALB/c
mice bearing subcutaneous 4T1 mammary carcinoma. In another study
by Li et al (36)
investigated the synergistic antitumor effect by combining
liposome-encapsulated mouse survivinT34A (20 µg/one) and
hyperthermia in mouse colon cancer models. In this study, the gene
delivery system we utilized was the same as previously reported
(35,36), which proved to efficiently increase
the therapeutic efficacy of the survivinT34A plasmid. Our previous
biodistribution studies have showed that cationic lipid-DNA
complexes was accumulated mainly in tumor tissues (37). This is due to the leaky
microvasculature in tumors that could facilitate extravasation, and
thus, increase the permeability of tumor vessels to liposomes.
In summary, the present study showed that the
combination therapy of survivinT34A and arsenic trioxide exhibited
synergistic induction of apoptosis in hepatocellular carcinoma
cells both in vitro and in vivo, and this synergy
might result from elevation of intracellular ROS level and
mitochondrial damage. Therefore, this study particularly indicated
not only a rational basis for combination therapy with survivin
suppression plus arsenic trioxide, but also a novel therapeutic
regimens for the treatment of some cancers that show failure to
chemotherapy and/or radiotherapy in clinical setting.
Acknowledgements
This study was supported by the Science and
Technology Support Program of Sichuan (2014SZ0122) and the National
Natural Science Foundation of China (grant no. 81101728).
Glossary
Abbreviations
Abbreviations:
|
ATO
|
arsenic trioxide
|
|
APL
|
acute promyelocytic leukemia
|
|
HCC
|
hepatocellular carcinoma
|
|
ATRA
|
all-trans-retinoic acid
|
|
TAL
|
tachypleusamebocytelysate
|
|
DOTAP
|
dioleyltrimethylammonium propane
|
|
ROS
|
reactive oxygen species
|
|
DCFH-DA
|
2,7-dichlorodihydrofluorescein
diacetate
|
|
NAC
|
N-acetyl-l-cysteine
|
|
DCF
|
2,7-dichlorofluorescein
|
|
PARP
|
poly(ADP)-ribose polymerase
|
References
|
1
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Altieri DC: The molecular basis and
potential role of survivin in cancer diagnosis and therapy. Trends
Mol Med. 7:542–547. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gąsowska-Bodnar A, Bodnar L, Dąbek A,
Cichowicz M, Jerzak M, Cierniak S, Kozłowski W and Baranowski W:
Survivin expression as a prognostic factor in patients with
epithelial ovarian cancer or primary peritoneal cancer treated with
neoadjuvant chemotherapy. Int J Gynecol Cancer. 24:687–696. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu JL, Gao W, Kang QM, Zhang XJ and Yang
SG: Prognostic value of survivin in patients with gastric cancer: A
systematic review with meta-analysis. PLoS One. 8:e719302013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deveraux QL and Reed JC: IAP family
proteins - suppressors of apoptosis. Genes Dev. 13:239–252. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kanwar JR, Shen WP, Kanwar RK, Berg RW and
Krissansen GW: Effects of survivin antagonists on growth of
established tumors and B7-1 immunogene therapy. J Natl Cancer Inst.
93:1541–1552. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pennati M, Colella G, Folini M, Citti L,
Daidone MG and Zaffaroni N: Ribozyme-mediated attenuation of
survivin expression sensitizes human melanoma cells to
cisplatin-induced apoptosis. J Clin Invest. 109:285–286. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang G, Li J, Zeng Z and Xian L:
Lentivirus-mediated gene therapy by suppressing survivin in BALB/c
nude mice bearing oral squamous cell carcinoma. Cancer Biol Ther.
5:435–440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pisarev V, Yu B, Salup R, Sherman S,
Altieri DC and Gabrilovich DI: Full-length dominant-negative
survivin for cancer immunotherapy. Clin Cancer Res. 9:6523–6533.
2003.PubMed/NCBI
|
|
10
|
Grossman D, Kim PJ, Schechner JS and
Altieri DC: Inhibition of melanoma tumor growth in vivo by survivin
targeting. Proc Natl Acad Sci USA. 98:635–640. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Verdecia MA, Huang H, Dutil E, Kaiser DA,
Hunter T and Noel JP: Structure of the human anti-apoptotic protein
survivin reveals a dimeric arrangement. Nat Struct Biol. 7:602–608.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Muchmore SW, Chen J, Jakob C, Zakula D,
Matayoshi ED, Wu W, Zhang H, Li F, Ng SC and Altieri DC: Crystal
structure and mutagenic analysis of the inhibitor-of-apoptosis
protein survivin. Mol Cell. 6:173–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
OConnor DS, Grossman D, Plescia J, Li F,
Zhang H, Villa A, Tognin S, Marchisio PC and Altieri DC: Regulation
of apoptosis at cell division by p34cdc2 phosphorylation of
survivin. Proc Natl Acad Sci USA. 97:13103–13107. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sanz MA, Grimwade D, Tallman MS, Lowenberg
B, Fenaux P, Estey EH, Naoe T, Lengfelder E, Büchner T, Döhner H,
et al: Management of acute promyelocytic leukemia: Recommendations
from an expert panel on behalf of the European LeukemiaNet. Blood.
113:1875–1891. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Momeny M, Zakidizaji M, Ghasemi R, Dehpour
AR, Rahimi-Balaei M, Abdolazimi Y, Ghavamzadeh A, Alimoghaddam K
and Ghaffari SH: Arsenic trioxide induces apoptosis in NB-4, an
acute promyelocytic leukemia cell line, through up-regulation of
p73 via suppression of nuclear factor kappa B-mediated inhibition
of p73 transcription and prevention of NF-kappaB-mediated induction
of XIAP, cIAP2, BCL-XL and survivin. Med Oncol. 27:833–842. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ghaffari SH, Bashash D, Dizaji MZ,
Ghavamzadeh A and Alimoghaddam K: Alteration in miRNA gene
expression pattern in acute promyelocytic leukemia cell induced by
arsenic trioxide: a possible mechanism to explain arsenic
multi-target action. Tumour Biol. 33:157–172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gazitt Y and Akay C: Arsenic trioxide: An
anticancer missile with multiple warheads. Hematology. 10:205–213.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seol JG, Park WH, Kim ES, Jung CW, Hyun
JM, Kim BK and Lee YY: Effect of arsenic trioxide on cell cycle
arrest in head and neck cancer cell line PCI-1. Biochem Biophys Res
Commun. 265:400–404. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dizaji MZ, Malehmir M, Ghavamzadeh A,
Alimoghaddam K and Ghaffari SH: Synergistic effects of arsenic
trioxide and silibinin on apoptosis and invasion in human
glioblastoma U87MG cell line. Neurochem Res. 37:370–380. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin CC, Hsu C, Hsu CH, Hsu WL, Cheng AL
and Yang CH: Arsenic trioxide in patients with hepatocellular
carcinoma: A phase II trial. Invest New Drugs. 25:77–84. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vuky J, Yu R, Schwartz L and Motzer RJ:
Phase II trial of arsenic trioxide in patients with metastatic
renal cell carcinoma. Invest New Drugs. 20:327–330. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim KB, Bedikian AY, Camacho LH,
Papadopoulos NE and McCullough C: A phase II trial of arsenic
trioxide in patients with metastatic melanoma. Cancer.
104:1687–1692. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kumar P, Gao Q, Ning Y, Wang Z, Krebsbach
PH and Polverini PJ: Arsenic trioxide enhances the therapeutic
efficacy of radiation treatment of oral squamous carcinoma while
protecting bone. Mol Cancer Ther. 7:2060–2069. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu YC, Yen WY, Lee TC and Yih LH: Heat
shock protein inhibitors, 17-DMAG and KNK437, enhance arsenic
trioxide-induced mitotic apoptosis. Toxicol Appl Pharmacol.
236:231–238. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang N, Wu ZM, McGowan E, Shi J, Hong ZB,
Ding CW, Xia P and Di W: Arsenic trioxide and cisplatin synergism
increase cytotoxicity in human ovarian cancer cells: Therapeutic
potential for ovarian cancer. Cancer Sci. 100:2459–2464. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li JX, Shen YQ, Cai BZ, Zhao J, Bai X, Lu
YJ and Li XQ: Arsenic trioxide induces the apoptosis in vascular
smooth muscle cells via increasing intracellular calcium and ROS
formation. Mol Biol Rep. 37:1569–1576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park MT, Kang YH, Park IC, Kim CH, Lee YS,
Chung HY and Lee SJ: Combination treatment with arsenic trioxide
and phytosphingosine enhances apoptotic cell death in arsenic
trioxide-resistant cancer cells. Mol Cancer Ther. 6:82–92. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jing Y, Dai J, Chalmers-Redman RM, Tatton
WG and Waxman S: Arsenic trioxide selectively induces acute
promyelocytic leukemia cell apoptosis via a hydrogen
peroxide-dependent pathway. Blood. 94:2102–2111. 1999.PubMed/NCBI
|
|
29
|
Li YM and Broome JD: Arsenic targets
tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res.
59:776–780. 1999.PubMed/NCBI
|
|
30
|
Yoo DR, Chong SA and Nam MJ: Proteome
profiling of arsenic trioxide-treated human hepatic cancer cells.
Cancer Genomics Proteomics. 6:269–274. 2009.PubMed/NCBI
|
|
31
|
Tirrò E, Consoli ML, Massimino M, Manzella
L, Frasca F, Sciacca L, Vicari L, Stassi G, Messina L, Messina A,
et al: Altered expression of c-IAP1, survivin, and Smac contributes
to chemotherapy resistance in thyroid cancer cells. Cancer Res.
66:4263–4272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Konduri S, Colon J, Baker CH, Safe S,
Abbruzzese JL, Abudayyeh A, Basha MR and Abdelrahim M: Tolfenamic
acid enhances pancreatic cancer cell and tumor response to
radiation therapy by inhibiting survivin protein expression. Mol
Cancer Ther. 8:533–542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu JL, Zhang XJ, Zhang Z, Zhang AH, Wang
W and Dong JH: Meta-analysis: Prognostic value of survivin in
patients with hepatocellular carcinoma. PLoS One. 8:e833502013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Burdon RH: Control of cell proliferation
by reactive oxygen species. Biochem Soc Trans. 24:1028–1032. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peng XC, Yang L, Yang LP, Mao YQ, Yang HS,
Liu JY, Zhang DM, Chen LJ and Wei YQ: Efficient inhibition of
murine breast cancer growth and metastasis by gene transferred
mouse survivin Thr34➝Ala mutant. J Exp Clin Cancer Res. 27:462008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li ZM, Zhao YW, Zhao CJ, Zhang XP, Chen
LJ, Wei YQ and Yang HS: Hyperthermia increases the therapeutic
efficacy of survivinT34A in mouse tumor models. Cancer Biol Ther.
12:523–530. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang AL, Wan Y, Liao DY, Hu HZ, Wei L,
Wang XH, Wen YJ, Li J, Chen LJ, Kan B, et al: Suppression of human
MDA-MB-435S tumor by U6 promoter-driven short hairpin RNAs
targeting focal adhesion kinase. J Cancer Res Clin Oncol.
136:1229–1242. 2010. View Article : Google Scholar : PubMed/NCBI
|