Introduction
Apigenin (4′,5,7-trihydroxy-flavone) is a
plant-derived flavonoid compound, and is ubiquitously found in both
fruits and vegetables. Besides its anti-inflammatory, anti-oxidant
and anticancer properties (1,2),
apigenin has received particular interest as a cancer
chemopreventive agent for a variety of cancers in recent years.
Apigenin inhibits the proliferation of the A2780 ovarian cancer
cells in a dose-time-dependent manner through overexpression of
ATF3 to suppress Id1 expression (3). Apigenin possesses anti-growth activity
on both the prostate (DU-145) and breast (MDA-MB-231) cancer cells
through caspase-3 activation mediated by estrogen receptor (ER)-β
(4). Apigenin at 60 µM induces
apoptosis in the human promyelocytic leukaemia HL-60 cells via
mitochondria dysfunction releasing cytochrome c to active
caspase-3, caspase-9 and cleavage of poly-(ADP-ribose) polymerase
(PARP) (5).
In the eukaryotic cells, protein molecules
spontaneously fold during or after biosynthesis to form
biologically functional conformation. Failure to fold into native
three-dimensional structure generally leads to inactive proteins
(6,7). The endoplasmic reticulum (ER) is the
major organelle for protein biosynthesis, folding, maturation and
translocation (8). When cells are
stimulated, unfolded proteins accumulate in the lumen of ER.
Dis-homeostasis of ER results in a pathological response known as
ER stress. The organelle has its unique signaling pathways to
overcome ER stress, such as i) slowing down translational rate to
prevent further accumulation of the mis-folded proteins; ii)
upregulating the genes capable of increasing the protein-folding
capacity in ER; iii) activating the NFκB to trigger immune and
anti-apoptotic responses; and iv) inducing apoptosis (9). When ER organelle is severely impaired,
apoptosis is provoked to eliminate the damaged cells notably
through upregulating the expression of a sensor protein, C/EBP
homologous protein (CHOP) (9).
Under non-stressed conditions, CHOP expresses at low level;
however, when cells are under adverse conditions (e.g. glucose
deprivation, amino acid starvation and ER stress), CHOP will be
overexpressed to disturb cell cycle progression (10). CHOP can activate downstream protein
expression by upregulating the pro-apoptotic proteins (e.g. Bax and
Bak) and by downregulating the anti-apoptotic proteins (e.g. Bcl-2
and Bcl-xl) to induce the intrinsic pathway, and can also
upregulate downstream the pro-apoptotic death receptor 5 (DR5) to
induce the extrinsic pathway (11).
Intracellular-free radicals such as reactive oxygen
species (ROS) and reactive nitrogen species (RNS) are related with
ER stress, mitochondria dysfunction and inflammation (12). In general, intracellular ROS levels
are relatively low. When cells are exposed to stimuli,
intracellular ROS generation increases. Although ROS can promote
cell proliferation and differentiation, excessive ROS cause
oxidative stress, which is harmful to cells (13). However, in cancer cells, increase of
ROS generation is related with apoptosis (14–16).
ROS at the same level can kill cancer cells without any significant
toxicity to their normal counterparts (17). Increasing evidence shows that
overload ROS will cause ER stress. For example, auranofin can
induce ROS-mediated ER stress and mitochondrial dysfunction via
upregulating CHOP expression and cleaving caspase-3 and PARP in the
human gastric cancer cells, BGC-823 and SGC-7901 (18). Sarsasapogenin from a Chinese medical
herb Anemarrhena asphodeloides Bunge causes both significant
intracellular ROS generation and CHOP expression in the HeLa cells
(19). However, if an anti-oxidant
N-acetyl cysteine (NAC) is used to treat the cells before
sarsasapogenin application, the amounts of ROS (together with ER
stress and apoptosis) decrease, implying that
sarsasapogenin-induced ER stress and mitochondria dys-function are
triggered by ROS generation (19).
Pre-treatment of the MIA PaCa-2 cells with NAC will
markedly decrease intracellular ROS generation induced by a
synthesized polyphenol conjugate
(E)-3-(3,5-dimethoxyphenyl)-1-(2-methoxyphenyl)prop-2-en-1-one
(DPP-23), along with reduced expression of the UPR (unfolded
protein response) proteins (GRP78/BiP, IRE1a, and CHOP) (20). This indicates that intracellular ROS
production is upstream of the ER stress-mediated apoptosis
(20). Iso-obtusilactone A isolated
from Cinnamomum kotoense can increase intracellular ROS
generation, and upregulate CHOP and DR5 protein levels in the
hepatoma Hep G2 cells; however, pre-treatment the cells with NAC
can decrease ROS generation associated with lower expression levels
of the two apoptotic-related proteins (21). It is thus proved that much ROS
production is able to provoke ER stress-mediated apoptosis towards
cancer cells.
To obtain more evidence on anticancer potential of
apigenin, this study aimed to verify its in vitro effects on
a human colon carcinoma line (HCT-116 cells), and to outline a
possible apoptotic mechanism involving both intracellular ROS
generation and ER stress.
Materials and methods
Chemicals and reagents
Apigenin with purity >99% was purchased from
Shanghai Yousi Biotechnology Co. Ltd. (Shanghai, China). Cell
Counting Kit-8 (CCK-8), cell cycle analysis kit, Annexin V-FITC
apoptosis detection kit, Hoechst 33258, ROS assay kit,
mitochondrial membrane potential assay kit with JC-1, Fura-2
pentakis (acetoxymethyl) ester (Fura-2 Am), radio
immunoprecipitation assay (RIPA) lysis buffer and BCA protein assay
kit were purchased from Beyotime Institute of Biotechnology
(Shanghai, China). TRNzol Universal Reagent, TIANScript RT kit and
Real Master Mix (SYBR Green) were purchased form Tiangen Biotech,
Co. Ltd. (Beijing, China). Other chemicals used were of analytical
grade. Water used was generated from Milli-Q Plus system
(Millipore, New York, NY, USA).
Cells and antibodies
The cell line (HCT-116) used in this study was
obtained from the Cell Bank of Shanghai Institute of Biochemistry
and Cell Biology (Shanghai, China). The cells were cultured in
McCoy's 5A medium (Sigma-Aldrich, Co. St. Louis, MO, USA)
supplemented with 10% of fetal bovine serum (Hyclone, Logan, UT,
USA) at 37°C in 5% CO2 atmosphere, as recommended by the
cell supplier.
Primary antibodies CHOP, DR5, cytochrome c oxidase
IV (COX IV), cytochrome c, Bax, BID, cleaved caspase-3, −8, and −9,
as well as secondary antibodies were provided by Cell Signaling
Technology (Shanghai) Biological Reagents Co., Ltd. (Shanghai,
China).
Assay of cell viability
The cells were seeded at a density of
1×104 cells per 100 µl per well onto the 96-well plates.
After cell attachment, the medium was discarded. Dimethyl
sulphoxide (DMSO, negative control) of 0.1%, 5-fluorouracil (5-Fu,
positive control) of 100 µM, and apigenin of 40–160 µM were added
to treat the cells for 24, 48 and 72 h, respectively. After that,
the solutions were discarded, and the cells were washed twice by a
phosphate buffer saline (PBS, 0.01 µM, pH 7.0). CCK-8 solution of
10 µl was added into each well to make a final concentration of
10%. The plates were then incubated at 37°C for another 4 h. A
microplate reader (Bio-Rad Laboratories, Hercules, CA, USA) was
used to measure the absorbencies at 570 nm. The vehicle-treated
cells were taken as 100% viable. The growth inhibition of the cells
was thus calculated as previously described (22).
Morphological observation
The cells (1×104 cells per 100 µl per
well) were seeded onto 6-well plates for attachment, treated by
0.1% DMSO, 40–160 µM apigenin for 24 h, rinsed with the PBS twice,
followed by the treatment of 0.5 ml of paraformaldehyde (4%) in the
PBS at 4°C overnight to fix the cells. The cells were washed with
the PBS twice, stained with Hoechst 33258 of 0.5 ml for 5 min at
room temperature in the dark, and then rinsed with the PBS twice.
The stained cells were observed and photographed under a
fluorescence microscope (Olympus, Tokyo, Japan) with respective
excitation and emission wavelengths of 350 and 460 nm.
Assay of cell cycle progression
After 24 h of treatment with 0.1% DMSO or 60–160 µM
apigenin, the cells were harvested and washed with the PBS.
Ice-cold 70% ethanol was added to fix the cells at 4°C overnight.
After that, the cells were rinsed with the ice-cold PBS, and
incubated with 25 µl propidium iodide (PI, 50 µg/ml) and 10 µl
RNase (100 µg/ml) for 30 min at 37°C in the dark. Flow cytometric
cell analysis was performed using a BD FACSort flow cytometry
(Becton Dickson Immunocytometry-Systems, San Jose, CA, USA).
CellQuest software (ModFit software, Verity Software House, Inc.,
Topsham, ME, USA) was used to determine the portions of the cells
in different cell stages of cell cycle progression
(G0/G1, S, and G2/M phases).
Apoptosis analysis by flow
cytometry
The cells treated with 0.1% DMSO or 60–160 µM
apigenin in the 6-well plates were harvested after 24 h, and washed
twice with the ice-cold PBS. Double staining with FITC-Annexin V
and PI was carried out using the Annexin V-FITC Apoptosis Detection
kit according to the manufacturer's protocol. Briefly, the cells
were incubated with 5 µl of the Annexin V-FITC and 10 µl of PI (20
µg/ml) at room temperature for 20 min in the dark. The cells were
then discriminated into viable, necrotic, early apoptotic, and late
apoptotic cells, using flow cytometry and CellQuest software as
above.
Detection of mitochondrial membrane
potential, intracellular ROS and Ca2+
The cells (1×106 per chamber) were
treated with DMSO (0.1%, control) and apigenin of 60–160 µM for 24
h. Afterward, the cells were re-suspended in fresh medium, and
incubated with the JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl
benzimidazolocarbocyanine iodide) of 1 ml at 37°C for 20 min. The
cells were rinsed with Dulbecco's phosphate-buffered saline (DPBS)
twice, and re-suspended in 2 ml medium. The loss of mitochondrial
membrane potential (MMP) was evaluated by the flow cytometry with
respective excitation and emission wavelengths of 485 and 590 nm as
previously described (23).
For the assay of intracellular ROS, the cells were
treated with 0.1% DMSO or 60–160 µM apigenin for 24 h at 37°C,
followed by two washes with the PBS. Then, 1 ml of DCF-DA
(2′,7′-dichlorofluorescein, 10 µM) were added into each well, and
the cells were re-incubated at 37°C for another 20 min. The cells
were rinsed with the fresh medium three times. Fluorescence
intensities were detected by a fluorescence spectrophotometer
(F-4500, Hitachi, Tokyo, Japan) at 488/525 nm with a 525 nm cut-off
(24).
The cells were seeded overnight and then applied
with 0.1% DMSO or 60–160 µM apigenin at 37°C for 24 h. The cells
were collected and rinsed with the Krebs-Ringer buffer (pH 7.4),
which contained 137 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5
mM CaCl2, 10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 25
mM D-glucose. The cells were collected and incubated with the
Fura-2 AM (5 µM) at 37°C for 60 min. After that, the cells were
washed twice and re-suspended in the Krebs-Ringer buffer, and
measured for fluorescence (Fs). The used emission
and excitation wavelengths were 510 and 340–380 nm, respectively.
The cells treated with 0.1% of Triton X-100 (v/v) were used to
determine the maximal fluorescence (Fmax),
followed by addition of 10 mM EGTA (ethylene glycol tetra-acetic
acid, pH 9.0) to determine the minimal fluorescence
(Fmin). Intracellular Ca2+
([Ca2+]i) was calculated using following
equation as [Ca2+]i=kd
(Fs-Fmin)/(Fmax-Fs),
in which kd has a value of 224 nM (25).
Isolation of mRNAs and quantitative
real-time PCR (qRT-PCR)
Total RNA of the HCT-116 cells was extracted with
the TRNzol Universal Reagent (Tiangen Biotech, Co. Ltd.), and
complementary DNA (cDNA) was then reverse transcribed using the
TIANScript RT kit and the protocol provided by the kit
manufacturer. The qRT-PCR was performed using a 7500 Real-Time PCR
System (Applied Biosystems, Foster City, CA, USA). cDNA of 1 µl was
added to 9 µl of 2.5X Real Master Mix (20X SYBR Green) containing 5
µl of each of the corresponding primer pairs to make a final system
volume of 20 µl for each well. Thermo-cycling conditions were used
as follows: initial activation for 1 min at 95°C, followed by 40
cycles of denaturation at 95°C for 15 sec, annealing for 20 sec at
60°C and extension for 32 sec at 68°C. The fluorescence was
measured during the extension step. Relative expression levels of
the target genes were determined using the 2−∆∆Ct method
(26). The β-actin housekeeping
gene was used as an internal control. The used primers were
designed with the sequences below, and synthesized by Sangon
Biotech (Shanghai) Co., Ltd. (Shanghai, China). i) human β-actin
forward, 5′-AACACCCCAGCCATGTACG-3′ and reverse,
5′-ATGTCACGCACGATTTCCC-3′; ii) CHOP forward,
5′-GCCAATGATGTGACCCTCAAT-3′ and reverse,
5′-CCTGGAAATGAAGAGGAAGAA-3′; iii) DR5 forward,
5′-AAGACCCTTGTGCTCGTTGT-3′ and reverse,
5′-GACACATTCGATGTCACTCCA-3′.
Western blot assay
After 24 h treatment with 0.1% DMSO or 60–160 µM
apigenin at 37°C for 24 h, the cells were harvested and lysed with
the RIPA lysis buffer, followed by a centrifugation at 14,000 × g
at 4°C for 10 min. The supernatants were collected after boiling
for 5 min. Protein contents were measured using the BCA Protein
Assay kit. Proteins (50 µg) were separated on 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, and electro-transferred
onto nitrocellulose membranes. The membranes were blocked in 5%
non-fat milk at 37°C for 1 h, and then incubated with the primary
antibodies at 4°C overnight. Afterward, the membranes were washed
with the TBST buffer (containing 10 mM Tris-HCl, pH 7.6, 150 mM
NaCl, and 0.1% Tween-20) three times, and incubated with the
secondary antibodies at 37°C for 1 h. Images of the blots were
captured, and densitometric analysis was performed using an
ImageQuant LAS 500 (Fujifilm, Tokyo, Japan).
Statistical analysis
All values are expressed as mean values or mean
values ± standard derivations from three independent experiments
and analyses. Statistical significance between different groups was
analyzed by one-way analysis of variance (ANOVA) with Duncan's
multiple range tests using the SPSS version 13.0 (SPSS Inc.,
Chicago, IL, USA). Statistical significance was defined at
P<0.05.
Results
Cytotoxic effect of anpigenin on the
cells
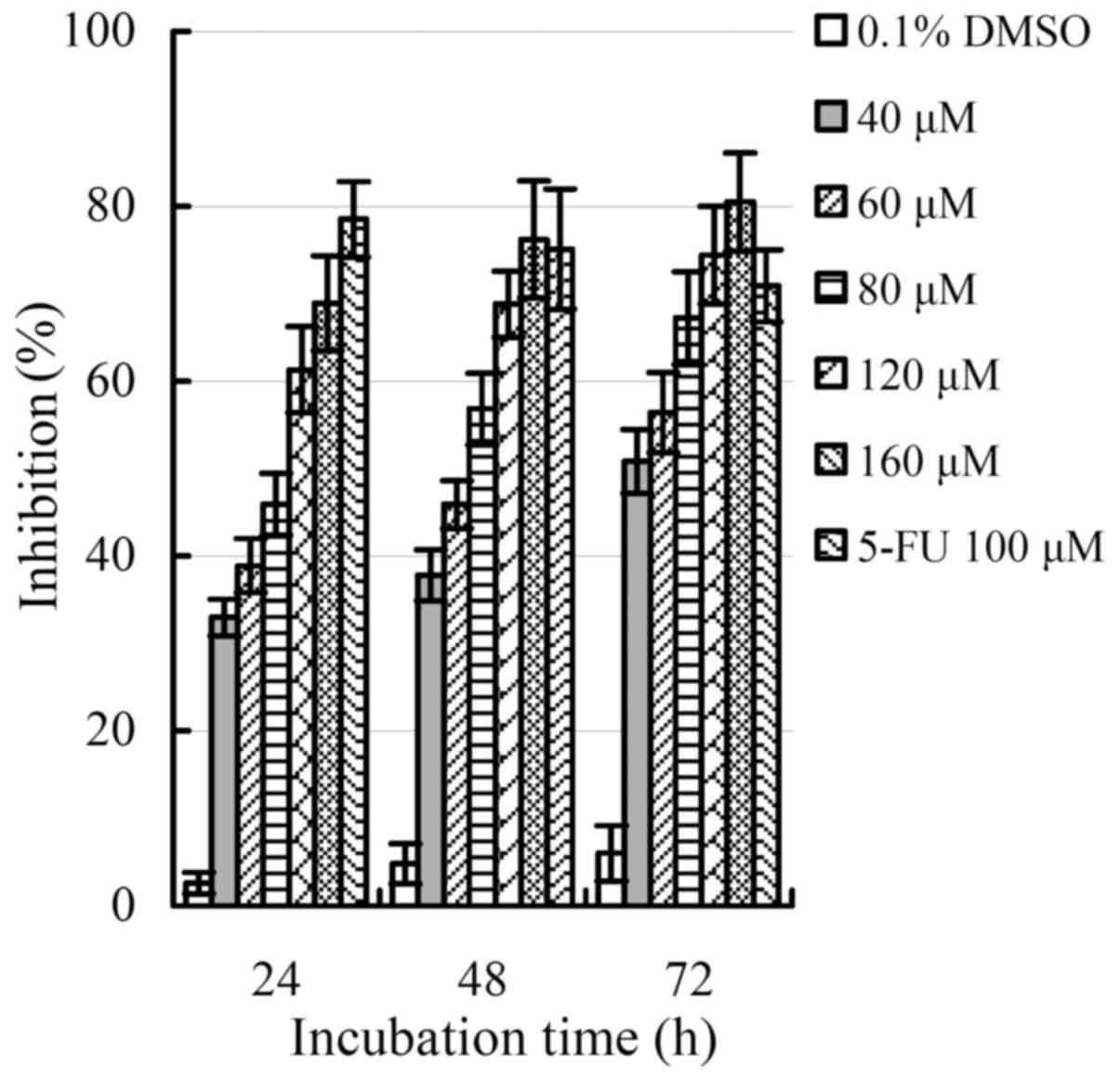
In vitro effect of apigenin on the HCT-116
cells is described in Fig. 1.
Apigenin exerted cytotoxic effect on the cells, and inhibited cell
growth (i.e. decreased cell viability) dose- and time-dependently.
Apigenin of 40 µM only inhibited cell growth by 9.3–23.1% when the
cells were treated for 24–72 h. Thus this dose was not used in
other assays except morphological observation. However, higher
apigenin dose showed higher cytotoxic effect and thereby resulted
in lower cell viability (i.e. greater inhibition). If apigenin was
used at 160 µM and the cells were treated for 72 h, growth
inhibition reached to a maximum value of 80.5%. The calculated
IC50 values of apigenin with treating times of 24, 48
and 72 h were approximately 98.2, 83.3 and 77.9 µM, respectively.
These data demonstrated clearly that longer treatment time also
resulted in apigenin stronger inhibition (i.e. cytotoxic effect) on
the cells.
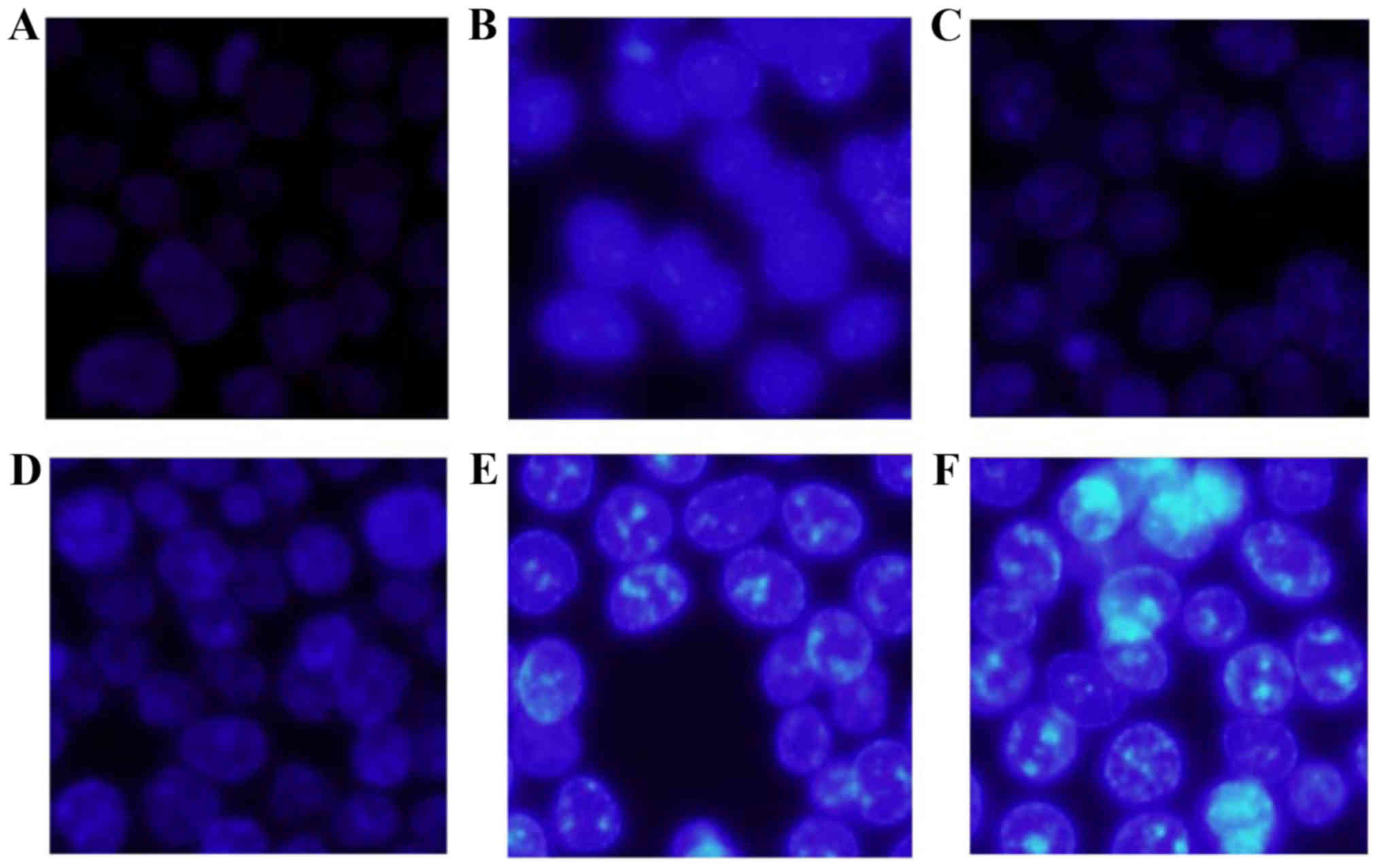
In addition, when the HCT-116 cells were treated
with apigenin of various doses, nuclei morphological changes were
observed in the treated cells after Hoechst 33258 staining
(Fig. 2), especially when apigenin
of 160 µM was used. High chromatin condensation and visible
formation of apoptotic bodies were found in the treated cells
(Fig. 2F). This phenomenon
indicated that apigenin was able to induce apoptosis besides growth
inhibition on the cells.
G0/G1 phase cell
cycle arrest and apoptosis induction of apigenin
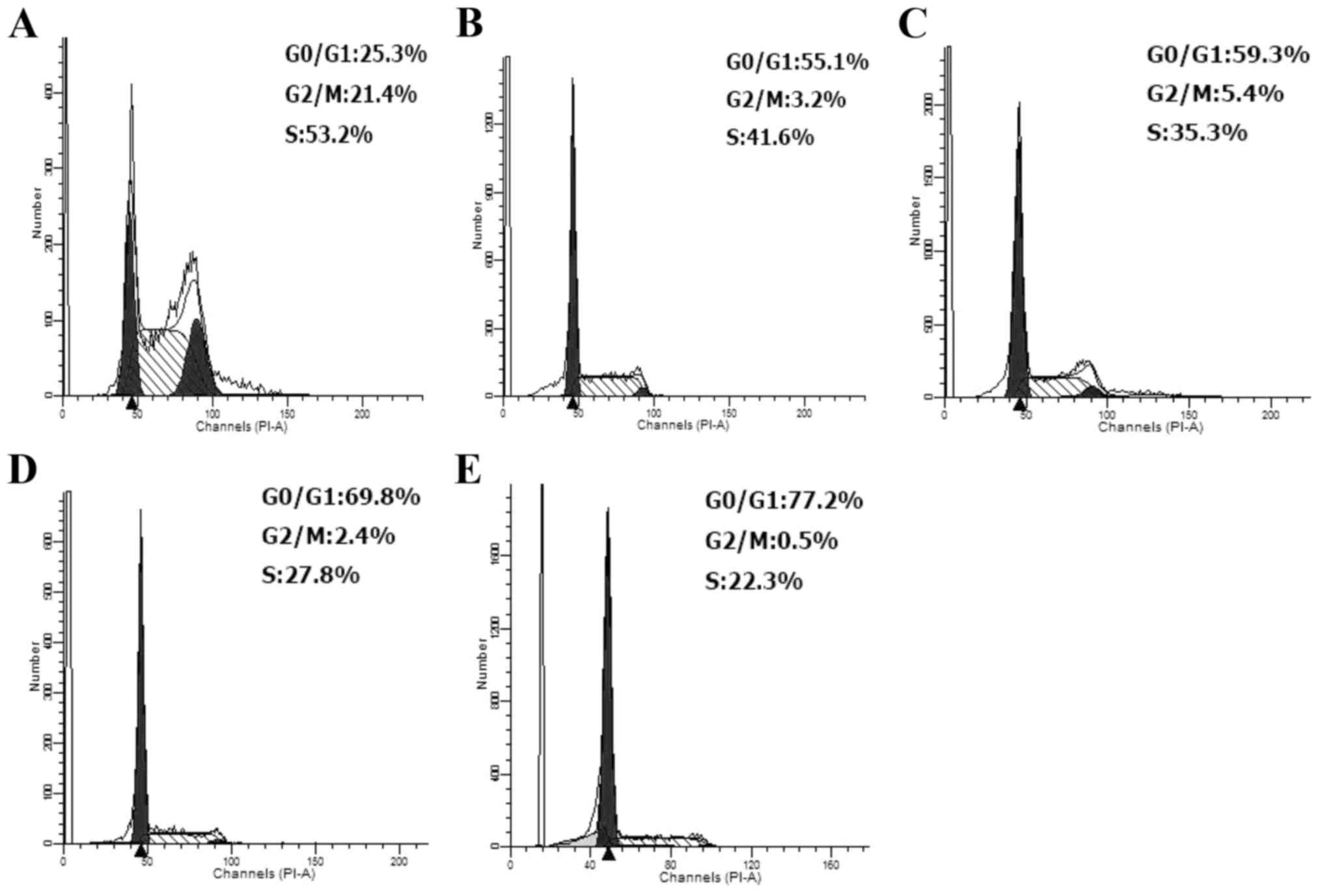
To understand if apigenin had effect on cell cycle
progression of the HCT-116 cells, the distribution of the cells in
different cell cycle phases was assessed using flow cytometry.
Consistent changes in the cell cycle at 24 h were observed along
with increased apigenin doses (60–160 µM). In the control cells,
the respective portions of S, G0/G1 and
G2/M phases were 52.2, 25.3 and 21.4% (Fig. 3). As apigenin dose increased into
higher level, the treated cells showed decreasing trend in the
portions of both G2/M and S phases but increasing trend
in the portion of G0/G1 phase. This
demonstrated that apigenin was dose-dependent in arresting the cell
cycle at G0/G1 phase.
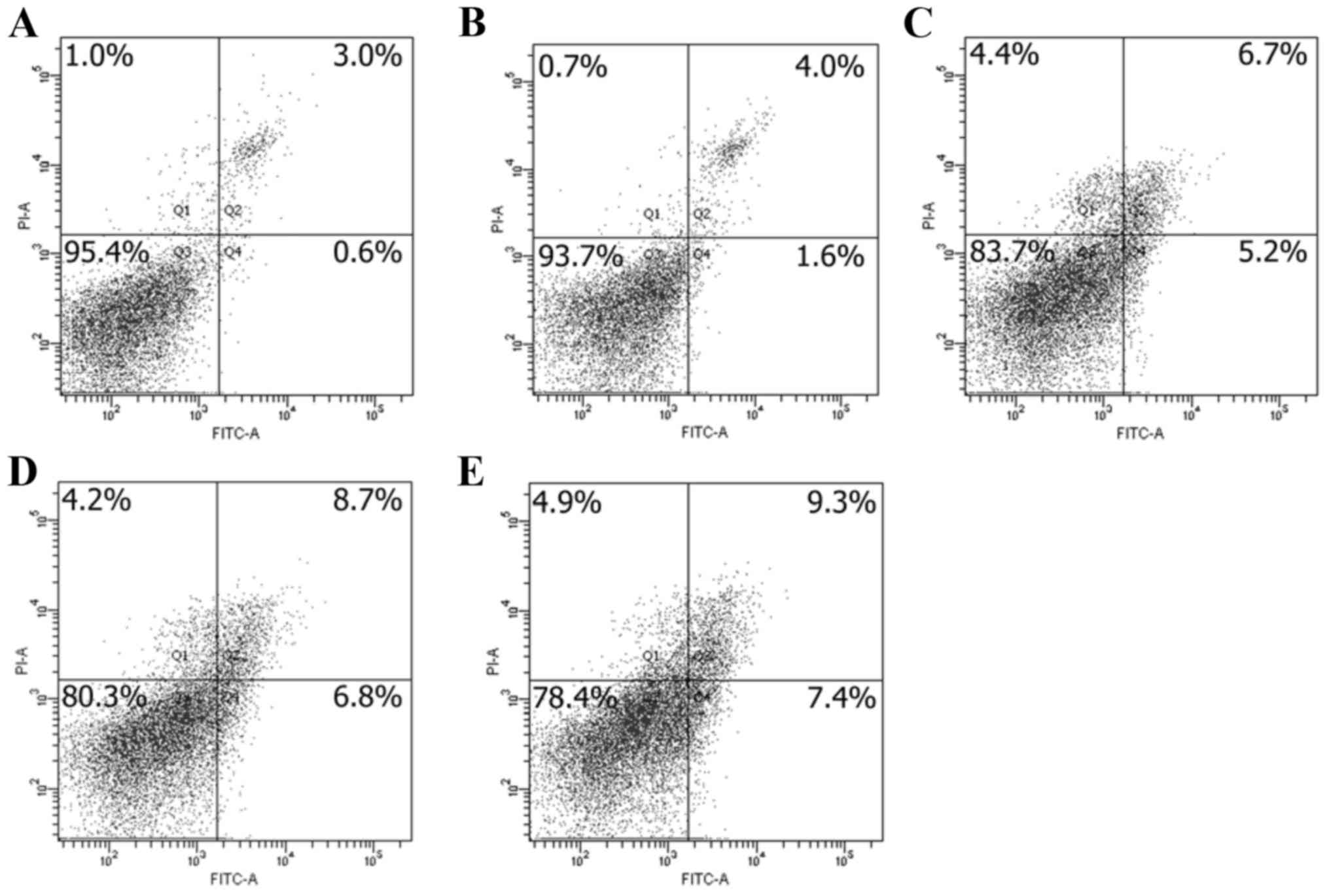
After 24 h exposure to apigenin of 60–160 µM, the
cells were collected and detected to show potential apoptosis
induction of apigenin. The results shown in Fig. 4 demonstrate that the portion of
apoptotic cells (early plus late apoptotic cells) was enhanced with
increased apigenin dose. Treatment of the cells with 60–160 µM
apigenin caused 5.6, 11.9, 15.5, and 16.7% Annexin V-FITC positive
cells (Fig. 4B-E). However, the
control cells were detected to only have 3.6% apoptotic cells
(Fig. 4A). These results implied
that apigenin indeed had apoptosis induction, which was dependent
on the used apigenin dose.
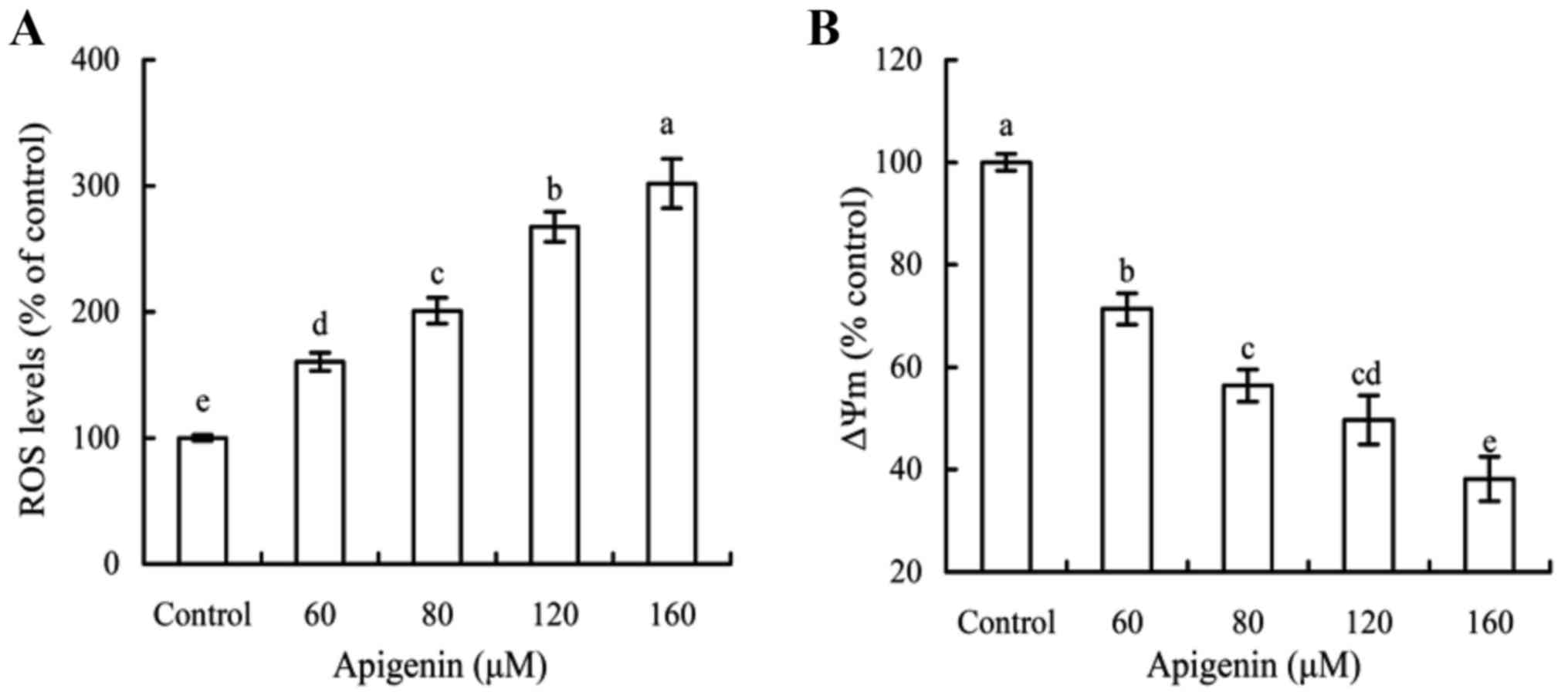
Intracellular ROS and Ca2+
as well as MMP loss in response to apigenin treatment
The cells were treated with apigenin of various
doses for 24 h, and assayed for their intracellular ROS levels. The
results (Fig. 5A) showed that ROS
generation in the cells was significantly increased after apigenin
exposure. In comparison with the control cells, the cells treated
with 160 µM apigenin had increased ROS level up to 301.8%. Other
three apigenin doses (60–120 µM) resulted in the cells with
increased ROS levels of 160.5, 200.8 and 267.4%. Enhancement of ROS
generation depended on the used apigenin doses. In addition, the
effect of apigenin on MMP loss of the treated cells was also
measured. After 24 h treatment with apigenin of four dose levels,
the treated cells showed significant decrease in MMP in comparison
with the control cells (Fig. 5B).
Apigenin was thus proved able to damage mitochondrial membrane.
Moreover, MMP loss of the treated cells was also observed in an
apigenin dose-dependent manner. These results pointed out an
important fact; that is, apigenin had potential toxic effects on
the cells via both mitochondria and ER organelles.
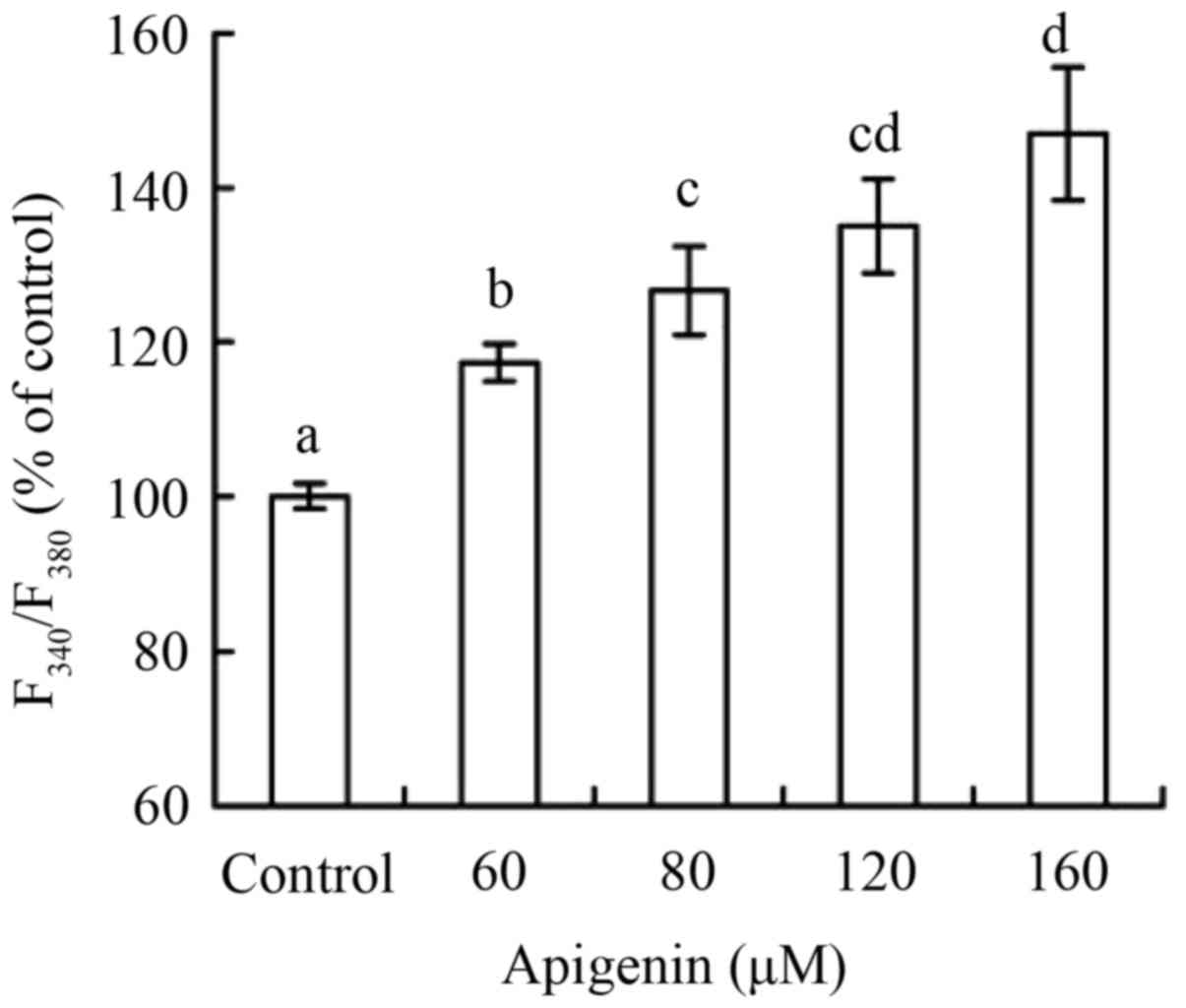
As the results demonstrated in Fig. 6, apigenin at 60–160 µM with treating
time of 24 h caused the treated cells increased intracellular
Ca2+ levels in a dose-dependent manner (P<0.05).
Intracellular Ca2+ levels of the treated cells were
117.3, 126.7, 135.1 and 147.4% of the control cells. These results
of Ca2+ release were consistent with those of ROS
generation and MMP loss. It is thus verified that higher ROS
generation induced ER stress, which consequently resulted in
abundant Ca2+ release into the cytosol.
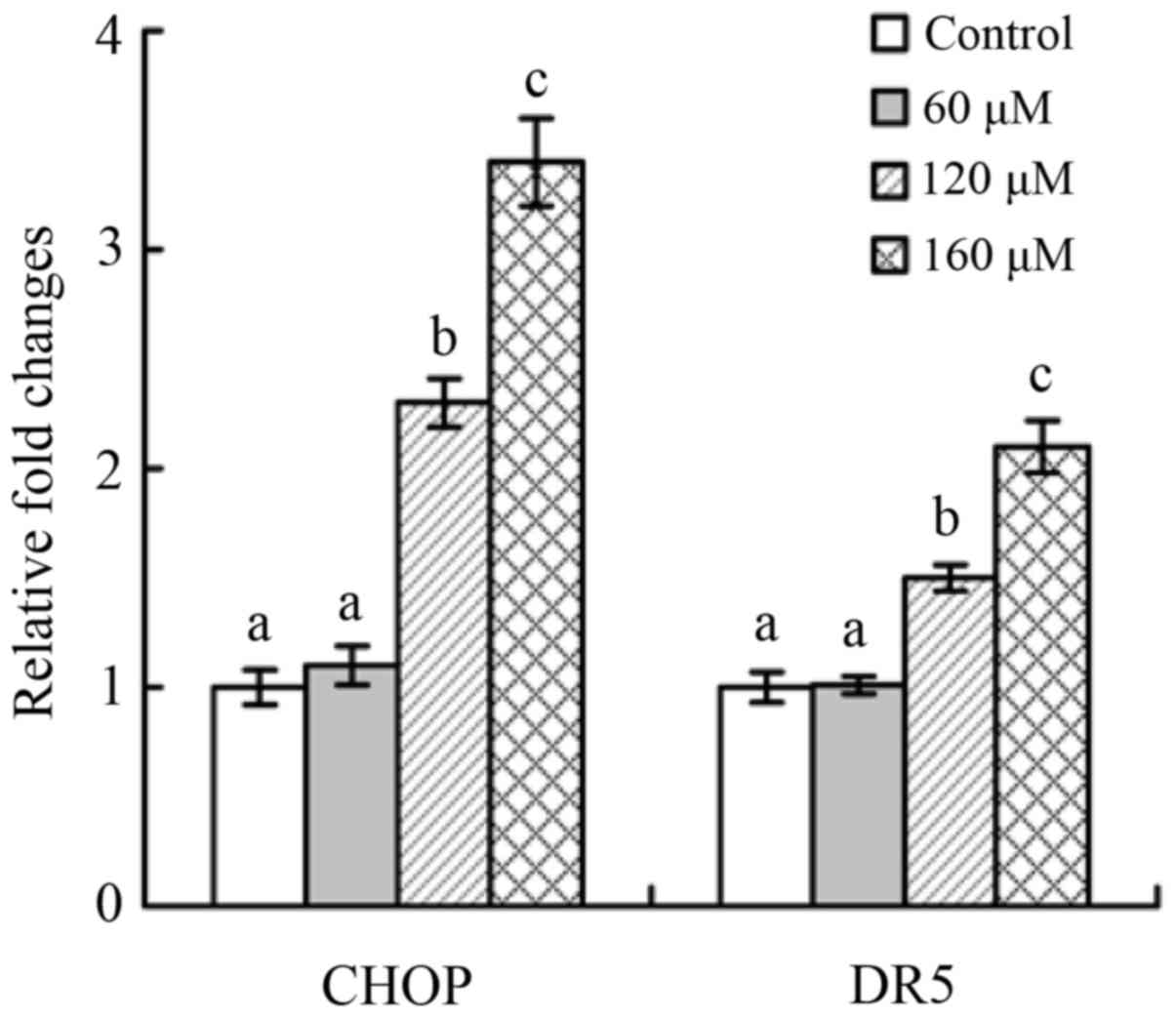
Increased mRNA expression of CHOP and
DR5 in the treated cells
Real-time RT-PCR results showed that apgenin at dose
levels of 60, 120, and 160 µM could upregulate both CHOP (1.2-,
2.3-, and 3.4-fold) and DR5 (1.1-, 1.5-, and 2.1-fold) mRNA
expression in the treated cells (Fig.
7). Higher apigenin dose clearly resulted in greater
upregulation of CHOP and DR5. These results proved that apigenin
might induce ER stress and activate the death receptor signaling
pathway, which should be underlined to reveal the related
mechanisms.
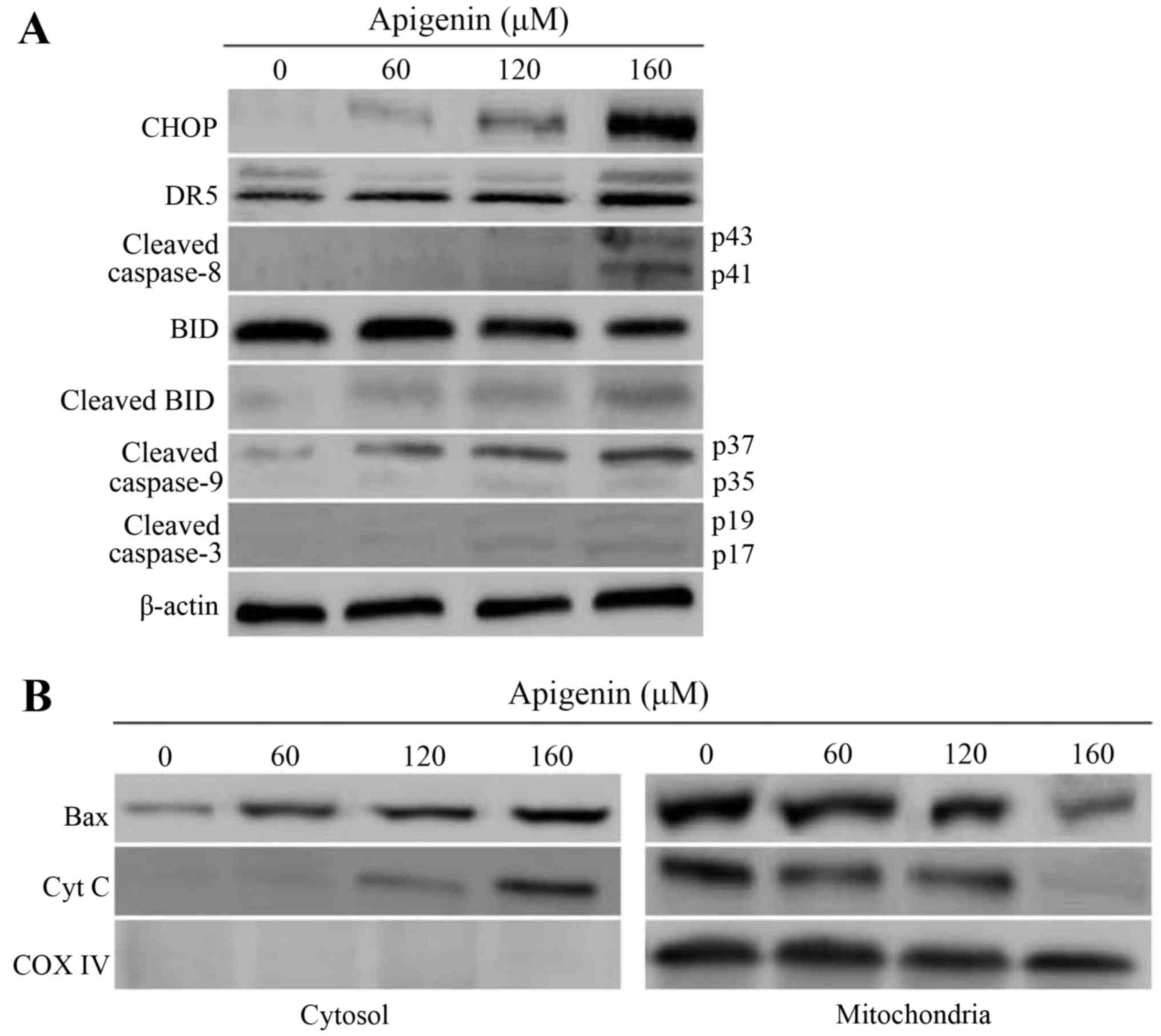
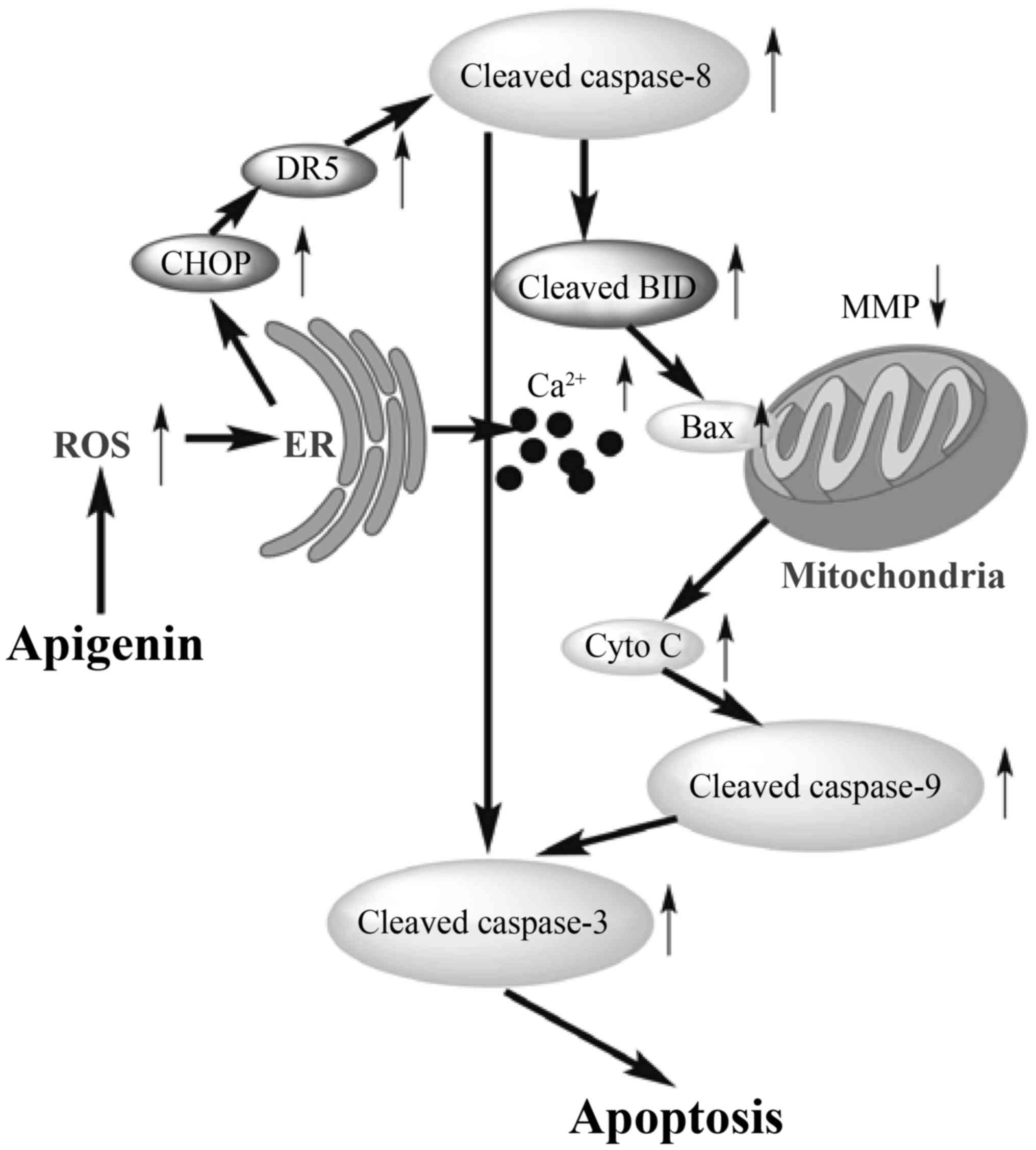
Apoptotic mechanism is involved in ROS
generation and ER stress
To reveal the underlying mechanism responsible for
apigenin-induced apoptosis in the HCT-116 cells, expression levels
of these associated proteins were evaluated. The results given in
Fig. 8 indicated that apigenin
increased protein expression levels of CHOP (1.1–3.5-fold), DR5
(1.2–2.4-fold), cleaved BID (1.1–1.6 fold), cleaved caspase-3
(1.0–1.3-fold), cleaved caspase-8 (1.1–1.6-fold), and cleaved
caspase-9 (1.3–1.7-fold) (Fig. 8A),
Bax (1.6–3.5-fold), and cytochrome c (1.0–2.2-fold) (Fig. 8B) in the cytosol, but decreased
protein expression level of BID (1.0–2.9-fold) (Fig. 8A). Higher apigenin dose led to
greater expression changes of these proteins. Based on these
results, it is thus verified that apigenin-induced apoptosis was
mediated by ROS generation and ER stress, through upregulating CHOP
and DR5 and therefore triggering both extrinsic and intrinsic
pathways. A molecular mechanism for the apoptosis induction of
apigenin on the cells is outlined in Fig. 9. Apigenin conferred the treated
cells with ER stress via greater ROS generation, increased CHOP and
DR5 mRNA levels, regulated the expression of these pro-apoptotic
proteins (cleaved caspase-3, cleaved caspase-8, and cleaved
caspase-9, cleaved BID, and Bax), increased cytochrome c release
from the mitochondria into the cytosol, and finally initiated
apoptosis of the cells.
Discussion
Several studies have reported that apigenin has
anticancer activities via anti-proliferation, angiogenesis and
apoptosis induction (1,27,28).
The involved mechanisms have also been revealed. Apigenin can
inhibit the growth of the HT-29 cells dose- and time-dependently,
cause DNA fragmentation, and increase mRNA expression levels of
CASP3 (late apoptosis, effector) and CASP8 (early apoptosis,
initiator) (29). Apigenin inhibits
the proliferation of the T-24 cells, and induces apoptosis via the
mitochondrial pathway (30).
Apigenin can activate PKCδ, which then activate caspase pathway to
induce apoptosis in the leukemia THP-1 cells (31). Apigenin is capable of inducing
apoptosis via a caspase-dependent pathway in the MDA-MB-453 cells
as well as inhibiting the well-known JAK2-STAT3-VEGF signaling
pathway (32). Apigenin is also
able to upregulate DR5 expression in the human acute lymphoblastic
leukemic cell line Jurkat, prostate cancer cell line DU145, and
colon cancer cell line DLD-1 to trigger TRAIL-induced apoptosis
(33). In this study, apigenin was
also observed to inhibit the growth of the HCT-116 cells, to arrest
cell cycle at G0/G1 phase, to increase both
intracellular ROS and Ca2+ levels, and to decrease MMP.
Finally, both extrinsic and intrinsic pathways were triggered.
Clearly, the outlined apoptotic mechanism involved ROS generation
and ER stress in this study is different to those reported
mechanisms mentioned above.
In normal cells, apigenin processes anti-oxidation.
However, in cancer cells, apigenin exhibits pro-oxidation rather
than anti-oxidation (34,35). In this study, intracellular ROS
level was suggested to play an important role in the ER
stress-mediated signaling pathway. This finding is supported by the
results from other studies. Apigenin increases ROS level in the
human lung cancer A549 cells, which leads to high Bax/Bcl-2 ratio
to cause mitochondria dysfunction and to trigger caspase-dependent
apoptosis (36). Apigenin can block
cell cycle progression at G2/M stage, increase
intracellular ROS production, and therefore cause AKT
hypophosphorylation in the triple-negative MDA-MB-468 breast cancer
cells (37). In addition, apigenin
can trigger intracellular ROS generation, activate
mitogen-activated protein kinase (MAPK) ERK1/2 in the human
fibroblast-like synoviocyte MH7A cells, and finally cause apoptosis
via activation of caspase-3 and −7 (38). These results support that apigenin
caused apoptosis induction towards the HCT-116 cells through
enhancing ROS generation and resultant ER stress.
The execution of apoptosis is accomplished by
caspase family via two major pathways, intrinsic pathway and
extrinsic pathway (39). In the
intrinsic pathway, cells are stimulated by various toxic compounds,
which causes cytochrome c release from the mitochondria into the
cytosol. A so-called complex, apoptosome, is thus formed to
activate the cleaved caspase-9 as well as downstream caspases
(40). The Bcl-2 protein family
contains both anti- and pro-apoptotic members. Bcl-2 and its
closest Bcl-xL and Bcl-w, can protect cells from apoptosis, whereas
other Bcl-2 relatives (e.g. Bax, Bak and Bok) are pro-apoptotic
proteins (41). Once the Bax/Bcl-2
ratio increases, apoptosis is induced, resulting in the release of
apoptogenic factors from the mitochondria (42). On the contrary, in the extrinsic
pathway, the caspases are activated by death receptors such as FAS,
TNFR1, DR3, DR4, and DR5 (43).
These cell surface proteins recruit adaptor proteins (e.g. FADD) to
form a death-inducing signaling complex with procaspase-8 that
induces caspase-8 activation. Downstream effector caspases (such as
caspase-3, −7 and −9) are thus cleaved (44). However, the extrinsic apoptotic
pathway has crosstalk with the intrinsic apoptotic pathway via
caspase-8 cleavage of the BID, which then activates the
mitochondrial pathway to amplify the apoptotic signal (45).
When the HCT-116 cells were treated with apigenin,
they showed decreased MMP and upregulated Bax protein. As a
consequence, cytochrome c was released from the mitochondria to the
cytosol, which activated cleaved caspase-9 and cleaved caspase-3 to
induce apoptosis. In addition, accumulation of intracellular ROS
led to continuous release of Ca2+ from the ER lumen to
the cytosol, which consequently caused ER stress. Overload
Ca2+ levels in the cytoplasm accelerated Ca2+
influx into the mitochondria, which resulted in greater ROS
generation and finally caused the opening of the permeability
transition pore. Increased ROS generation within the mitochondria
was thus released, and as a feedback to simulate the
Ca2+ release channels on the ER (46). CHOP as protein indicator of ER
stress was thus activated to upregulate its downstream protein DR5.
DR5 overexpression activated cleaved caspase-8. Cleaved caspase-8
has two roles: to activate cleaved caspase-3 and to cleave BID to
generate cleaved BID. Cleaved BID in turn induced Bax activation.
Thus, the mitochondrial pathway was amplified by the ER stress
signaling pathway.
Other studies have assessed apoptosis induction of
some natural products, and shown apoptotic mechanism similar to
this study. Hesperidin from Citrus seed can induce apoptosis
in the human hepatocellular carcinoma HepG2 cells via both
mitochondrial and death receptor pathways, evidenced by the
upregulation of Bax and Bak protein levels, downregulation of
Bcl-xl protein level, and activation of tBID and caspase-3, −8, and
−9 (47). Licochalcone B, isolated
from the roots of Chinese licorice, exhibits apoptosis induction
towards the oral squamous cell carcinoma HN22 and HSC4 cells via
increasing ROS levels, as well as CHOP, DR4 and DR5 protein
expression (48). Accompanying by
upregulation of these apoptotic proteins, expression levels of
anti-apoptotic proteins (e.g. Mcl-1) decrease, but pro-apoptotic
proteins (e.g. Bax) level is upregulated (48). Emodin, a natural anthraquinone-type
compound from Rheum palmatum L, can induce apoptosis in the
human cervical cancer HeLa cells (49). In the emodin-treated cells, protein
expression levels of cytochrome c, Apaf-1, FAS, FasL, and FADD are
all upregulated whilst those levels of procaspase-3, −8 and −9 are
all downregulated (49). These
findings state that emodin can induce both intrinsic mitochondrial
and extrinsic death receptor apoptotic pathways (49), and therefore provide support to the
outlined mechanism of this study.
Based on the present results, it is concluded that
apigenin is a promising anticancer compound capable of inhibiting
proliferation of the colon carcinoma HCT-116 cells, disturbing cell
cycle progression to arrest the cells at
G0/G1 phase, causing abundant intracellular
ROS generation and Ca2+ release, destroying
mitochondrial membrane, and inducing apoptosis. Apigenin can
upregulate the expression of CHOP, DR5, cleaved caspase-3, cleaved
caspase-8, and cleaved caspase-9, cleaved BID, and Bax, and can
also enhance cytochrome c release. The outlined apoptotic mechanism
is thus, for the first time, suggested to involve ROS-induced and
ER stress-mediated intrinsic and extrinsic pathways.
Acknowledgements
This study was funded by the Key Research Project in
Science and Technology of the Education Department of Heilongjiang
Province (project no. 11551z018).
Glossary
Abbreviations
Abbreviations:
|
AKT
|
RAC-α serine/threonine-protein
kinase
|
|
Apaf-1
|
apoptotic peptidase activating factor
1
|
|
ATF3
|
activating transcription factor 3
|
|
Bak
|
Bcl-2 antagonist/killer
|
|
Bax
|
Bcl-2 associated × protein
|
|
Bcl-2
|
B-cell cll/lymphoma 2
|
|
Bcl-w
|
Bcl-2-like protein 2
|
|
Bcl-xl
|
B-cell lymphoma-extra large
|
|
BID
|
BH3 interacting domain death
agonist
|
|
Bok
|
Bcl-2-related ovarian killer
|
|
CCK-8
|
Cell Counting Kit-8
|
|
cDNA
|
complementary DNA
|
|
CHOP
|
C/EBP homologous protein
|
|
COX IV
|
cytochrome c oxidase IV
|
|
DCF-DA
|
2′,7′-dichloro-fluorescein
|
|
DMSO
|
dimethyl sulphoxide
|
|
DPBS
|
Dulbecco's phosphate-buffered
saline
|
|
DR3
|
death receptor 3
|
|
DR4
|
death receptor 4
|
|
DR5
|
death receptor 5
|
|
EGTA
|
ethylene glycol tetraacetic acid
|
|
ER
|
endoplasmic reticulum
|
|
FADD
|
FAS-associating death
domain-containing protein
|
|
FAS
|
cell surface death receptor
|
|
FasL
|
fas ligand
|
|
5-Fu
|
5-fluorouracil
|
|
Fura-2 Am
|
Fura-2 pentakis (acetoxymethyl)
ester
|
|
HEPES
|
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
|
|
Id1
|
inhibitor of DNA binding 1
|
|
JAK2
|
janus kinase 2
|
|
JC-1
|
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl
benzimidazolocarbocyanine iodide
|
|
MAPK
|
mitogen-activated protein kinase
|
|
Mcl-1
|
myeloid cell leukemia 1
|
|
MMP
|
mitochondrial membrane potential
|
|
NAC
|
N-acetyl cysteine
|
|
NFκB
|
nuclear factor κB subunit
|
|
PARP
|
poly-(ADP-ribose) polymerase
|
|
PBS
|
phosphate buffer saline
|
|
PI
|
propidium iodide
|
|
PKCδ
|
C Protein kinase C δ type
|
|
PTP
|
permeability transition pore
|
|
qRT-PCR
|
quantitative real-time PCR
|
|
RIPA
|
radio immunoprecipitation assay
|
|
RNS
|
reactive nitrogen species
|
|
ROS
|
reactive oxygen species
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
TNFR1
|
tumor necrosis factor receptor-1
|
|
tBID
|
truncated BID
|
|
TRAIL
|
tumor necrosis factor-related
apoptosis-inducing ligand
|
|
UPR
|
unfolded protein response
|
|
VEGF
|
vascular endothelial growth factor
A
|
References
|
1
|
Yang CS, Landau JM, Huang MT and Newmark
HL: Inhibition of carcinogenesis by dietary polyphenolic compounds.
Annu Rev Nutr. 21:381–406. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patel D, Shukla S and Gupta S: Apigenin
and cancer chemoprevention: Progress, potential and promise
(Review). Int J Oncol. 30:233–245. 2007.PubMed/NCBI
|
|
3
|
Li ZD, Hu XW, Wang YT and Fang J: Apigenin
inhibits proliferation of ovarian cancer A2780 cells through Id1.
FEBS Lett. 583:1999–2003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mak P, Leung YK, Tang WY, Harwood C and Ho
SM: Apigenin suppresses cancer cell growth through ERbeta.
Neoplasia. 8:896–904. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang IK, Lin-Shiau SY and Lin JK:
Induction of apoptosis by apigenin and related flavonoids through
cytochrome c release and activation of caspase-9 and caspase-3 in
leukaemia HL-60 cells. Eur J Cancer. 35:1517–1525. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: Cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim R, Emi M, Tanabe K and Murakami S:
Role of the unfolded protein response in cell death. Apoptosis.
11:5–13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malhotra JD and Kaufman RJ: The
endoplasmic reticulum and the unfolded protein response. Semin Cell
Dev Biol. 18:716–731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meares GP, Mines MA, Beurel E, Eom TY,
Song L, Zmijewska AA and Jope RS: Glycogen synthase kinase-3
regulates endoplasmic reticulum (ER) stress-induced CHOP expression
in neuronal cells. Exp Cell Res. 317:1621–1628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamaguchi H and Wang HG: CHOP is involved
in endoplasmic reticulum stress-induced apoptosis by enhancing DR5
expression in human carcinoma cells. J Biol Chem. 279:45495–45502.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gorman AM, Healy SJ, Jäger R and Samali A:
Stress management at the ER: Regulators of ER stress-induced
apoptosis. Pharmacol Ther. 134:306–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fruehauf JP and Meyskens FL Jr: Reactive
oxygen species: A breath of life or death? Clin Cancer Res.
13:789–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rigas B and Sun Y: Induction of oxidative
stress as a mechanism of action of chemopreventive agents against
cancer. Br J Cancer. 98:1157–1160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schumacker PT: Reactive oxygen species in
cancer cells: Live by the sword, die by the sword. Cancer Cell.
10:175–176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trachootham D, Zhou Y, Zhang H, Demizu Y,
Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, et
al: Selective killing of oncogenically transformed cells through a
ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer
Cell. 10:241–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zou P, Chen M, Ji J, Chen W, Chen X, Ying
S, Zhang J, Zhang Z, Liu Z, Yang S, et al: Auranofin induces
apoptosis by ROS-mediated ER stress and mitochondrial dysfunction
and displayed synergistic lethality with piperlongumine in gastric
cancer. Oncotarget. 6:36505–36521. 2015.PubMed/NCBI
|
|
19
|
Shen S, Zhang Y, Zhang R and Gong X:
Sarsasapogenin induces apoptosis via the reactive oxygen
species-mediated mitochondrial pathway and ER stress pathway in
HeLa cells. Biochem Biophys Res Commun. 441:519–524. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shin SY, Lee JM, Lee MS, Koh D, Jung H,
Lim Y and Lee YH: Targeting cancer cells via the reactive oxygen
species-mediated unfolded protein response with a novel synthetic
polyphenol conjugate. Clin Cancer Res. 20:4302–4313. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen CY, Yiin SJ, Hsu JL, Wang WC, Lin SC
and Chern CL: Isoobtusilactone A sensitizes human hepatoma Hep G2
cells to TRAIL-induced apoptosis via ROS and CHOP-mediated
up-regulation of DR5. J Agric Food Chem. 60:3533–3539. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johnson JL and de Mejia E Gonzalez:
Interactions between dietary flavonoids apigenin or luteolin and
chemotherapeutic drugs to potentiate anti-proliferative effect on
human pancreatic cancer cells, in vitro. Food Chem Toxicol.
60:83–91. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Salvioli S, Ardizzoni A, Franceschi C and
Cossarizza A: JC-1, but not DiOC6(3) or rhodamine 123, is a
reliable fluorescent probe to assess delta psi changes in intact
cells: Implications for studies on mitochondrial functionality
during apoptosis. FEBS Lett. 411:77–82. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boissy RE, Trinkle LS and Nordlund JJ:
Separation of pigmented and albino melanocytes and the concomitant
evaluation of endogenous peroxide content using flow cytometry.
Cytometry. 10:779–787. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vega A, Chacón P, Monteseirín J, El Bekay
R, Alba G, Martín-Nieto J and Sobrino F: Expression of the
transcription factor NFAT2 in human neutrophils: IgE-dependent,
Ca2+- and calcineurin-mediated NFAT2 activation. J Cell
Sci. 120:2328–2337. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−∆∆CT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Van Dross R, Xue Y, Knudson A and Pelling
JC: The chemopreventive bioflavonoid apigenin modulates signal
transduction pathways in keratinocyte and colon carcinoma cell
lines. J Nutr. 133:(Suppl 1). S3800–S3804. 2003.
|
|
28
|
Shukla S and Gupta S: Dietary agents in
the chemoprevention of prostate cancer. Nutr Cancer. 53:18–32.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Turktekin M, Konac E, Onen HI, Alp E,
Yilmaz A and Menevse S: Evaluation of the effects of the flavonoid
apigenin on apoptotic pathway gene expression on the colon cancer
cell line (HT29). J Med Food. 14:1107–1117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi MD, Shiao CK, Lee YC and Shih YW:
Apigenin, a dietary flavonoid, inhibits proliferation of human
bladder cancer T-24 cells via blocking cell cycle progression and
inducing apoptosis. Cancer Cell Int. 15:33–45. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vargo MA, Voss OH, Poustka F, Cardounel
AJ, Grotewold E and Doseff AI: Apigenin-induced-apoptosis is
mediated by the activation of PKCdelta and caspases in leukemia
cells. Biochem Pharmacol. 72:681–692. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seo HS, Ku JM, Choi HS, Woo JK, Jang BH,
Shin YC and Ko SG: Induction of caspase-dependent apoptosis by
apigenin by inhibiting STAT3 signaling in HER2-overexpressing
MDA-MB-453 breast cancer cells. Anticancer Res. 34:2869–2882.
2014.PubMed/NCBI
|
|
33
|
Horinaka M, Yoshida T, Shiraishi T, Nakata
S, Wakada M and Sakai T: The dietary flavonoid apigenin sensitizes
malignant tumor cells to tumor necrosis factor-related
apoptosis-inducing ligand. Mol Cancer Ther. 5:945–951. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miyoshi N, Naniwa K, Yamada T, Osawa T and
Nakamura Y: Dietary flavonoid apigenin is a potential inducer of
intracellular oxidative stress: The role in the interruptive
apoptotic signal. Arch Biochem Biophys. 466:274–282. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Andueza A, García-Garzón A, de Galarreta M
Ruiz, Ansorena E, Iraburu MJ, López-Zabalza MJ and Martínez-Irujo
JJ: Oxidation pathways underlying the pro-oxidant effects of
apigenin. Free Radic Biol Med. 87:169–180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu HF, Chie YJ, Yang MS, Lee CS, Fu JJ,
Yang JS, Tan TW, Wu SH, Ma YS, Ip SW, et al: Apigenin induces
caspase-dependent apoptosis in human lung cancer A549 cells through
Bax- and Bcl-2-triggered mitochondrial pathway. Int J Oncol.
36:1477–1484. 2010.PubMed/NCBI
|
|
37
|
Harrison ME, Coombs MR Power, Delaney LM
and Hoskin DW: Exposure of breast cancer cells to a subcytotoxic
dose of apigenin causes growth inhibition, oxidative stress, and
hypophosphorylation of Akt. Exp Mol Pathol. 97:211–217. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shin GC, Kim C, Lee JM, Cho WS, Lee SG,
Jeong M, Cho J and Lee K: Apigenin-induced apoptosis is mediated by
reactive oxygen species and activation of ERK1/2 in rheumatoid
fibroblast-like synoviocytes. Chem Biol Interact. 182:29–36. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hale AJ, Smith CA, Sutherland LC, Stoneman
VE, Longthorne VL, Culhane AC and Williams GT: Apoptosis: Molecular
regulation of cell death. Eur J Biochem. 236:1–26. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang C and Youle RJ: The role of
mitochondria in apoptosis. Annu Rev Genet. 43:95–118. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Inoue S, Browne G, Melino G and Cohen GM:
Ordering of caspases in cells undergoing apoptosis by the intrinsic
pathway. Cell Death Differ. 16:1053–1061. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cory S, Huang DCS and Adams JM: The Bcl-2
family: Roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ashkenazi A: Targeting the extrinsic
apoptosis pathway in cancer. Cytokine Growth Factor Rev.
19:325–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nagata S: Apoptosis by death factor. Cell.
88:355–365. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
46
|
Berridge MJ: The endoplasmic reticulum: A
multifunctional signaling organelle. Cell Calcium. 32:235–249.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Banjerdpongchai R, Wudtiwai B, Khaw-On P,
Rachakhom W, Duangnil N and Kongtawelert P: Hesperidin from Citrus
seed induces human hepatocellular carcinoma HepG2 cell apoptosis
via both mitochondrial and death receptor pathways. Tumour Biol.
37:227–237. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oh H, Yoon G, Shin JC, Park SM, Cho SS,
Cho JH, Lee MH, Liu K, Cho YS, Chae JI, et al: Licochalcone B
induces apoptosis of human oral squamous cell carcinoma through the
extrinsic- and intrinsic-signaling pathways. Int J Oncol.
48:1749–1757. 2016.PubMed/NCBI
|
|
49
|
Yaoxian W, Hui Y, Yunyan Z, Yanqin L, Xin
G and Xiaoke W: Emodin induces apoptosis of human cervical cancer
heLa cells via intrinsic mitochondrial and extrinsic death receptor
pathway. Cancer Cell Int. 13:71–78. 2013. View Article : Google Scholar : PubMed/NCBI
|