Introduction
Oral cancers are the sixth most common malignancies
and they affect more than 300,000 new patients being diagnosed
every year worldwide (1,2). Of oral cancers 90% are oral squamous
cell carcinoma, the most common malignant neoplasm of the oral
cavity (1,3). In fact, in spite of the many
advancements made in the field of oral cancer prevention and
multimodality treatments, the 5-year survival rate for OSCC remains
at a disappointingly stable level, almost unchanged over the past
20 years (4–6). The poor prognosis of OSCC is mainly
due to a low response rate to current therapeutic strategies.
Altered molecular expressions might be potential
markers for a diagnosis and prognosis of OSCC (7). The susceptibility of an individual to
oral cancer is mediated by genetic and environmental factors
(3,8). Epigenetics is another major player in
multistep carcinogenesis of oral cancers (4). Histone deacetylases (HDACs) play a key
role in the epigenetic regulation of genes by catalyzing the
removal of acetyl groups. Eighteen HDACs have been characterized in
humans and are subdivided into four groups based on their homology
to yeast HDACs (9). Class I HDACs
are homologous to yeast Rpd3 and consist of HDAC1, 2, 3 and 8
(10). Class II HDACs are
homologous to yeast Hda1 and have been subdivided into class IIa
(HDAC4, 5, 7 and 9) and IIb (HDAC6 and 10) based on domain
organization (11,12). Class III HDACs is composed of the
Sirtuins (SIRT) proteins 1–7 and HDAC11 is classified separately as
class IV (11). Recently,
overexpression of HDACs has been observed in many cancers and
inhibition of specific HDAC has emerged as a new target for cancer
therapy. Class I HDACs are the most thoroughly investigated with
respect to function and relevance for tumor formation and
progression (13). HDAC1, HDAC2 and
HDAC3 expression were associated with advanced-stage disease and
poor prognosis (14,15). High HDAC8 expression was also
observed in advanced stage of neuroblastoma (16). Previous studies showed that HDAC2
expression was overexpressed in paraffin-embedded biopsies from
OSCC patients. However, the correlation between HDAC8 expression
and oral cancer has not been reported. The present study examined
the expression of HDAC8 and the inhibitory effect of HDAC8 in
OSCC.
Materials and methods
Cell culture and reagent
The human OSCC YD-8 cells, YD-10B cells and SNU-1076
cells were purchased from the Korea Cell Line Bank (Seoul, Korea).
The human OSCC FaDu cells were purchased from the American Type
Culture Collection (ATCC; Rockville, MD, USA). YD-8, YD-10B and
SNU-1076 cell lines were incubated in RPMI-1640 medium (Gibco,
Rockville, MD, USA) containing 10% fetal bovine serum (FBS; Gibco)
and 100 U/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA,
USA). FaDu cell lines were incubated in MEM medium (Gibco)
containing 10% FBS (Gibco) and 100 U/ml penicillin-streptomycin
(Invitrogen). Immortalized normal oral keratinocytes (INOK) was
used for normal control, as described in a previous study (17) and was incubated in Dulbeccos
modified Eagles medium (DMEM) containing 10% FBS (Gibco) and 100
U/ml penicillin-streptomycin (Invitrogen). All cells were
maintained as monolayers at 37°C in an atmosphere containing 5%
CO2/air.
Tissue sample preparation
Tissue samples from patients were collected during
surgery or biopsy after obtaining informed consents from patients
in Wonkwang Dental Hospital. Fourteen OSCC patients confirmed by
pathological diagnosis were included in the present study. Median
age of these patients was 57 years, 9 patients were male and 5
females. Eight inflammatory gingival inflammatory hyperplasia or
fibrous hyperplasia of the buccal mucosa were examined as benign
controls. Median age of these patients was 53 years, 4 patients
were male and 4 females. Samples were fixed in 10% buffered
formalin and then embedded in paraffin. All surgically resected
specimens were sectioned for histopathological examination and
immunohistochemical detections. The protocol for the present study
was approved by the ethics committee of Wonkwang University, School
of Dentistry.
Immunohistochemistry
Paraffin-embedded tissues were used to identify
HDAC8 expression. Deparaffinization was achieved with xylene
followed by a descending series of ethanol concentrations. Antigen
retrieval was carried out in a microwave-heated citrate buffer (pH
6.0) for 20 min. The endogenous peroxidases were blocked with 3%
H2O2/methanol for 15 min at room temperature.
The non-specific epitopes were blocked with 1% normal goat serum
for 30 min at room temperature. The tissue sections were incubated
overnight at 4°C with HDAC8 antibody (1:100; Abcam, Cambridge, MA,
USA). The immunoreactions were visualized using a
streptavidin-biotin complex method followed by a diaminobenzidine
reaction (Invitrogen). The tissue sections were counterstained with
hematoxylin in order to visualize the nuclei. The immunoreactions
were viewed under an optical microscope (magnification, ×400;
Leica) and the images recorded on a digital camera (Olympus
Optical, Co., Ltd., Tokyo, Japan).
Trypan blue exclusion assay
The trypan blue exclusion assay was based on the
capability of viable cells to exclude the dye. Five minutes later
0.4% trypan blue (Gibco) was added to cells, they were loaded into
a hematocytometer and counted for the dye uptake. The number of
viable cells was calculated as the percentage of the total cell
population.
siRNA transfection
The siRNA oligonucleotides for HDAC8 were purchased
from Genolution Pharmaceuticals (Seoul, Korea), and the
non-targeting control siRNA (NC. siRNA) was used as the negative
control. The human HDAC8 siRNA sequences are as follows: HDAC8
siRNA #1, sense 5-CGAG UAUGUCAGUAUGUGU(UU)-3 and antisense 5-ACACAU
ACUGACAUACUCG(UU)-3; HDAC8-siRNA #2, sense
5-CAUAUGCACUGCAUAAGCA(UU)-3 and antisense
5-UGCUUAUGCAGUGCAUAUG(UU)-3. The cells were transfected with 100 nM
siRNA for 24 h using Lipofectamine RNAiMAX reagent (Invitrogen).
The cells were harvested for protein analysis after 48 h. Western
blot analysis was used to validate the silencing of protein
expression.
Western blot analysis
The cells were washed with phosphate-buffered saline
(PBS) and harvested in lysis buffer. Samples containing equal
amounts of protein were resolved on SDS-polyacrylamide gel in a
6–15% gel, transferred to a polyvinylidene difluoride (PVDF)
membrane (NEN Life Science, Inc., Boston, USA), and probed
sequentially with antibodies against HDAC8 (Millipore, Bedford, MA,
USA), cleaved caspase-9, procaspase-7, procaspase-3, PARP, LC3B,
Beclin-1, ATG5, ATG12, p62 (Cell Signaling Technology, Danvers, MA,
USA) and actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The
blots were developed using an enhanced chemiluminescence (ECL) kit
(Amersham, Cardiff, UK).
Apoptosis analysis by flow
cytometry
The cells were fixed in chilled 75% methanol and
stained with a propidium iodine (PI) solution (100 µg/ml RNase and
10 µg/ml PI in PBS) for cell cycle analysis. The cells were stained
to the Vybrant® apoptosis assay kit (Molecular Probes,
Eugene, OR, USA), followed by labeling Alexa Fluor® 488
Annexin V and PI for apoptosis analysis. Data acquisition and
analysis was carried out using Cell Llab Quanta™ SC flow cytometry
(Beckman Coulter, Inc., Miami, FL, USA) and software.
Detection of acidic vesicular
organelles (AVOs)
Autophagy is characterized by the formation and
promotion of AVO. To detect the development of AVOs, the cells were
stained with acridine orange as previously described (18). In acridine orange stained cells, the
cells fluoresce bright green, whereas acidic compartments fluoresce
bright red. Briefly, the treated cells were stained with acridine
orange (1 µg/ml) for 15 min. To quantify the development of AVOs,
the stained cells were analyzed using FACScan flow cytometer and
CellQuest software (Beckman Coulter, Inc.).
Statistical analysis
Data are expressed as the mean ± SEM of at least
three individual experiments. Statistical comparisons between
groups were performed using two-tailed Students t-test (Microsoft
Excel). Statistical significance was set at *P<0.05, **P<0.01
and ***P<0.001.
Results
Expression level of HDAC8 in OSCC
tissues and OSCC cell lines
To identify whether HDAC8 is highly expressed in
OSCC, we examined the expression of HDAC8 in benign controls and
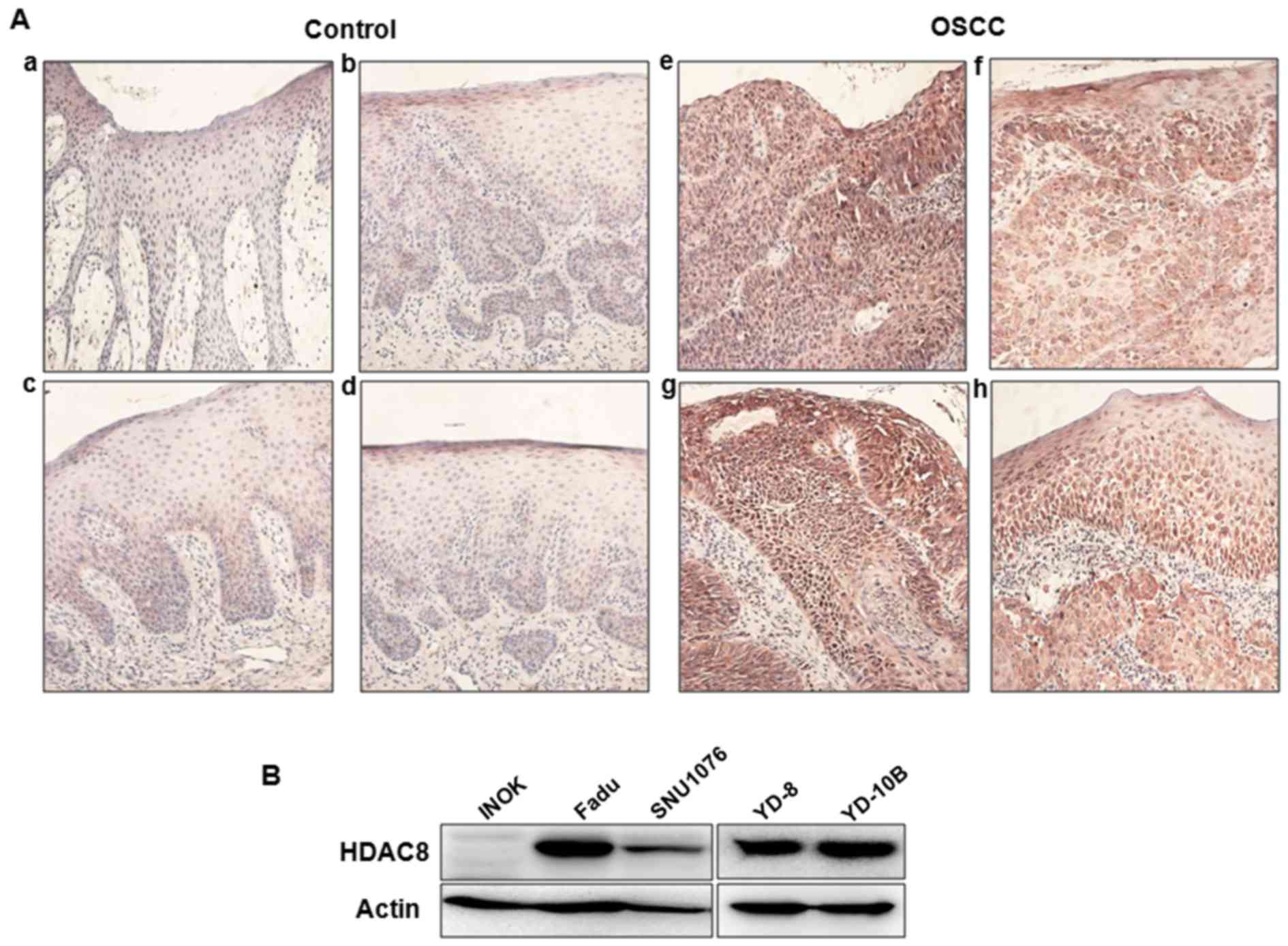
tumor tissues using immunohistochemstry. As shown in Fig. 1A, the level of HDAC8 expression was
markedly overexpressed in OSCC tissues compared to benign control
tissues. HDAC8 expression was mainly observed in the cytoplasm of
tissues, especially epithelial cells. To confirm overexpression of
HDAC8 in OSCC, we compared the levels of HDAC8 expression between
several human OSCC cell lines and INOK cells using western blot
analysis. Overexpression of HDAC8 were observed in several OSCC
cell lines, whereas there was no expression in INOK cells (Fig. 1B). Especially, HDAC8 was highly
expressed in YD-10B and FaDu cells than other cell lines.
Therefore, YD-10B and FaDu cell lines were selected for the
subsequent experiments.
HDAC8 knockdown inhibits the OSCC cell
proliferation
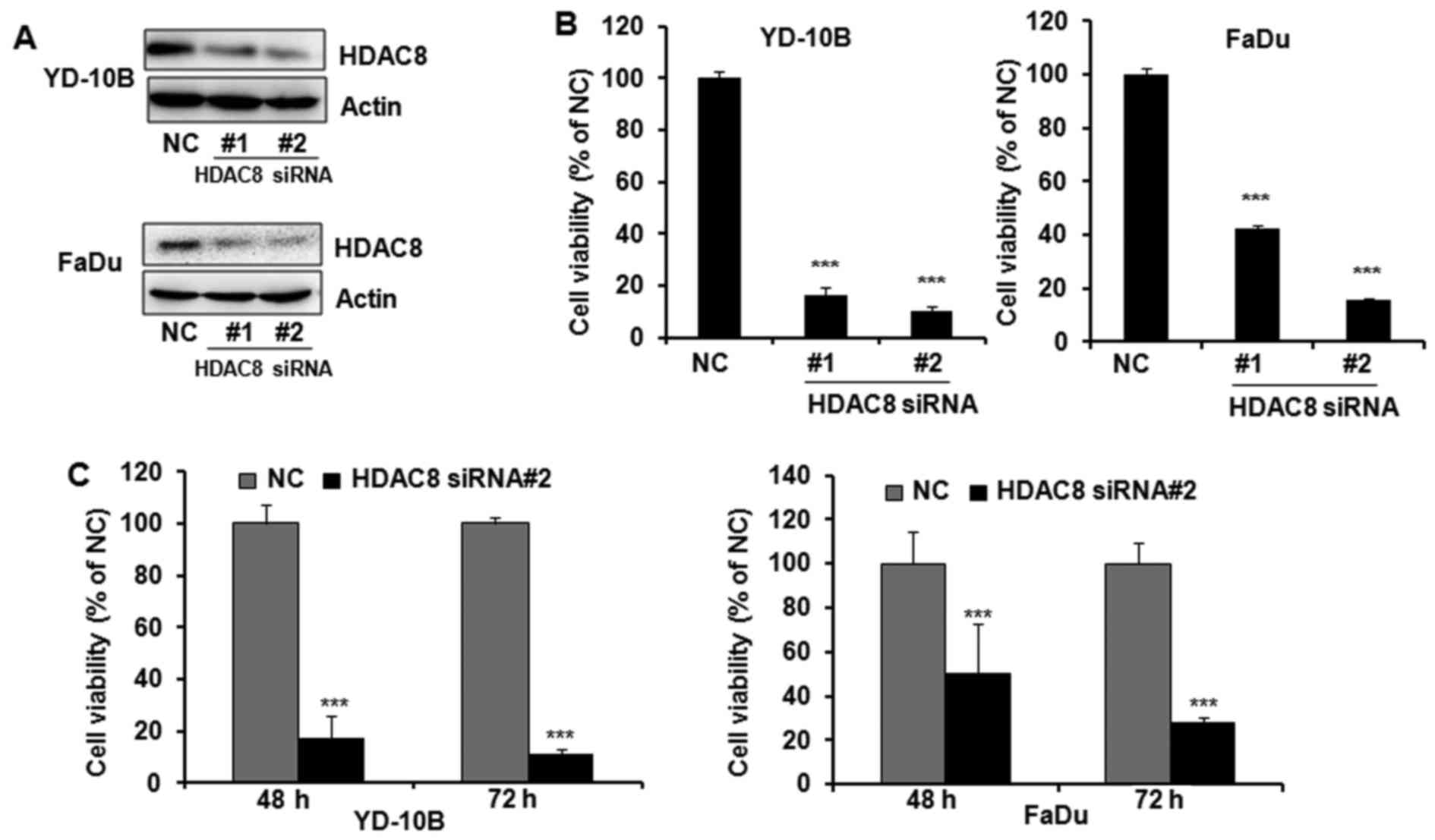
To determine whether the expression of HDAC8 is
associated with cell proliferation in OSCC, HDAC8 was knocked down
in YD-10B and FaDu cells by transfection of HDAC8 siRNAs. YD-10B
and FaDu cells were transfected with two different siRNAs specific
for HDAC8 and the knockdown effect of HDAC8 siRNAs was determined
by western blotting. Transfection with HDAC8 siRNAs reduced
targeted HDAC8 expression in both YD-10B and FaDu cells, whereas
the non-specific targeting control did not affect HDAC8 expression
(Fig. 2A). As shown in Fig. 2B, HDAC8 silencing significantly
inhibited the proliferation of OSCC cells by both HDAC8 siRNA
transfections. HDAC8 siRNA#2 was shown to more efficiently silence
HDAC8 expression and inhibit cell proliferation than HDAC8 siRNA#1
in both OSCC cells. Therefore, HDAC8 siRNA #2 was used for further
HDAC8 knockdown study. Furthermore, HDAC8 siRNA#2 supressed cell
proliferation in a time-dependent manner (Fig. 2C).
HDAC8 silencing induces apoptotic cell
death
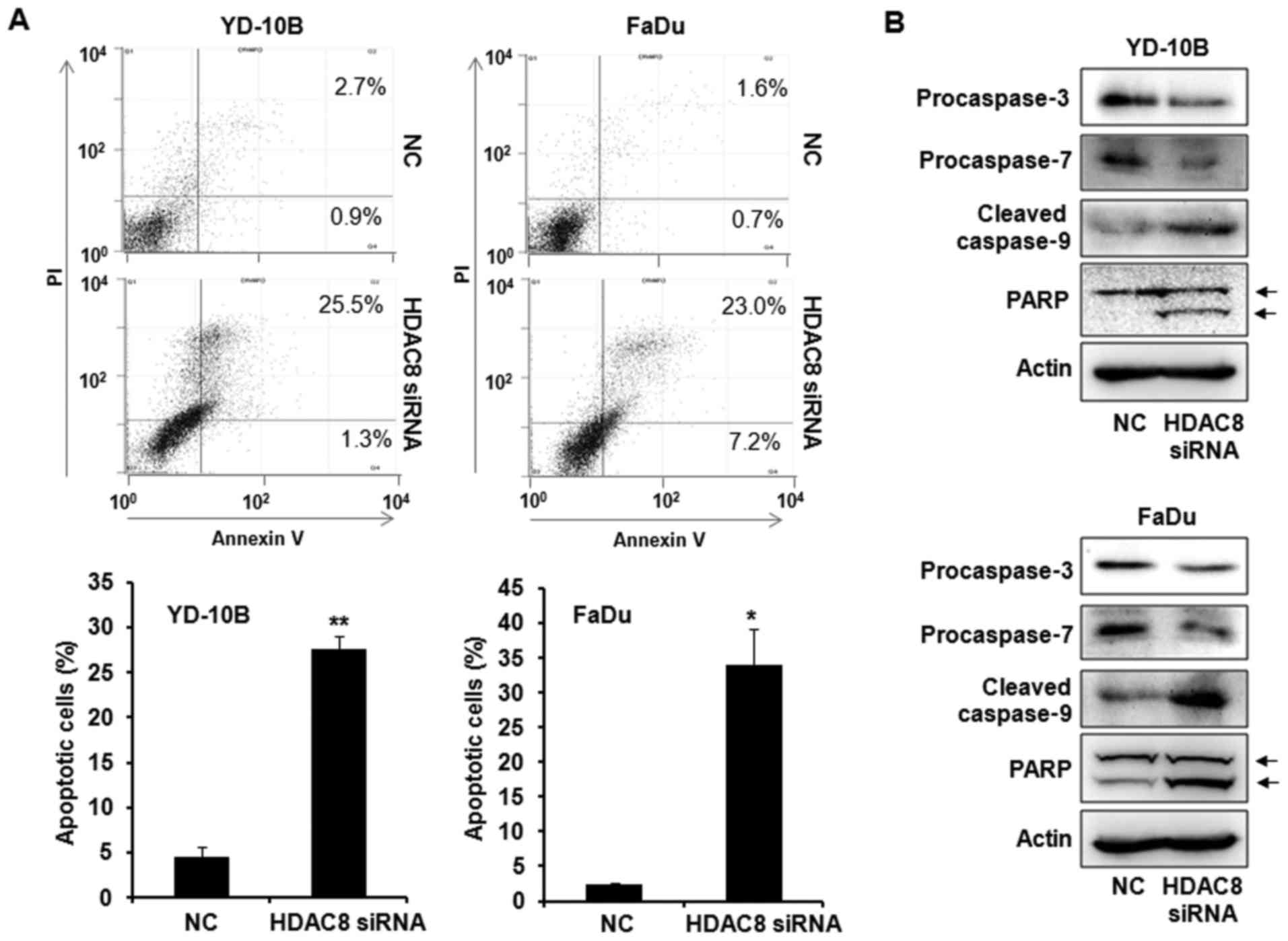
To assess whether the growth inhibitory effects by
HDAC8 knockdown were associated with the induction of apoptosis, we
first examined Annexin V/PI staining by flow cytometry assay. As
shown in Fig. 3A, HDAC8 knockdown
by HDAC8 siRNA transfection significantly increased the number of
Annexin V positive apoptotic cells compared with the NC siRNA
transfected control. The expression of apoptosis-related proteins
was next measured to determine the mechanism of apoptosis using
western blot analysis. The transfection with HDAC8 siRNA markedly
induced the decrease of procaspase-3, −7 and the increase of
cleaved caspase-9 in OSCC cells. HDAC8 knockdown also increased the
level of PARP cleavage, a known endogenous substrate for caspases
(Fig. 3B). These data indicated
that HDAC8 knockdown induced apoptosis by activating caspases,
which cleaved PARP in OSCC cells.
Autophagy induction by HDAC8
silencing
We next determined whether HDAC8 silencing can
induce autophagy in OSCC cells. The levels of autophagy-related
gene expression were examined in HDAC8 siRNA trasfected OSCC cells.
As shown in Fig. 4A, the
transfection with HDAC8 siRNA markedly increased the levels of
Beclin-1, ATG5 and ATG12 which are involved in the early stage of
autophagosome formation. The level of the microtubule-associated
protein 1 light change 3 (LC3)-II, a marker for autophagic vesicles
was also increased in HDAC8-silenced cells compared with control
cells. In addition, reduction of p62 as a marker for autophagic
flux was observed in HDAC8 siRNA transfected cells. The autophagy
response to HDAC8 silencing was next confirmed by acridine orange
staining with flow cytometry assay. Acridine orange staining showed
that HDAC8 knockdown significantly increased the number of AVOs,
red-positive cells, compared to control. Indicated cell percentage
over the line showed acidic red positive cell ratios (Fig. 4B). To evaluate the function of
autophagy by HDAC8 silencing, specific autophagy inhibitor
chloroquine (CQ) was co-treated with HDAC8 siRNA in OSCC cells. We
examined the cell viability using trypan blue exclusion assay.
Combined treatment with HDAC8 knockdown and CQ significantly
reduced cell viability as compared to HDAC8 knockdown without CQ in
both OSCC cells (Fig. 4C). The
expression levels of PARP, caspase-3 and LC3B-II were next examined
using western blot analysis. The accumulation of LC3B-II by CQ was
observed in combined treatment compared to HDAC8 siRNA transfection
alone as expected. Furthermore, the level of PARP cleavage was
markedly increased and procaspase-3 expression was decreased in
combined treatment with HDAC8 knockdown and CQ compared to HDAC8
knockdown alone, these results indicated that inhibition of
autophagy enhanced HDAC8 silencing-induced apoptosis (Fig. 4D).
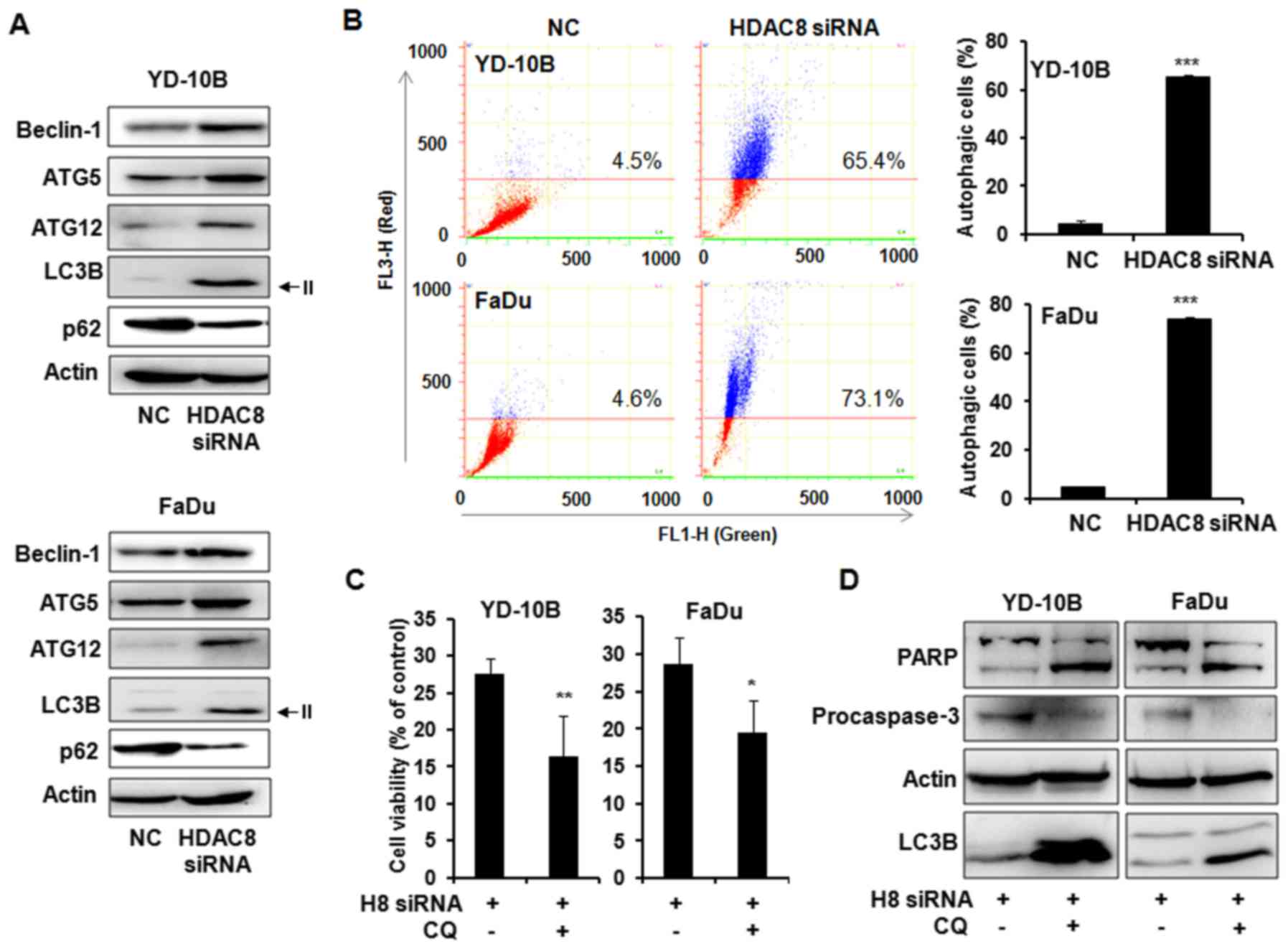 | Figure 4.Autophagy in HDAC8 silencing YD-10B
and FaDu cells. Cells were transfected with HDAC8 siRNA for 24 h
and harvested after 48 h. (A) Expression of the autophagy-related
proteins in YD-10B and FaDu cells. Cell lysates were subjected to
western blot analysis using a series of antibodies against
Beclin-1, ATG5, ATG12, LC3B and p62. The protein levels were
normalized by a comparison to the actin levels. The representative
bands from three independent experiments are shown. (B)
Fluorescence-activated cell sorting analysis using acridine orange.
The cells were stained with acridine orange (1 µg/ml) and then
subjected to flow cytometric analysis. FL1-H, green color
intensity; FL3-H, red color intensity. Indicated cell percentage
over the line show acidic red positive cell ratios. The data are
reported as the mean ± SD of three independent experiments.
***P<0.001 compared to the NC siRNA transfected control. (C) The
cells were transfected with HDAC8 siRNA for 24 h and were harvested
after 48 h incubation with or without chloroquine (CQ, 50 µmol/l).
Cell viability was determined by trypan blue exclusion assay. The
representative results from three independent experiments are
shown. *P<0.05, **P<0.01 compared to the HDAC8 siRNA
transfected cells. (D) Western blot analysis of PARP, procaspase-3
and LC3B expressions in HDAC8 siRNA transfected cells with or
without CQ (50 µmol/l). H8 siRNA, HDAC8 siRNA; NC. siRNA,
non-targeting control siRNA. The protein levels were normalized by
a comparison to the actin levels. The representative bands from
three independent experiments are shown. |
Discussion
Class I HDACs have been most widely studied in their
classical role as histone modifiers and transcriptional repressors
(19). The most frequently studied
and best-characterized human HDACs are HDAC1 and HDAC2. Also, HDAC8
is most recently identified class I HDAC and the role of HDAC8 in
normal and cancer cells remains unclear (20,21).
The expression of HDAC8 has been determined in several cancer
tissues. Upregulation of HDAC8 was detected in colon, urothelial,
ovarian and endometrial cancers (13,20,22,23),
and high HDAC8 expression was associated with markers of poor
prognosis and poor overall survival in neuroblastoma (16). Likewise, our results also showed
that HDAC8 were markedly upregulated in both OSCC tissues and OSCC
cell lines. However, there was no expression in INOK cells and weak
expression in benign control tissues. This study first determined
the expression of HDAC8 in OSCC, therefore, HDAC8 upregulation can
be a diagnostic marker for OSCC and may be associated with oral
carcinogenesis.
HDACs were first identified as enzymes that function
to remove acetyl groups from histones (24,25).
To exert their function, HDACs need to be in the nucleus (26). However, recent studies suggest that
the primary substrates of HDAC enzymes are not histone proteins,
but non-histone proteins (24,27).
It could be demonstrated that the subcellular localization of HDAC
can be nuclear as well as cytoplasmic. The class I HDACs are found
primarily in the nucleus of most cell types and form nuclear
multiprotein complexes that interact with other chromatin modifiers
and transcription factors (10,20).
Unlike other class I HDACs, the subcellular localization of HDAC8
seems to vary with cell types (20,28).
Nakagawa et al (22)
reported that HDAC8 expression was detected in both the nucleus and
the cytoplasm for gastric adenocarcinoma, esophageal squamous cell
carcinoma, prostate carcinoma and breast papillotubular carcinoma,
whereas nuclear localization of HDAC8 was detected in non-cancerous
gastric, esophageal or prostate epithelium. However, cytosolic
expression of HDAC8 was described in differentiating smooth muscle
cells and prostate stromal cells (29,30).
In this study, the immunohistochemistry results showed that
overexpressed HDAC8 was mainly distributed in the cytoplasm of OSCC
tissues. Although the expression levels of HDAC8 were very weak in
benign control tissues, the localization of HDAC8 was also observed
in the cytoplasm. HDAC localization is connected to modulate key
cellular processes, including transcriptional regulatory function
(31). Previous studies reported
that HDAC8 knockdown did not affect global histone H4 acetylation
(16,20), but induced cytoplasm protein
α-tubulin acetylation (20,32), which provided a strong indication
for the cytosolic localization of HDAC8. The results indicated that
HDAC8 is localized in the cytoplasm of oral tissues and, therefore,
we carefully suggest that it may be associated with regulating the
non-histone protein response, rather than histone deactylation
function. Further study will be needed to investigate the
correlation of HDAC8 localization and its function in OSCC.
HDAC appears to be important in the regulation of
proliferation in cancer cells. However, the biological function of
specific HDACs is still unknown. Although the present study showed
upregulation of HDAC8 in OSCC, it is not sure that HDAC8 inhibition
might be a suitable therapeutic target for OSCC. Therefore, we next
examined whether siRNA-mediated HDAC8 knockdown induce cell growth
inhibition in OSCC cells. The results showed that HDAC8 siRNA
markedly reduced the levels of HDAC8 and HDAC8 knockdown
significantly inhibited the cell proliferation in both OSCC cell
lines. Although there was the same effect of HDAC8
knockdown-induced cell growth inhibition in several cancer cells,
the apoptosis induction by HDAC8 inhibition was different. HDAC8
inhibition by pharmacological drug or siRNA induced apoptosis in
human and murine-derived maligant peripheral nerve sheath tumors
and hepatocellular carcinoma, whereas there was a limited induction
of apoptosis after either HDAC8 knockdown or pharmacological
inhibitor treatment in urothelical cancer cell lines (20,21,33).
Consistent with the former findings, the present study showed that
HDAC8 knockdown markedly induced apoptosis through caspases
activation in OSCC cells. The results implied that HDAC8 might play
an important role in regulating cell proliferation and apoptosis in
OSCC cells.
Apoptosis plays a major controling role in cancer
cell death, but recent reports have demonstrated that autophagy is
also an important molecular mechanism for cancer cell death
(34,35). While autophagy is constitutively
important for intracellular quality control and maintenance of
cellular homeostasis from the cytoplasm to lysosomes, autophagy is
frequently activated to much higher levels in cancer cells in
response to a variety of chemotherapeutic treatments (36–39).
In recent studies, HDAC6 is dispensable for starvation-induced
autophagy and HDAC8 is degraded via autophagy and the
ubiqitin-proteasesome system in lung cancer cells (40,41).
We previously determined that HDAC inhibitor apicidin induced both
apoptosis and autophagy in OSCC cells (42), but the correlation between autophagy
induction and specific HDAC inhibition has not been reported in
cancer cells. In the present results, HDAC8 silencing markedly
increased the levels of major autophagic related proteins and
autophagic vesicles in OSCC cells. In addition, autophagy
inhibition by CQ increased apoptotic cell death in HDAC8 siRNA
transfected OSCC cells, which means autophagy by HDAC8 knockdown
have pro-survival effect in OSCC cells. These results are similar
to our previous study for HDAC inhibitor apicidin-treated OSCC
cells (42). We wondered whether
HDAC8 is decreased in apicidin-treated OSCC cells. The expression
of HDAC8 was also markedly inhibited in apicidin treated OSCC cells
(data not shown). Overall, HDAC8 inhibition by siRNA or
pharmacological drug may be highly related with autophagy induction
as a pro-survival function and inhibitors of autophagy could
increase the antitumor effect of HDAC8 inhibition in OSCC
cells.
In conclusion, HDAC8 overexpressed in OSCC tissues
and OSCC cell lines are mainly localized in the cytoplasm. HDAC8
knockdown inhibited cancer cell proliferation and activated both
caspase-dependent apoptotic cell death and pro-survival autophagy
in OSCC cells. Moreover, combined treatment with HDAC8 knockdown
and autophagy inhibitor enhanced cell death through apoptosis
induction. Taken together, these findings provide new and important
information on the diagnosis of OSCC and HDAC8 could be an
effective molecular target of antitumor therapy for patients with
OSCC.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(NRF-2011-0023907, NRF-2016R1D1A3B03931034).
References
|
1
|
Effiom OA, Adeyemo WL, Omitola OG, Ajayi
OF, Emmanuel MM and Gbotolorun OM: Oral squamous cell carcinoma: a
clinicopathologic review of 233 cases in Lagos, Nigeria. J Oral
Maxillofac Surg. 66:1595–1599. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zushi Y, Narisawa-Saito M, Noguchi K,
Yoshimatsu Y, Yugawa T, Egawa N, Fujita M, Urade M and Kiyono T: An
in vitro multistep carcinogenesis model for both HPV-positive and
-negative human oral squamous cell carcinomas. Am J Cancer Res.
1:869–881. 2011.PubMed/NCBI
|
|
3
|
Weng CJ, Hsieh YH, Chen MK, Tsai CM, Lin
CW and Yang SF: Survivin SNP-carcinogen interactions in oral
cancer. J Dent Res. 91:358–363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mascolo M, Siano M, Ilardi G, Russo D,
Merolla F, De Rosa G and Staibano S: Epigenetic disregulation in
oral cancer. Int J Mol Sci. 13:2331–2353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mydlarz WK, Hennessey PT and Califano JA:
Advances and perspectives in the molecular diagnosis of head and
neck cancer. Expert Opin Med Diagn. 4:53–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen YJ, Chang JT, Liao CT, Wang HM, Yen
TC, Chiu CC, Lu YC, Li HF and Cheng AJ: Head and neck cancer in the
betel quid chewing area: recent advances in molecular
carcinogenesis. Cancer Sci. 99:1507–1514. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Agrawal D, Gupta S, Agarwal D, Gupta OP
and Agarwal M: Role of GSTM1 and GSTT1 polymorphism: susceptibility
to oral submucous fibrosis in the North Indian population.
Oncology. 79:181–186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marks PA and Dokmanovic M: Histone
deacetylase inhibitors: discovery and development as anticancer
agents. Expert Opin Investig Drugs. 14:1497–1511. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Delcuve GP, Khan DH and Davie JR:
Targeting class I histone deacetylases in cancer therapy. Expert
Opin Ther Targets. 17:29–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lucio-Eterovic AK, Cortez MA, Valera ET,
Motta FJ, Queiroz RG, Machado HR, et al: Differential expression of
12 histone deacetylase (HDAC) genes in astrocytomas and normal
brain tissue: class II and IV are hypoexpressed in glioblastomas.
BMC Cancer. 8:243–253. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martin M, Kettmann R and Dequiedt F: Class
IIa histone deacetylases: regulating the regulators. Oncogene.
26:5450–5467. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weichert W, Denkert C, Noske A,
Darb-Esfahani S, Dietel M, Kalloger SE, Huntsman DG and Köbel M:
Expression of Class I histone deacetylases indicates poor prognosis
in endometrioid subtypes of ovarian and endometrial carcinomas.
Neoplasia. 10:1021–1027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iglesias-Linares A, Yañez-Vico RM and
González-Moles MA: Potential role of HDAC inhibitors in cancer
therapy: insights into oral squamous cell carcinoma. Oral Oncol.
46:323–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang HH, Chiang CP, Hung HC, Lin CY, Deng
YT and Kuo MY: Histone deacetylase 2 expression predicts poorer
prognosis in oral cancer patients. Oral Oncol. 45:610–614. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oehme I, Deubzer HE, Wegener D, Pickert D,
Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von
Deimling A, et al: Histone deacetylase 8 in neuroblastoma
tumorigenesis. Clin Cancer Res. 15:91–99. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim SA, Kwon SM, Yoon JH and Ahn SG: The
antitumor effect of PLK1 and HSF1 double knockdown on human oral
carcinoma cells. Int J Oncol. 36:867–872. 2010.PubMed/NCBI
|
|
18
|
Kanematsu S, Uehara N, Miki H, Yoshizawa
K, Kawanaka A, Yuri T and Tsubura A: Autophagy inhibition enhances
sulforaphane-induced apoptosis in human breast cancer cells.
Anticancer Res. 30:3381–3390. 2010.PubMed/NCBI
|
|
19
|
Shakespear MR, Halili MA, Irvine KM,
Fairlie DP and Sweet MJ: Histone deacetylases as regulators of
inflammation and immunity. Trends Immunol. 32:335–343. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lehmann M, Hoffmann MJ, Koch A, Ulrich SM,
Schulz WA and Niegisch G: Histone deacetylase 8 is deregulated in
urothelial cancer but not a target for efficient treatment. J Exp
Clin Cancer Res. 10:592014. View Article : Google Scholar
|
|
21
|
Lopez G, Bill KL, Bid HK, Braggio D,
Constantino D, Prudner B, Zewdu A, Batte K, Lev D and Pollock RE:
HDAC8, a potential therapeutic target for the treatment of
malignant peripheral nerve sheath tumors (MPNST). PLoS One.
10:e01333022015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakagawa M, Oda Y, Eguchi T, Aishima S,
Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, et al:
Expression profile of class I histone deacetylases in human cancer
tissues. Oncol Rep. 18:769–774. 2007.PubMed/NCBI
|
|
23
|
Niegisch G, Knievel J, Koch A, Hader C,
Fischer U, Albers P and Schulz WA: Changes in histone deacetylase
(HDAC) expression patterns and activity of HDAC inhibitors in
urothelial cancers. Urol Oncol. 31:1770–1779. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim HJ and Bae SC: Histone deacetylase
inhibitors: molecular mechanisms of action and clinical trials as
anti-cancer drugs. Am J Transl Res. 3:166–179. 2011.PubMed/NCBI
|
|
25
|
Gray SG and Ekstrom TJ: The human histone
deacetylase family. Exp Cell Res. 262:75–83. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
de Ruijter AJ, van Gennip AH, Caron HN,
Kemp S and van Kuilenburg AB: Histone deacetylases (HDACs):
characterization of the classical HDAC family. Biochem J.
370:737–749. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gregoretti IV, Lee YM and Goodson HV:
Molecular evolution of the histone deacetylase family: functional
implications of phylogenetic analysis. J Mol Biol. 338:17–31. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oehme I, Deubzer HE, Lodrini M, Milde T
and Witt O: Targeting of HDAC8 and investigational inhibitors in
neuroblastoma. Expert Opin Investig Drugs. 18:1605–1617. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Waltregny D, De Leval L, Glénisson W, Ly
Tran S, North BJ, Bellahcène A, Weidle U, Verdin E and Castronovo
V: Expression of histone deacetylase 8, a class I histone
deacetylase, is restricted to cells showing smooth muscle
differentiation in normal human tissues. Am J Pathol. 165:553–564.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Waltregny D, North B, Van Mellaert F, de
Leval J, Verdin E and Castronovo V: Screening of histone
deacetylases (HDAC) expression in human prostate cancer reveals
distinct class I HDAC profiles between epithelial and stromal
cells. Eur J Histochem. 48:273–290. 2004.PubMed/NCBI
|
|
31
|
Mathias RA, Guise AJ and Cristea IM:
Post-translational modifications regulate class IIa histone
deacetylase (HDAC) function in health and disease. Mol Cell
Proteomics. 14:456–470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Quan P, Moinfar F, Kufferath I, Absenger
M, Kueznik T, Denk H, Zatloukal K and Haybaeck J: Effects of
targeting endometrial stromal sarcoma cells via histone deacetylase
and PI3K/AKT/mTOR signaling. Anticancer Res. 34:2883–2897.
2014.PubMed/NCBI
|
|
33
|
Wu J, Du C, Lv Z, Ding C, Cheng J, Xie H,
Zhou L and Zheng S: The up-regulation of histone deacetylase 8
promotes proliferation and inhibits apoptosis in hepatocellular
carcinoma. Dig Dis Sci. 58:3545–3553. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu JJ, Lin M, Yu JY, Liu B and Bao JK:
Targeting apoptotic and autophagic pathways for cancer
therapeutics. Cancer Lett. 300:105–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hannigan AM and Gorski SM: Macroautophagy:
the key ingredient to a healthy diet? Autophagy. 5:140–151. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Q, Yang W, Man N, Zheng F, Shen Y,
Sun K, Li Y and Wen LP: Autophagy-mediated chemosensitization in
cancer cells by fullerene C60 nanocrystal. Autophagy. 5:1107–1117.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Levine B: Cell biology: Autophagy and
cancer. Nature. 446:745–747. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin SK and White E: Tumor suppression by
autophagy through the management of metabolic stress. Autophagy.
4:563–566. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee JY and Yao TP: Quality control
autophagy: a joint effort of ubiquitin, protein deacetylase and
actin cytoskeleton. Autophagy. 6:555–557. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Park JY and Juhnn YS: cAMP signaling
increases histone deacetylase 8 expression by inhibiting
JNK-dependent degradation via autophagy and the proteasome system
in H1299 lung cancer cells. Biochem Biophys Res Commun.
470:336–342. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ahn MY, Ahn SG and Yoon JH: Apicidin, a
histone deaceylase inhibitor, induces both apoptosis and autophagy
in human oral squamous carcinoma cells. Oral Oncol. 47:1032–1038.
2011. View Article : Google Scholar : PubMed/NCBI
|