Introduction
Melanoma is one of the most aggressive human cancers
and its incidence continues to increase worldwide (1). Although melanoma is one of the most
aggressive human cancers, the identification of multiple melanoma
driven mutations and improved understanding of cancer immune
tolerance checkpoints has led to identification of new therapeutic
opportunities for patients (1,2). The
most common of these mutations is BRAFV600E. It is found
in 50–60% of melanomas and successful targeting of the
BRAFV600E, such as vemurafenib, has produced amazing
clinical responses in patients with melanoma harboring this
mutation (3,4). Vemurafenib is the first drug approved
for the treatment of BRAF-mutant cancer (3). Unfortunately, the majority of
responding patients eventually develop resistance and disease
progression, typically within 5–7 months after starting the
treatment (5). Therefore,
overcoming the acquired resistance remains a considerable
therapeutic challenge to achieve durable responses and prolonged
survival in these patients.
A variety of molecular mechanisms have been
identified to be involved in acquired resistance to vemurafenib
(5). Unlike acquired resistance to
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors
are mainly generated by secondary mutations in EGFR, multiple
mechanisms account for the resistance to vemurafenib (6). These mechanisms include the
loss/inactivation of phosphatase and tensin homolog (PTEN)
function, deletion of the retinoblastoma protein (RB), elevated
expression of the kinases CRAF, amplification of cyclin D1,
alternative splicing of BRAF mRNA, activating mutations in NRAS,
mitogen-activated protein kinase kinase (MEK), or AKT and
persistent activation of receptor tyrosine kinases, including EGFR,
IGF1R and platelet-derived growth factor β (PDGFRβ) (7). The diversity of resistance to
vemurafenib implies that BRAF inhibitor resistance can be overcome
through broadly targeted strategies that inhibit multiple pathways
simultaneously.
Histone deacetylases (HDACs) are a group of enzymes
that function by catalyzing the removal of the acetyl groups of
both histones and non-histone proteins (8). HDACs are considered to be among the
most promising targets in drug development for cancer therapy
(8). Among the 18 HDACs, HDAC6 has
recently sparked great interest as it deacetylates various
substrates involved in the regulation of protein trafficking and
degradation, autophagy, apoptosis, cell cycle, migration and
proliferation (9). HDAC6 is
required for the proliferation and metastasis of melanoma cells and
knockdown of HDAC6 decreases proliferation and induces cell cycle
arrest of melanoma cells (10,11).
HDAC6 has also been shown to confer resistance to chemotherapy in
many types of cancer (12–14). Many pathways involved in acquired
resistance to vemurafenib are known to be regulated by HDAC6
(7,15). However, the role of HDAC6 in
vemurafenib resistance is still unclear.
Here we report that overexpression of HDAC6 confers
resistance to vemurafenib in the BRAFV600E melanoma cell
line A375. HDAC6 deacetylase activity selective inhibitor,
ACY-1215, impairs proliferation and induces apoptosis of A375.
Combination use of vemurafenib with ACY-1215 displayed an additive
therapeutic effect in BRAF-mutant melanoma cells by inducing ER
stress and inactivation of extracellular signal-regulated kinase
(ERK). Our results suggest that inhibition of HDAC6 may be a
promising strategy for overcoming the resistance to vemurafenib in
melanoma harboring BRAFV600E mutation.
Materials and methods
Cell culture
The BRAF-mutant melanoma cell line A375 was obtained
from the American Type Culture Collection (Manassas, VA, USA). A375
was cultured in RPMI-1640 medium (HyClone) supplemented with 10%
FBS and 1% penicillin/streptomycin.
Reagents
ACY-1215 and Z-VAD-FMK were purchased from Medchem
Express (Monmouth Junction, NJ, USA). The anti-PARP (#9542),
anti-eIF2α (#5324), anti-p-eIF2α (#9721) and anti-CHOP (#2895)
antibodies were obtained from Cell Signaling Technology (Danvers,
MA, USA). The anti-caspase-3 (EAP0893) antibody was from
Elabscience (Wuhan, Hubei, China). The anti-p-ERK (ab76299)
antibody was purchased from Abcam (Cambridge, UK). The anti-ERK
(16443-1-AP) and anti-GFP (50430-2-AP) antibodies was obtained from
Proteintech (Chicago, IL, USA). The anti-β-actin mouse monoclonal
antibody (AM1021B) was purchased from Abgent (San Diego, CA,
USA).
Cell proliferation and colony
formation detection
A375 cell proliferation was measured at the
indicated times by Cell counting kit-8 (CCK-8) kit (Dojindo,
Kumamoto, Japan Japan). A375 colony formation was measured by
seeding cells in 6-well plates 1,000 per well and were cultured
over a 14-day period. Colonies were fixed in 4% paraformaldehyde
and stained with 0.1% crystal violet.
Annexin V assay of cell apoptosis
Effects of plasmids or drugs on apoptosis of A375
cells were evaluated by Annexin V/7-aminoactinomycin D (7-AAD)
assay. Flow cytometric analysis of A375 cells labeled with Annexin
V-phycoerythrin (PE) and 7-AAD apoptosis detection kit (BD
Biosciences, San Jose, CA, USA) was performed according to the
manufacturer's instructions. The rates of cellular apoptosis were
acquired immediately on a FACSArial flow cytometer (BD
Biosciences).
Plasmids and transfection
The plasmid encoding HDAC6 was kindly provided by
Professor Jun Zhou (16). The
plasmids have been validated by sequencing, transfection and
subsequent western blotting. The plasmids were transfected into
cells with Lipofectamine 2000 transfection reagent (Invitrogen,
Waltham, MA, USA) according to the manufacturer's protocol.
Protein extraction and western
blotting
A375 cells were lysed in RIPA lysis buffer (1%
Triton X-100, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5%
sodium deoxycholate and 50 mM Tris-HCl, pH 7.4) with protease
inhibitor cocktails and phosphatase inhibitor cocktails from Roche
(Mannheim, Germany) on ice for 30 min and centrifuged at 12,000 rpm
for 15 min to collect whole cell lysate. Protein extracts were
subjected to electrophoresis on a 10% SDS-PAGE gel and transferred
onto PVDF membrane (Roche). Then the membrane was then incubated in
blocking buffer for 2 h before the addition of the primary
antibodies. The secondary antibody used was the horseradish
peroxidase-conjugated goat anti-rabbit/mouse secondary antibody
(Proteintech). Signals were detected using WesternBright ECL HRP
substrate (Advansta, Menlo Park, CA, USA) and developed with Kodak
film.
Statistical analysis
Statistical analysis was performed with Student's
t-test. All analyses were realized by using the statistical
software SPSS19.0 (IBM Corp., Armonk, NY, USA). Data are expressed
as the mean ± standard deviation (SD). P<0.05 was considered a
statistically significant difference.
Results
HDAC6 confers resistance to
vemurafenib mediated killing of BRAF-mutant A375 cells
HDAC6 can confer resistance to chemotherapy in
several types of cancer (12–14),
but the role of HDAC6 in melanoma chemotherapy resistance is
largely unknown. HDAC6 has been shown to be overexpressed in
melanoma cell lines and tissues (10). We speculate that overexpression of
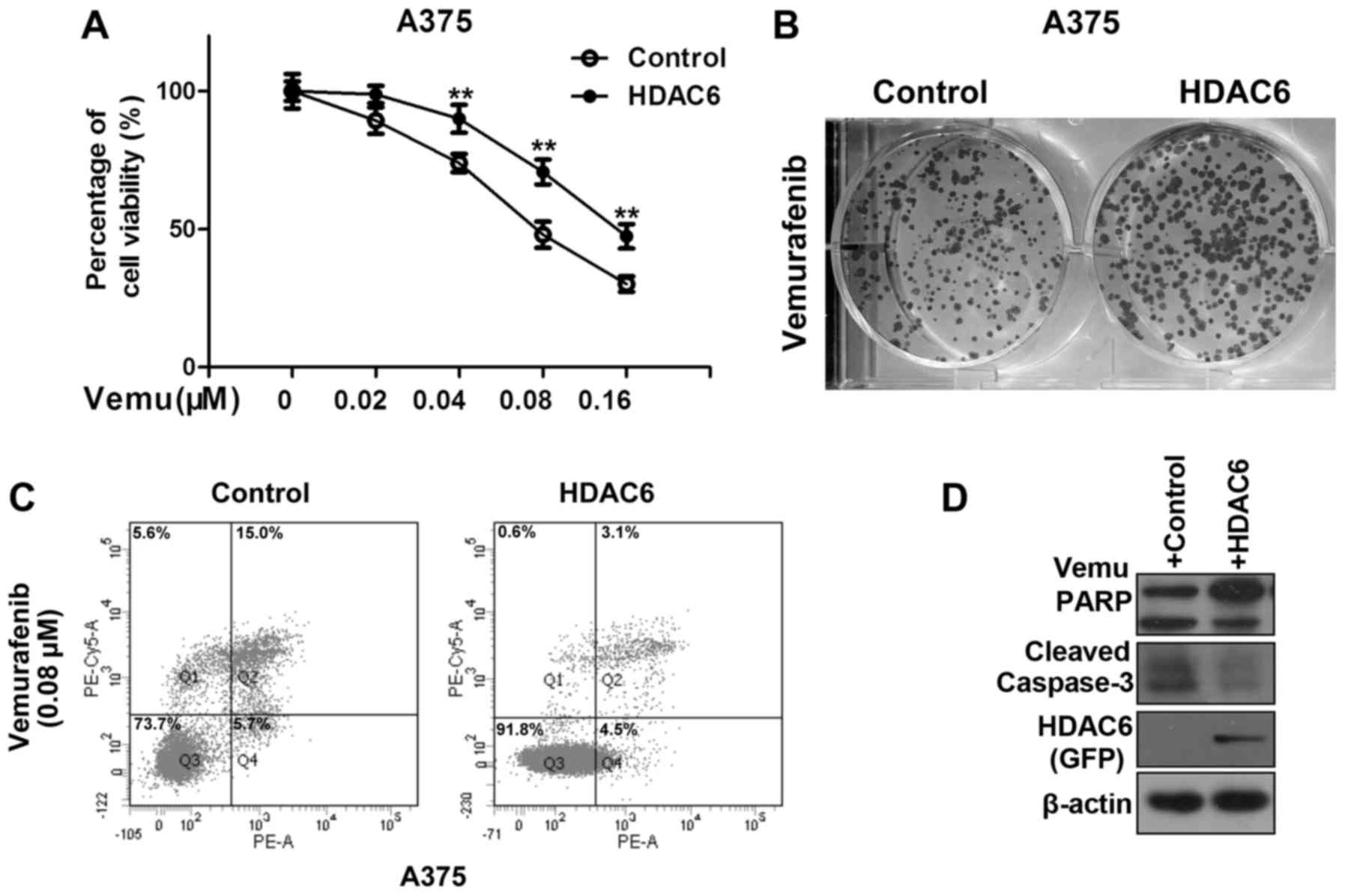
HDAC6 in melanoma might confer resistance to vemurafenib. As shown
in Fig. 1A, overexpression of HDAC6
significantly improved the percentage of viable A375 cells after
vemurafenib treatment. We also came to the same conclusion by the
colony formation assay (Fig. 1B).
Vemurafenib has been shown to cause apoptotic cell death at
elevated concentrations, thus we investigated whether HDAC6
regulates vemurafenib mediated cell apoptosis in melanoma cells.
Overexpression of HDAC6 significantly decreased the percentage of
apoptotic cells after vemurafenib treatment (Fig. 1C). A decrease in apoptosis was
further evidenced by detection of cleaved PARP and caspase-3
(Fig. 1D).
ACY-1215, a selective HDAC6 inhibitor,
sensitizes A375 cells to vemurafenib
Because HDAC6 confers resistance to vemurafenib, we
speculated that the inhibition of HDAC6 might contribute to an
increase in the efficiency of vemurafenib in melanoma. To further
assess this possibility, we tested the effect of HDAC6 inhibition
on A375 using the selective inhibitor ACY-1215. ACY-1215 is the
first oral, selective HDAC6 inhibitor in clinical trials and was
well tolerated as monotherapy ≤360 mg/day (17,18).
Unlike non-selective HDAC inhibitors, which are associated with
severe fatigue, vomiting, diarrhea and myelosuppression (18). ACY-1215 offers a therapeutic
advantage due to minimal toxicity. HDAC6 is required for the
proliferation and metastasis of melanoma cells (10,11).
However, few studies have focused on the function of ACY-1215 in
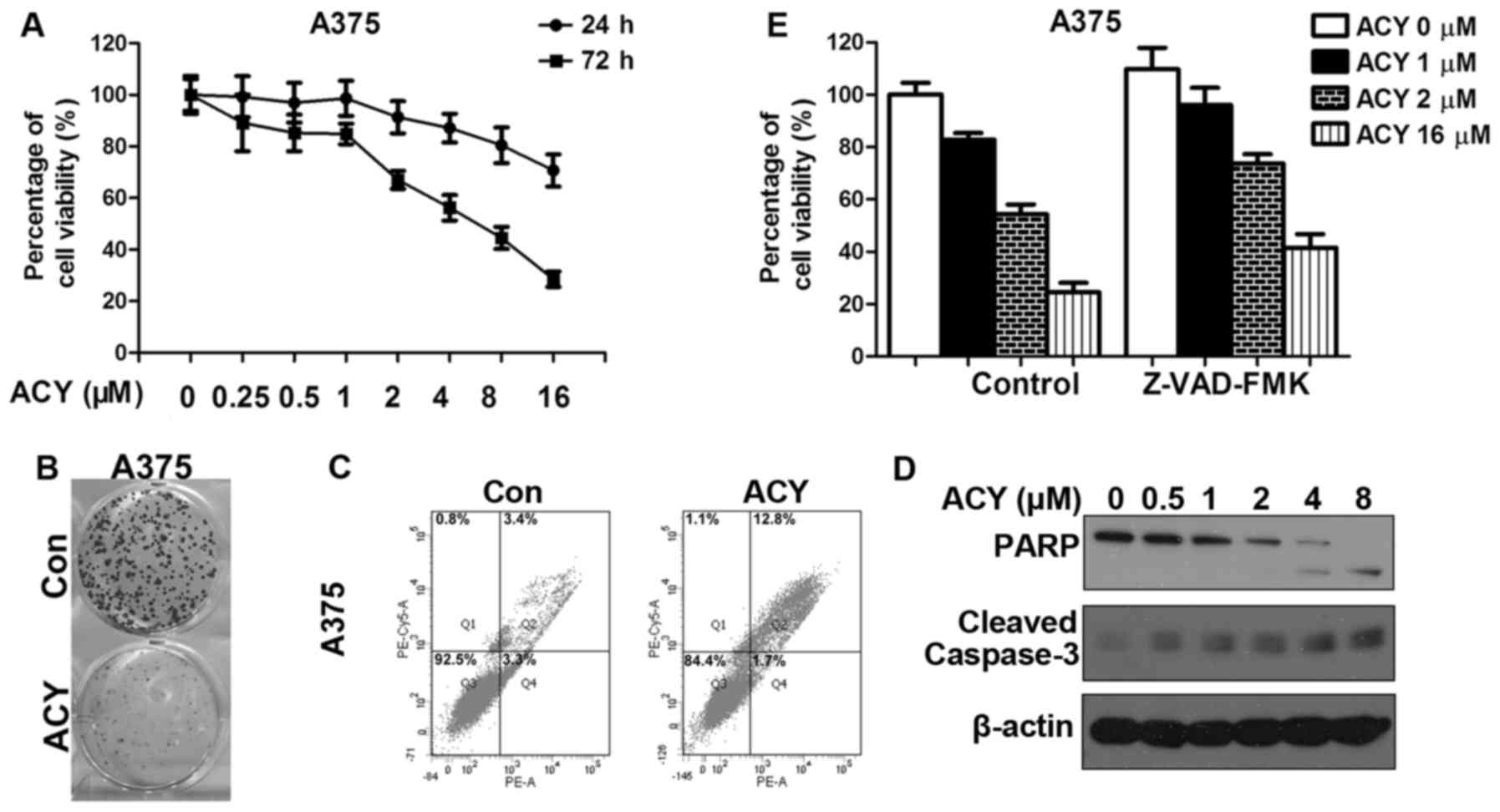
melanoma cells. We first assessed growth inhibition in response to
ACY-1215 treatment in A375. A dose-dependent decrease in cell
proliferation was observed (Fig.
2A). The same conclusion was drawn from the results of the
clone formation assay. We noted a significant decrease in clone
numbers after ACY-1215 treatment (Fig.
2B). ACY-1215 also induces apoptosis in A375, as evidenced by
increases in the number of Annexin V-positive cells and the
cleavage of PARP and caspase-3 (Fig. 2C
and E). Combination inhibition of apoptosis by pan caspase
inhibitor Z-VAD-FMK with ACY-1215 remarkablely improved the
percentage of cell viability compared to ACY-1215 alone (Fig. 2D). To examine whether a cooperative
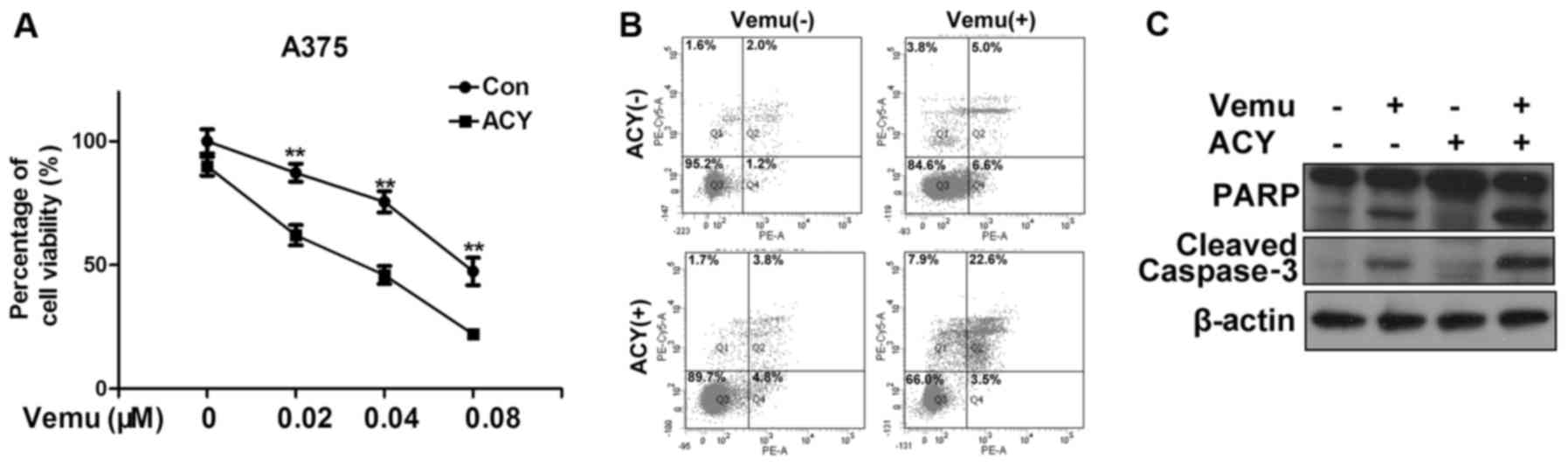
effect exists between ACY-1215 and vemurafenib in the
chemotherapeutic treatment of melanoma, we treated A375 cells with
ACY-1215 and vemurafenib either alone or in combination. In
agreement with our hypothesis, the co-treatment of ACY-1215 and
vemurafenib significantly reduced the cell viability compared to
vemurafenib alone (Fig. 3A). We
also observed a significant increase of the percentage of apoptotic
cells after cotreatment of vemurafenib with ACY-1215 compared to
vemurafenib alone (Fig. 3B). The
protein levels of cleaved PARP and caspase-3 also increased after
co-treatment (Fig. 3C).
ACY-1215-induced ER stress plays a
role in overcoming acquired resistance to vemurafenib
Next, we investigated the mechanism by which
ACY-1215 contribute to the vemurafenib-induced cell death of
melanoma cells. HDAC6 plays a vital role in regulation of degrading
misfolded protein through the ubiquitin proteasome system and
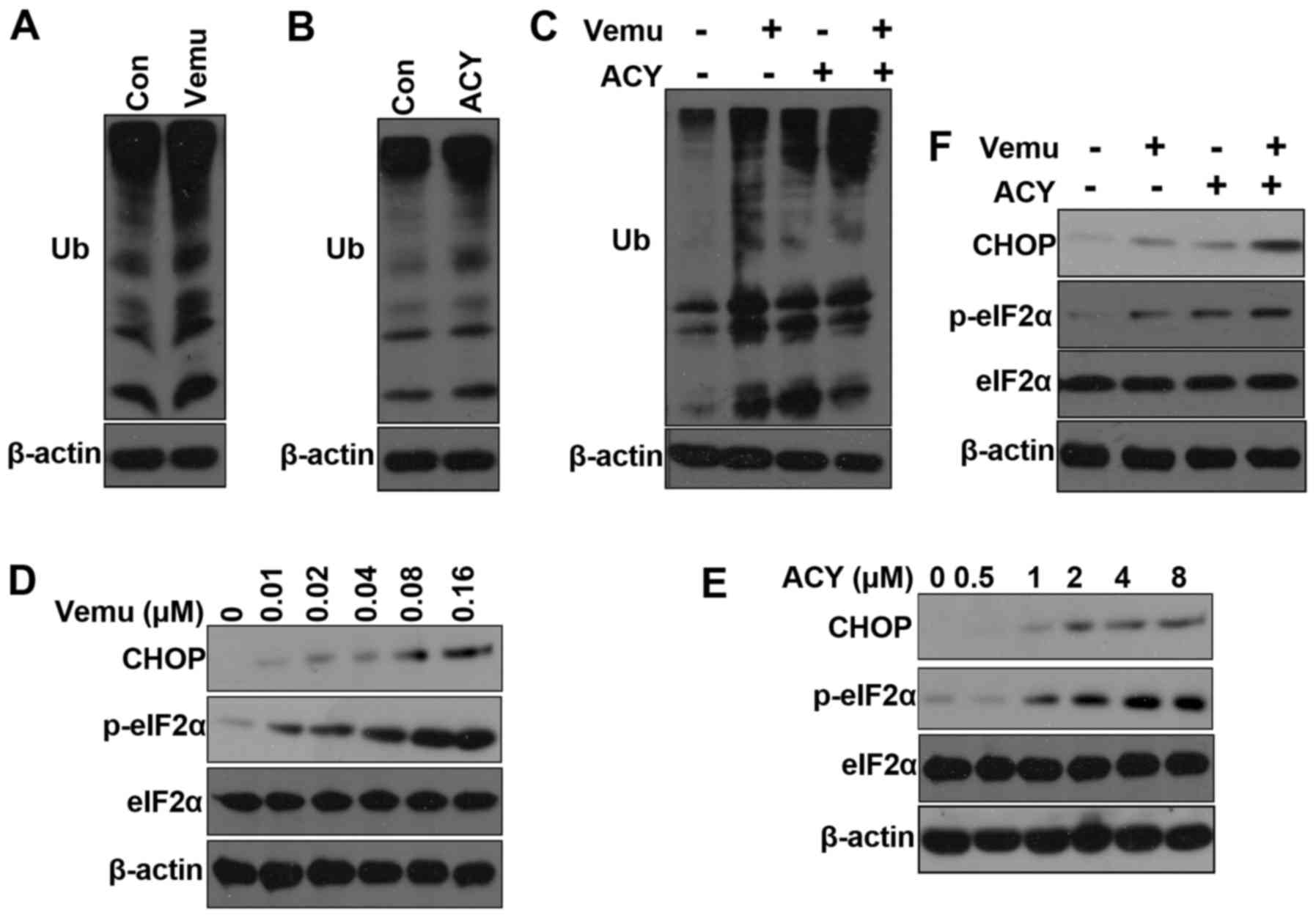
autophagy (19,20). We found that both vemurafenib and
ACY-1215 increased the accumulation of polyubiquitinated proteins
(Fig. 4A and B). Moreover, the
combination of ACY-1215 plus vemurafenib increased the accumulation
of polyubiquitinated proteins compared with either agent alone
(Fig. 4C). Accumulation of
polyubiquitinated proteins can induce ER stress (21). Vemurafenib has been shown to induce
endoplasmic reticulum (ER) stress-mediated apoptosis to kill
BRAFV600E melanoma cells (22). Upon ER stress, the sensor PERK is
activated and phosphorylates eIF2a (23,24).
During ER stress, the protein levels of CHOP are also elevated and
CHOP functions to mediate programmed cell death (24,25).
To determine the onset of ER stress in response to ACY-1215, we
examined the phosphorylation of eIF2α by western blotting and found
that it was rapidly phosphorylated after ACY-1215 treatment
(Fig. 4D). Vemurafenib can also
clearly induce ER stress as previously reported (Fig. 4C). Combination treatment of
vemurafenib and ACY-1215 significantly increased the
phosphorylation of eIF2α and protein levels of CHOP. Taken
together, these results indicate that ACY-1215 induced ER stress
plays a role in overcoming resistance to vemurafenib.
HDAC6-mediated ERK activation partly
accounts for its oncogenic role in melanoma
Hyperactivation of the RAF-MEK-ERK signaling pathway
plays a vital role in tumorigenesis of BRAF-mutant melanoma
(26). RAF kinase directly
phosphorylates MEK, which in turn phosphorylates ERK to promote
cell proliferation and to inhibit apoptosis (27). Vemurafenib blocks ERK
phosphorylation to inhibit proliferation of BRAF-mutant cells
(3,22), this was also confirmed in our
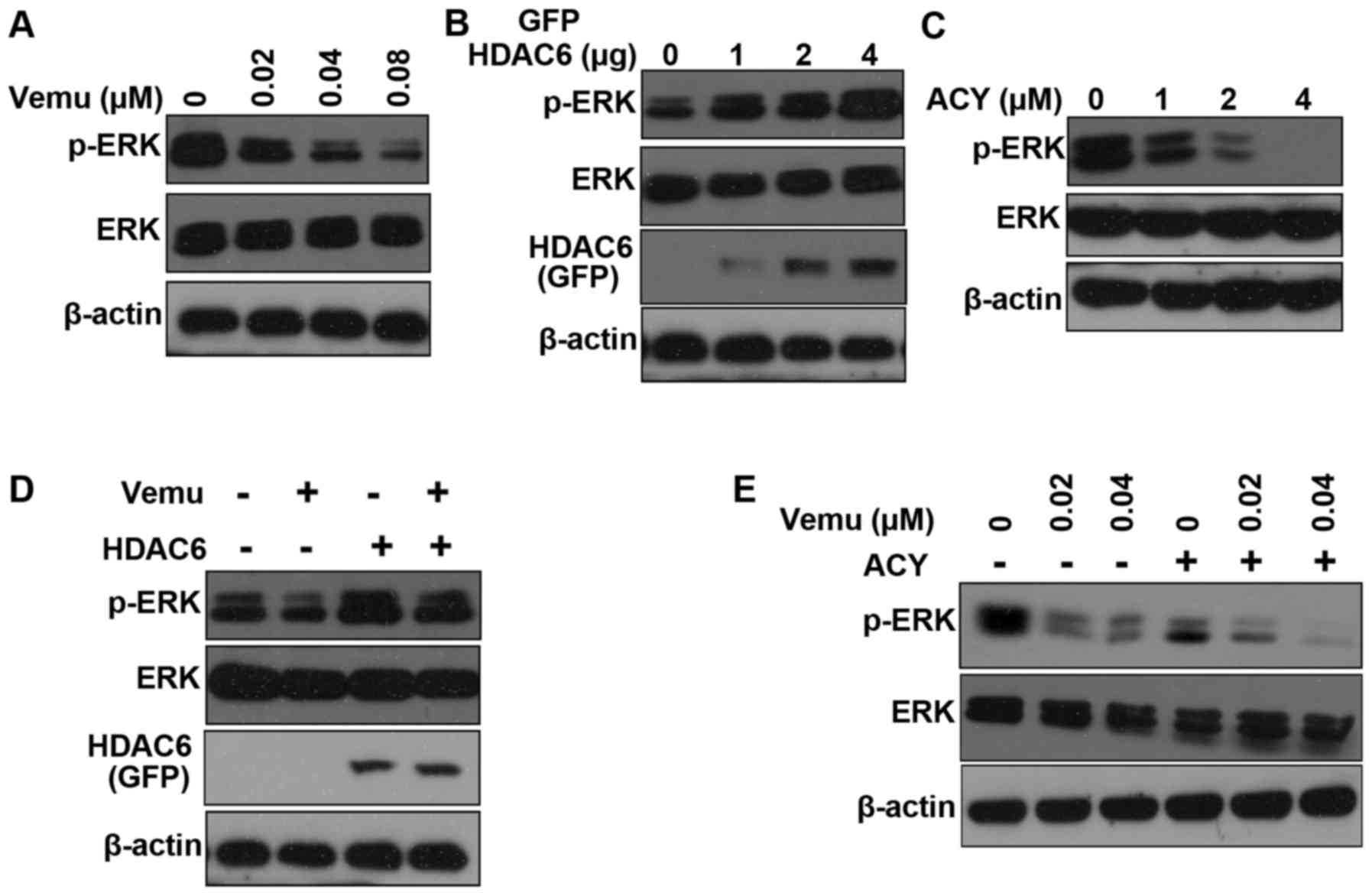
experiment (Fig. 5A). HDAC6 can
induce ERK hyper-activation in Madin-Darby canine kidney (MDCK) and
HEK293T cells (28). Overexpression
of HDAC6 also increased ERK activity in A375 cells (Fig. 5B). Conversely, ACY-1215 decreases
the phosphorylation of ERK in a dose-dependent manner (Fig. 5C). Moreover, overexpression of HDAC6
increased ERK activity after vemurafenib treatment (Fig. 5D). We found that the activation of
ERK was inhibited by the combination of ACY-1215 and vemurafenib
compared with the administration of either inhibitor alone
(Fig. 5E). Taken together, our
results show that inhibition of HDAC6 sensitizes A375 cells to
vemurafenib partly via inactivation of ERK.
Discussion
In this study, we addressed whether inhibition of
HDAC6 can sensitize BRAF-mutant melanoma cells to vemurafenib
(PLX4032). Overexpression of HDAC6 confers resistance to
vemurafenib in A375 cells. Inhibition of HDAC6 by a selective
inhibitor, ACY-1215, sensitizes A375 cells to vemurafenib mediated
proliferation inhibition and apoptosis induction. Moreover,
ACY-1215 mediated ER stress induction and ERK inactivation might
play a role in overcoming acquired resistance to vemurafenib.
Immune checkpoint blockade by directly targeting
programmed death 1 (PD-1) with monoclonal antibodies has gained
great success in clinic in patients with advanced melanoma
(29,30). PD-1 can negatively regulate the
effector phase of T-cell responses after ligation of PD-1 ligand 1
(PD-L1), which plays a vital role in the ability of tumor cells to
evade the immune system (31).
Therefore, cancer tissues can limit the host immune response via
upregulation of PD-1 ligand (PD-L1) and its ligation to PD-1 on
antigen-specific CD8+ cells (31). However, not all patients respond
equally to this treatment (32).
There is an urgent need to identify new potential therapeutic
adjuvants to improve immunotherapeutic efficiency towards melanoma
(33). Some HDACs have been shown
to function as modulators of the immune response (33). Among them, HDAC6 recently sparked
great interest as the inhibition of HDAC6 can downregulate the
expression of PD-L1 in primary melanoma samples and cell lines
(33). Woan et al also
showed that targeting HDAC6 can enhance antitumor immunity in
melanoma cells (11). Therefore,
HDAC6 might also exert oncogenic functions by mediating immune
escape in melanoma. Combination use of HDAC6 inhibitors and PD1
inhibitors might further benefit melanoma patients. HDAC6
inhibitors are being tested in clinical trials in combination with
nivolumab (PD1 blocking antibody) in patients with unresectable
non-small cell lung cancer (NSCLC) (https://www.clinicaltrials.gov/, ClinicalTrials.gov Identifier:NCT02635061).
Prahallad et al reported that feedback
activation of EGFR mediated by BRAFV600E inhibition
might contribute to unresponsiveness of colon cancer to vemurafenib
(34). Corcoran et al proved
that EGFR-mediated reactivation of MAPK signaling contributes to
resistance to vemurafenib in colorectal cancers (35). HDAC6 regulates EGFR endocytic
trafficking and degradation (36,37).
HDAC6 has been shown to confer resistance to chemotherapy through
stabilization and activation of EGFR (13,38,39).
Therefore, HDAC6 might also confer resistance to vemurafenib via
regulation of EGFR stabilization and activation. Pharmacologic
inhibition of heat shock protein 90 (HSP90) can abrogate both
acquired and intrinsic vemurafenib resistance by restoring
apoptotic response (7). Many
proteins involved in vemurafenib resistance, including mutated
BRAF, CRAF, cyclin D1, IGF1R, AKT and CDK4 are known to be
regulated by HSP90 (7). HDAC6 can
directly regulate HSP90 chaperone activity through deacetylation of
HSP90 (15). Therefore, HDAC6 can
also confer resistance to vemurafenib via regulation of HSP90
activity. Autophagy inhibition improves chemosensitivity in
BRAFV600E tumors (40).
HDAC6 regulates autophagosome maturation and inhibition of HDAC6
has been shown to sensitize cancer cells to chemotherapy by
inhibiting autophagy (20).
In conclusion, we found that HDAC6 confers
resistance to vemurafenib in BRAF-mutant melanoma and that
inhibition of HDAC6 impairs the proliferation of BRAF-mutant
melanoma. Moreover, the HDAC6 inhibitor sensitizes BRAF-mutant
melanoma cells to vemurafenib-induced cell proliferation inhibition
and apoptosis induction, which occur partly through induction of ER
stress and inactivation of ERK. Therefore, inhibition of HDAC6
might be a potential strategy for treating melanoma and overcoming
resistance to vemurfenib.
Acknowledgements
The HDAC6 overexpression plasmids were kindly
provided by Professor Jun Zhou of College of Life Sciences, Nankai
University. This study was supported by Hubei Province Natural
Sciences Foundation (no. 2013CFB263).
Glossary
Abbreviations
Abbreviations:
|
PARP
|
poly(ADP-ribose) polymerase
|
|
Raf
|
rapidly accelerated fibrosarcoma
|
References
|
1
|
Flaherty KT, Hodi FS and Fisher DE: From
genes to drugs: Targeted strategies for melanoma. Nat Rev Cancer.
12:349–361. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Simões MC, Sousa JJ and Pais AA: Skin
cancer and new treatment perspectives: A review. Cancer Lett.
357:8–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bollag G, Tsai J, Zhang J, Zhang C,
Ibrahim P, Nolop K and Hirth P: Vemurafenib: The first drug
approved for BRAF-mutant cancer. Nat Rev Drug Discov. 11:873–886.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holderfield M, Deuker MM, McCormick F and
McMahon M: Targeting RAF kinases for cancer therapy: BRAF-mutated
melanoma and beyond. Nat Rev Cancer. 14:455–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Strickland LR, Pal HC, Elmets CA and Afaq
F: Targeting drivers of melanoma with synthetic small molecules and
phytochemicals. Cancer Lett. 359:20–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Romano E, Pradervand S, Paillusson A,
Weber J, Harshman K, Muehlethaler K, Speiser D, Peters S, Rimoldi D
and Michielin O: Identification of multiple mechanisms of
resistance to vemurafenib in a patient with
BRAFV600E-mutated cutaneous melanoma successfully
rechallenged after progression. Clin Cancer Res. 19:5749–5757.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paraiso KH, Haarberg HE, Wood E, Rebecca
VW, Chen YA, Xiang Y, Ribas A, Lo RS, Weber JS, Sondak VK, et al:
The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance
mediated through diverse mechanisms. Clin Cancer Res. 18:2502–2514.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Falkenberg KJ and Johnstone RW: Histone
deacetylases and their inhibitors in cancer, neurological diseases
and immune disorders. Nat Rev Drug Discov. 13:673–691. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaliszczak M, Trousil S, Åberg O, Perumal
M, Nguyen QD and Aboagye EO: A novel small molecule hydroxamate
preferentially inhibits HDAC6 activity and tumour growth. Br J
Cancer. 108:342–350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu J, Gu J, Feng Z, Yang Y, Zhu N, Lu W
and Qi F: Both HDAC5 and HDAC6 are required for the proliferation
and metastasis of melanoma cells. J Transl Med. 14:72016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Woan KV, Lienlaf M, Perez-Villaroel P, Lee
C, Cheng F, Knox T, Woods DM, Barrios K, Powers J, Sahakian E, et
al: Targeting histone deacetylase 6 mediates a dual anti-melanoma
effect: Enhanced antitumor immunity and impaired cell
proliferation. Mol Oncol. 9:1447–1457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang L, Xiang S, Williams KA, Dong H, Bai
W, Nicosia SV, Khochbin S, Bepler G and Zhang X: Depletion of HDAC6
enhances cisplatin-induced DNA damage and apoptosis in non-small
cell lung cancer cells. PLoS One. 7:e442652012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Hu P, Tang F, Lian H, Chen X,
Zhang Y, He X, Liu W and Xie C: HDAC6 promotes cell proliferation
and confers resistance to temozolomide in glioblastoma. Cancer
Lett. 379:134–142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshida M, Furumai R, Nishiyama M, Komatsu
Y, Nishino N and Horinouchi S: Histone deacetylase as a new target
for cancer chemotherapy. Cancer Chemother Pharmacol. 48:(Suppl 1).
S20–S26. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krämer OH, Mahboobi S and Sellmer A:
Drugging the HDAC6-HSP90 interplay in malignant cells. Trends
Pharmacol Sci. 35:501–509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Y, Ran J, Liu M, Li D, Li Y, Shi X,
Meng D, Pan J, Ou G, Aneja R, et al: CYLD mediates ciliogenesis in
multiple organs by deubiquitinating Cep70 and inactivating HDAC6.
Cell Res. 24:1342–1353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Santo L, Hideshima T, Kung AL, Tseng JC,
Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et
al: Preclinical activity, pharmacodynamic, and pharmacokinetic
properties of a selective HDAC6 inhibitor, ACY-1215, in combination
with bortezomib in multiple myeloma. Blood. 119:2579–2589. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Raje N, Vogl DT, Hari PN, et al: ACY-1215,
a selective histone deacetylase (HDAC) 6 inhibitor: Interim results
of combination therapy with bortezomib in Patients with multiple
myeloma (MM). Blood. 122:7592013.PubMed/NCBI
|
|
19
|
Pandey UB, Nie Z, Batlevi Y, McCray BA,
Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA,
Schuldiner O, et al: HDAC6 rescues neurodegeneration and provides
an essential link between autophagy and the UPS. Nature.
447:859–863. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong
E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al: HDAC6
controls autophagosome maturation essential for ubiquitin-selective
quality-control autophagy. EMBO J. 29:969–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Senft D and Ronai ZA: UPR, autophagy, and
mitochondria crosstalk underlies the ER stress response. Trends
Biochem Sci. 40:141–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beck D, Niessner H, Smalley KS, Flaherty
K, Paraiso KH, Busch C, Sinnberg T, Vasseur S, Iovanna JL, Drießen
S, et al: Vemurafenib potently induces endoplasmic reticulum
stress-mediated apoptosis in BRAFV600E melanoma cells. Sci Signal.
6:ra72013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rodvold JJ, Mahadevan NR and Zanetti M:
Immune modulation by ER stress and inflammation in the tumor
microenvironment. Cancer Lett. 380:227–236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rutkowski DT and Kaufman RJ: A trip to the
ER: Coping with stress. Trends Cell Biol. 14:20–28. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rizos H, Menzies AM, Pupo GM, Carlino MS,
Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al:
BRAF inhibitor resistance mechanisms in metastatic melanoma:
Spectrum and clinical impact. Clin Cancer Res. 20:1965–1977. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Samatar AA and Poulikakos PI: Targeting
RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug
Discov. 13:928–942. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tien SC and Chang ZF: Oncogenic Shp2
disturbs microtubule regulation to cause HDAC6-dependent ERK
hyperactivation. Oncogene. 33:2938–2946. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ott PA, Hodi FS and Robert C: CTLA-4 and
PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable
clinical benefit in melanoma patients. Clin Cancer Res.
19:5300–5309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Merelli B, Massi D, Cattaneo L and Mandalà
M: Targeting the PD1/PD-L1 axis in melanoma: Biological rationale,
clinical challenges and opportunities. Crit Rev Oncol Hematol.
89:140–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tumeh PC, Harview CL, Yearley JH, Shintaku
IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu
V, et al: PD-1 blockade induces responses by inhibiting adaptive
immune resistance. Nature. 515:568–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hao M, Song F, Du X, Wang G, Yang Y, Chen
K and Yang J: Advances in targeted therapy for unresectable
melanoma: New drugs and combinations. Cancer Lett. 359:1–8. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lienlaf M, Perez-Villarroel P, Knox T,
Pabon M, Sahakian E, Powers J, Woan KV, Lee C, Cheng F, Deng S, et
al: Essential role of HDAC6 in the regulation of PD-L1 in melanoma.
Mol Oncol. 10:735–750. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Prahallad A, Sun C, Huang S, Di
Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A
and Bernards R: Unresponsiveness of colon cancer to BRAF(V600E)
inhibition through feedback activation of EGFR. Nature.
483:100–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Corcoran RB, Ebi H, Turke AB, Coffee EM,
Nishino M, Cogdill AP, Brown RD, Pelle P Della, Dias-Santagata D,
Hung KE, et al: EGFR-mediated re-activation of MAPK signaling
contributes to insensitivity of BRAF mutant colorectal cancers to
RAF inhibition with vemurafenib. Cancer Discov. 2:227–235. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao YS, Hubbert CC and Yao TP: The
microtubule-associated histone deacetylase 6 (HDAC6) regulates
epidermal growth factor receptor (EGFR) endocytic trafficking and
degradation. J Biol Chem. 285:11219–11226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Deribe YL, Wild P, Chandrashaker A, Curak
J, Schmidt MH, Kalaidzidis Y, Milutinovic N, Kratchmarova I,
Buerkle L, Fetchko MJ, et al: Regulation of epidermal growth factor
receptor trafficking by lysine deacetylase HDAC6. Sci Signal.
2:ra842009.PubMed/NCBI
|
|
38
|
Wang Z, Hu P, Tang F and Xie C:
HDAC6-mediated EGFR stabilization and activation restrict cell
response to sorafenib in non-small cell lung cancer cells. Med
Oncol. 33:502016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Z, Tang F, Hu P, Wang Y, Gong J, Sun
S and Xie C: HDAC6 promotes cell proliferation and confers
resistance to gefitinib in lung adenocarcinoma. Oncol Rep.
36:589–597. 2016.PubMed/NCBI
|
|
40
|
Sanduja S, Feng Y, Mathis RA, Sokol ES,
Reinhardt F, Halaban R and Gupta PB: AMPK promotes tolerance to Ras
pathway inhibition by activating autophagy. Oncogene. 35:5295–5303.
2016. View Article : Google Scholar : PubMed/NCBI
|