Introduction
A small proportion of breast and ovarian cancer is
explained by a mutation in BRCA1 and BRCA2. Mutations
in the BRCA1 or BRCA2 genes, respectively located on
chromosome 17q21 and chromosome 13q12-13, are responsible for
hereditary breast and ovarian cancer syndrome (1). In France, causal mutations in
BRCA1 and BRCA2 genes account for about 10% of this
syndrome (2,3). BRCA1 (breast cancer 1) gene
encodes a multifunctional protein that interacts with tumor
suppressors, DNA repair proteins, cell cycle regulators, RNA
polymerase II holoenzyme, transcription factors, co-repressors,
chromatin remodeling enzymes, and RNA processing factors (4). BRCA1 protein, therefore, has a
critical role in maintaining genomic stability with key functions
in DNA repair, cell cycle progression, transcriptional regulation
and ubiquitylation (4,5). BRCA2 (breast cancer 2) gene
encodes a protein with rather similar roles in the DNA damage
response and DNA repair pathways (5). The BRCA2 protein is a tumor
suppressor that mediates recruitment of the RAD51 recombinase
protein to DNA double-stand breaks (5). The primary function of the
BRCA2 protein is to facilitate homologous recombination, an
important DNA repair mechanism to maintain genomic integrity
(5,6). Loss or inactivation of one copy of
BRCA1 or BRCA2 results in accumulation of mutations
and structural changes in the genome, thereby increasing risk of
cancer (6). BRCA1 and
BRCA2 mutations are inherited in an autosomal dominant
condition with variable penetrance (7).
The present study reports the case of a French
patient with two synchronous different breast cancers metastatic at
presentation who was found to carry a double, never described
mutation, in BRCA1 and BRCA2 genes.
Case report
Clinical history
In July 2014, a 46-year-old woman was referred to
our medical oncology unit because she had noted at self-palpation a
mass in her left breast. Physical examination confirmed an
inflammatory indurated mass of the left breast measuring 12 cm.
Moreover, it revealed an areola retraction of the right breast
measuring 3 cm without any discharge. There were bilateral
clinically fixed axillary lymph nodes.
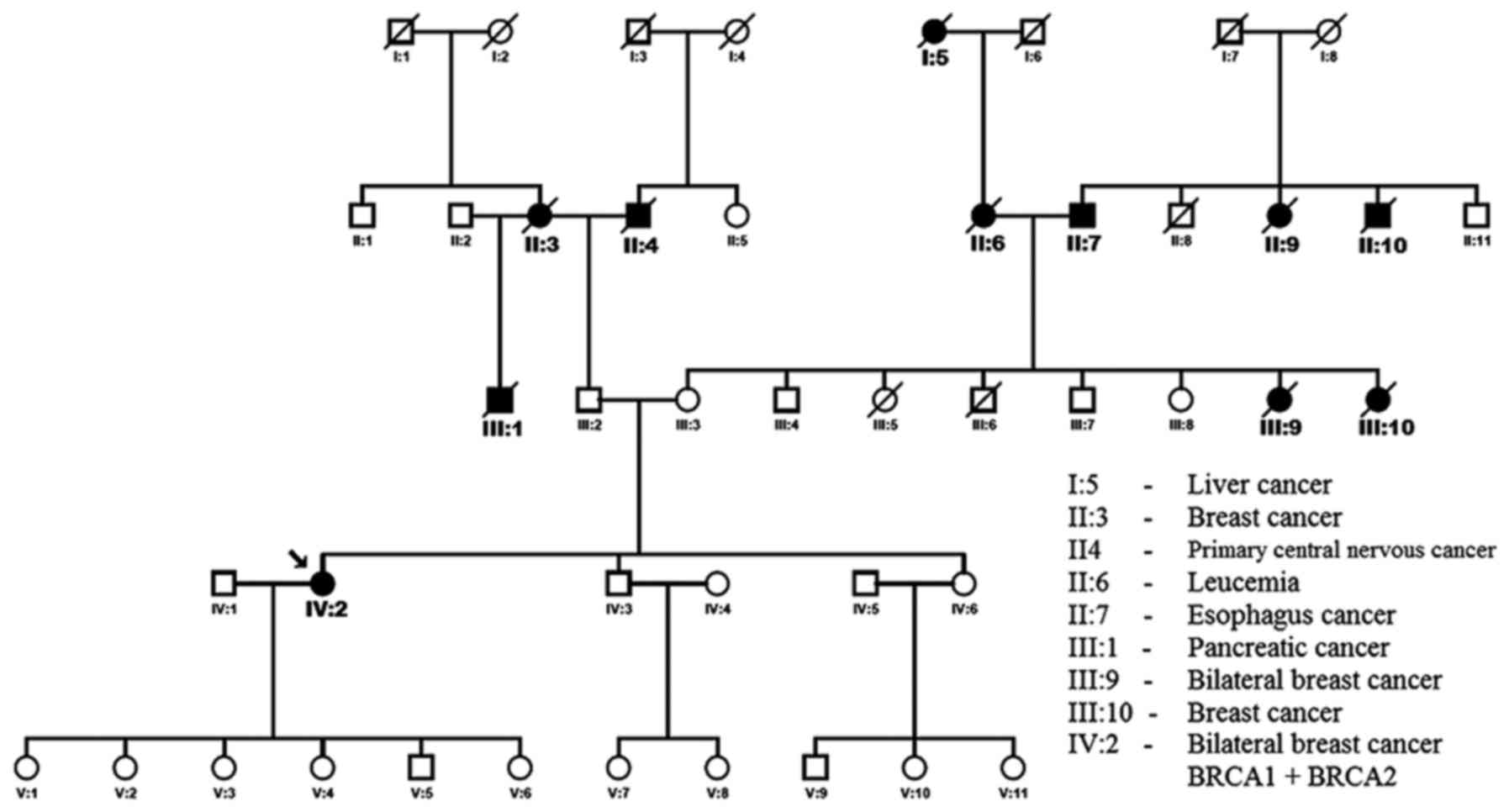
The proband has a positive familial history of
breast carcinomas and other cancers (Fig. 1). Her paternal grandmother (II:3)
died at the age of 72 years from breast cancer. One of her maternal
aunts (III:9) developed bilateral breast cancer at 40 years and
died at 59 years. Another maternal aunts (III:10) developed breast
cancer at 45 years and died at 66 years. Her father's half-brother
died from a pancreatic adenocarcinoma (III:1).
We noted in her personal medical history,
cholecystectomy and active tobacco intoxication measured at 21
pack-years. Initially she had no regular treatment. The patient
started her first period at the age of 14. She had 6 healthy
children and she breast-fed two of them. There were no abortion
rating, no miscarriages. After the birth of her sixth child, a
hormonal intrauterine device was inserted and then removed in July
2014 at breast cancer diagnosis. Chemotherapy induced a stop of
menses.
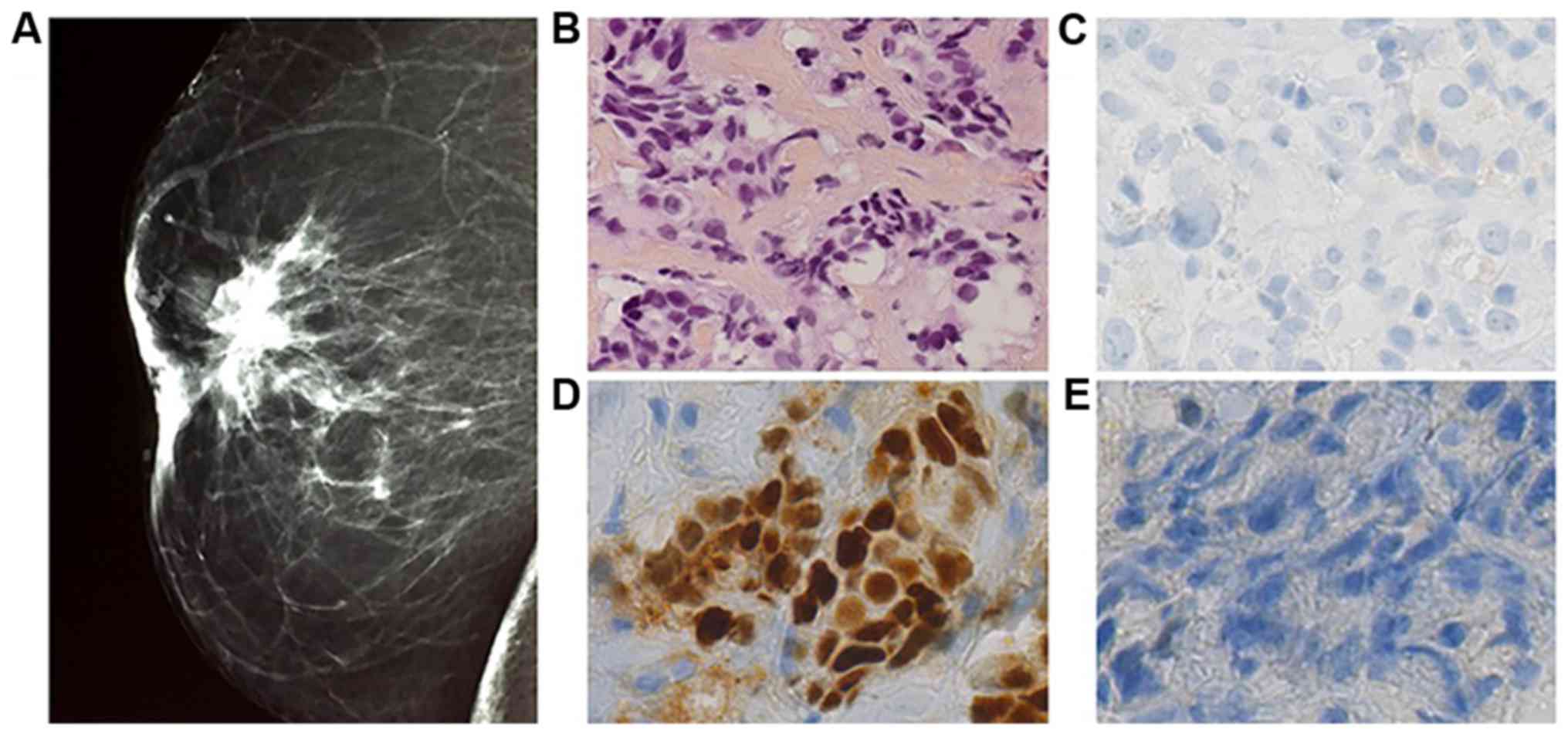
Bilateral digital mammography was performed. Right
breast was Bi Rads type II classified, with supra areolar
retractile opacity measuring 2.2 cm without any calcification
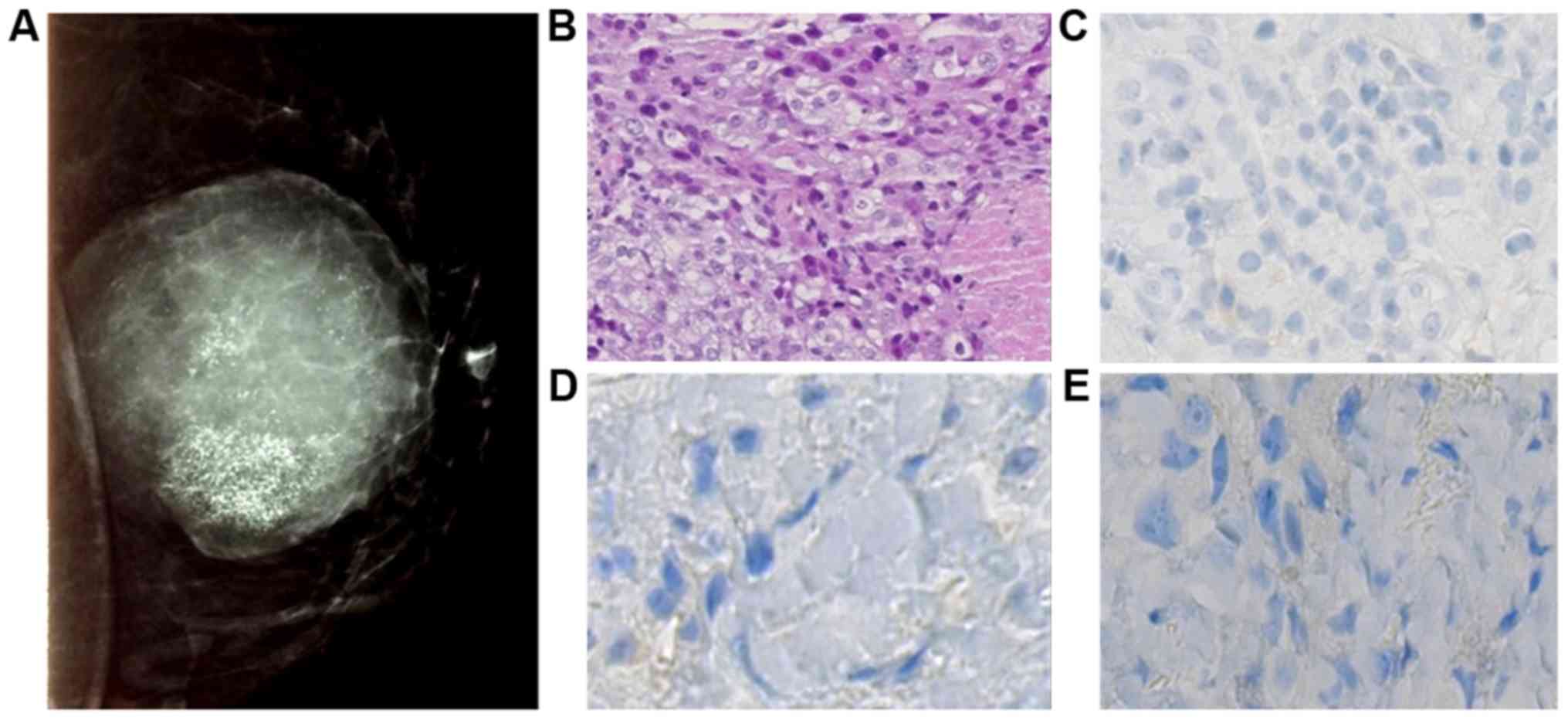
(Fig. 2). In left breast, opacity
in the supero-internal quadrant, measuring 10 cm, well limited with
diffuse micro-calcifications was observed (Fig. 3).
Biopsies confirmed the presence of two breast
cancers with distinct histology. The right breast biopsy diagnosed
infiltrating ductal carcinoma of no special type. Elston and Ellis
grade was scored at II (3.2.1). On immunohistochemical staining,
the tumor strongly expressed estrogen receptor (95%), with no
expression of progesterone receptor and no overexpression of HER 2.
Ki 67 expression was assessed at 14% (Fig. 2). It was T4bN2 classified.
The left breast biopsy diagnosed a triple negative
infiltrating ductal carcinoma of no special type (no expression of
estrogen receptor, progesterone receptor and HER2). Elston and
Ellis grade was scored at III (3.3.3) and cytokeratin 5 and 6 were
expressed in 40% of cells (Fig. 3).
It was T4dN2 classified.
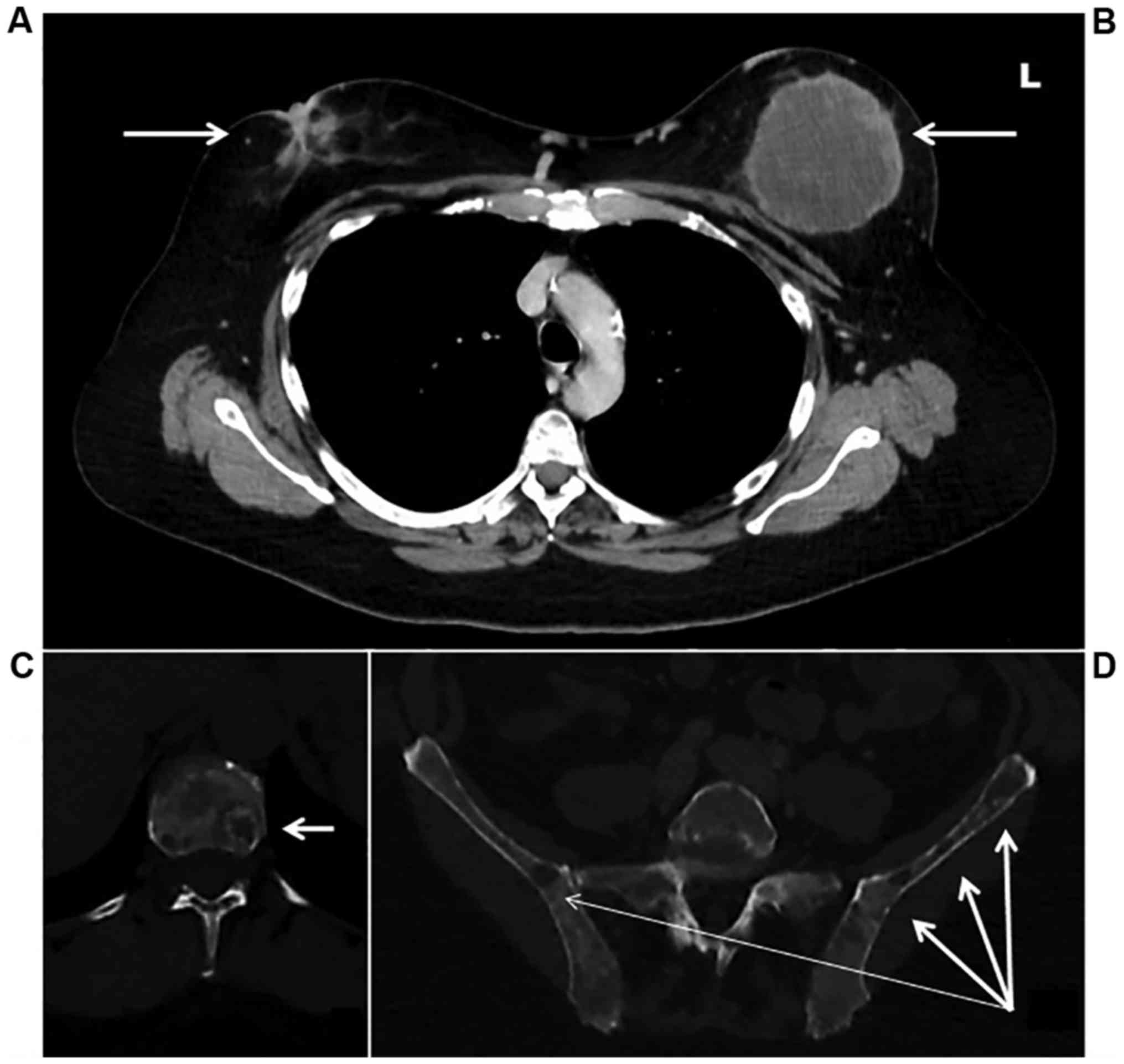
A staging computed tomography and an isotopic bone
scan detected diffuse bone lesions located on the entire spine,
pelvis, skull, and sternum without any visceral metastatic lesions
(Fig. 4). CA 15.3 marker was
measured at 2678 U/ml. Computed tomography and pelvic ultra sound
did not detect ovarian lesion.
Gene testing
The patient was tested for germline mutations in
BRCA1 and BRCA2 genes after written informed and
signed consent. Written informed consent was obtained from the
patient and her parents for publication of this case report and any
accompanying images. Ethics approval was not applicable. The BRCA
True™ test (Pathway Genomics Laboratories) is designed to analyze
the coding and flanking regions of BRCA1 and BRCA2
genes associated with hereditary breast and ovarian cancer by
next-generation sequencing-base (NGS) and Sanger sequencing.
Genomic DNA (gDNA) is extracted from the patient's specimen
(peripheral blood and saliva sample), and evaluated for quality and
quantity using standard procedures. The gDNA is processed to enrich
for the targeted exons and flanking regions in a PCR-based reaction
with target-specific primers. Massive parallel sequencing is
carried out on the enriched target DNA to detect variants in these
regions. Sanger DNA sequencing is utilized for targeted gene
regions that are insufficiently covered on NGS for variant
detection and to confirm specific findings when suspected
pathogenic or novel variants are detected. Gross deletions and
duplications in BRCA1 and BRCA2 genes are identified
by multiplex ligation-dependent probe amplification. Two pathogenic
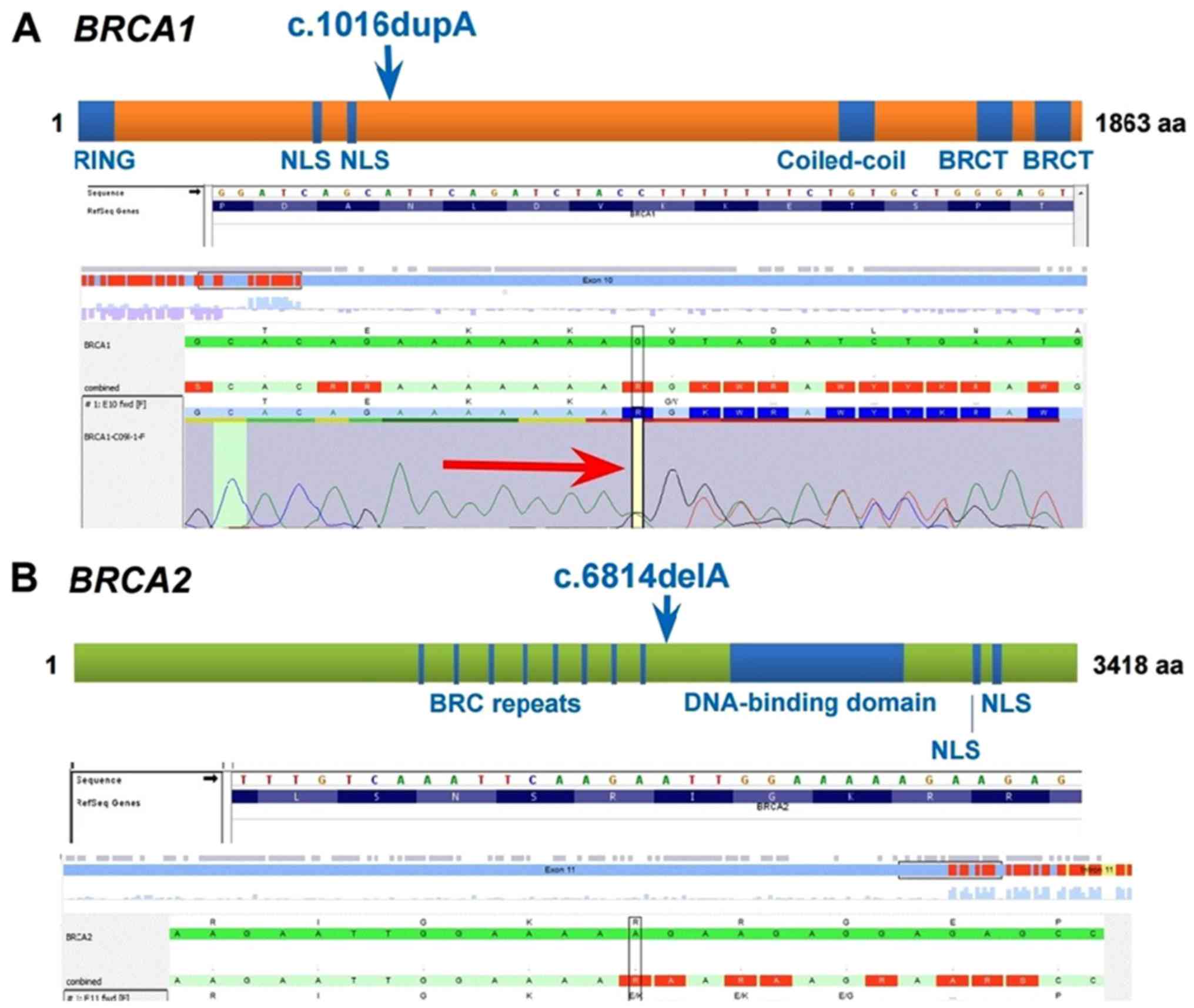
monoallelic mutations were detected, one in each gene: c.1016dupA
(p.V340Gfs*6) mutation in the BRCA1 gene and c.6814delA
(p.R2272Efs*8) mutation in the BRCA2 gene (Fig. 5). The BRCA1 c.1016dupA
(p.V340Gfs*6) pathogenic variant, also known as 1135insA or
1135dup, is predicted to truncate the BRCA1 protein. The
BRCA2 c.6814delA (p.R2272Efs*8) pathogenic variant, also
known as 7042delA, is predicted to truncate the BRCA2
protein. Mutation designation is according to the American College
of Medical Genetics (ACMG) guidelines.
Genetic management
After receiving these results, the proband was
referred to genetic counseling. Subsequent predictive testing was
offered to the proband's parents. The proband's asymptomatic mother
(Fig. 1, indicated by III:3) was
found to be positive for the same mutation in the BRCA1
gene. She is 69 years old. Bilateral digital mammography and pelvic
ultrasound was performed. To date she has had no cancer, and has
now entered into a specific follow-up program. The proband's father
(Fig. 1, indicated by III:2), aged
68 did not carry either mutation. He has had no cancer.
Patient management and outcomes
The patient received a first line of chemotherapy by
weekly paclitaxel (90 mg/m2) with bevacizumab every 2
weeks (10 mg/kg) for 6 months. Treatment was well tolerated and
associated with monthly injections of denosumab 120 mg. After 6
months of treatment, a good partial response according to RECIST
criteria 1.1 was observed with a decrease of the two breast lesions
(from 93 mm to 57 mm in the left breast and from 24 to 13 mm in the
right breast). Axillary lymph nodes were not any more palpable.
Computed tomography and bone scan revealed an improvement on bone
lesions associated with a decrease of CA 15.3 at 285 U/ml. We
decided to focus on systemic therapy, and to delay the two primary
breast cancer surgery. A maintenance therapy with capecitabine 2000
mg twice a day and bevacizumab 15 mg/kg each 21 days was started
(8). Twenty months later, the
patient is still alive with no criteria of progression of the
disease; she has recovered her regular activities and comes every 3
weeks for a maintenance treatment and follow-up.
Discussion
To the best of our knowledge, we described the first
case of a double heterozygosity for BRCA1 and BRCA2
pathogenic variants in a French metastatic breast cancer patient,
with mutation c.1016dupA in BRCA1 and mutation c.6814delA in
BRCA2 gene never described together so far. The co-existence
in an individual of mutations in the BRCA1 and BRCA2
genes is a very rare finding, particularly in non-Ashkenazi
individuals (1,9), and our patient has no known Ashkenazi
heritage.
Moreover, we determined that the patient inherited
the BRCA1 mutation from her mother. Regarding the
BRCA2 mutation, it could be a de novo mutation or
maybe the father is not the biological father. Ethical French rules
do not allow us to genetically test this last hypothesis.
Leegte et al presented a review of the
literature and described the phenotypic expression of double
heterozygosis for BRCA1 and BRCA2 in 34 women
(9). All cases presented an
Ashkenazi mutation. The highest risk of cancer appears to occur in
a combination of 5382insC and 6174delT for BRCA1 and BRAC2
genes, respectively (1,9,10). The
population carrier frequency of 185delAG, 5382insC (BRCA1),
and 6174delT (BRCA2) is estimated to be 0.92, 0.26, and
1.20, respectively, in the Ashkenazi population (11,12)
adding up to approximately 2.4%. Peto et al estimated the
prevalence of BRCA1 and BRCA2 mutation carriers to be
0.11% and 0.12%, respectively, in the non-Ashkenazi (UK)
population, which adds up to 0.23% (13). This indicates that the chance for an
occurrence of double heterozygosity in these populations is between
1 in 1,800 to 190,000, respectively.
Furthermore, the BRCA1 c.1016dupA variant is
considered a Norwegian founder mutation, but has also been observed
in individuals who are of French-Canadian, French, Italian or Dutch
ancestry (2,14–16).
The BRCA2 c.6814delA (p.R2272Efs*8) pathogenic variant, has
been identified in individuals with a personal or family history of
breast and/or ovarian cancer (17,18).
The clinical presentation was advanced and unfavorable, it may
raise up the possibility of a pejorative impact of either involved
mutations or resulting from their association. Nevertheless, review
of the literature suggests that the co-existence of BRCA1
and BRCA2 mutations likely not cause a more severe phenotype
of breast cancer (1,9).
In this patient, concomitant mutations of both
BRCA1 and BRCA2 might lead to two different breast
cancers with distinct histologic features. It might suggest that
even if initially all breast cells present the same genetic
characteristics, oncogenesis of the two tumors are different and
lead to two breast cancers of different subtypes: a triple-negative
and a luminal breast cancer. The next step could be to sequence the
two breast tumors to assess if cancer cells had lost both the wild-
type alleles of BRCA1 and BRCA2. It could explain
whether the oncogenesis of these tumors is driven by a loss of
function of BRCA1 and/or BRCA2. A comparative genomic
analysis of both tumors could prove extremely informative on early
and specific BRCA associated tumorigenesis mechanisms, but
unfortunately, it is not available in our center.
The key-information of this case can be summarized
in three points: we report a double heterozygosity with a never
described association of two BRCA mutations (c.1016dupA in
BRCA1 and c.6814delA in BRCA2 gene). These two
mutations were discovered with the diagnosis of two distinct
concomitant pathological type of breast cancer. We search for
BRCA1 and BRCA2 mutations in the parents; one
mutation is inherited from the mother and the other could be de
novo.
Glossary
Abbreviations
Abbreviations:
|
BRCA gene
|
breast cancer gene
|
|
HER2
|
human epidermal growth factor receptor
2
|
|
RECIST
|
response evaluation criteria in solid
tumors
|
References
|
1
|
Smith M, Fawcett S, Sigalas E, Bell R,
Devery S, Andrieska N and Winship I: Familial breast cancer: Double
heterozygosity for BRCA1 and BRCA2 mutations with differing
phenotypes. Fam Cancer. 7:119–124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Caputo S, Benboudjema L, Sinilnikova O,
Rouleau E, Béroud C and Lidereau R: French BRCA GGC Consortium:
Description and analysis of genetic variants in French hereditary
breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2
databases. Nucleic Acids Res. 40:D992–D1002. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oncogénétique en 2014 - Consultations,
laboratoires et prise en charge - reference: APPDECONCOG15.
Institut National Du Cancer. (In French).
|
|
4
|
Deng CX and Brodie SG: Roles of BRCA1 and
its interacting proteins. BioEssays. 22:728–737. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roy R, Chun J and Powell SN: BRCA1 and
BRCA2: Different roles in a common pathway of genome protection.
Nat Rev Cancer. 12:68–78. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Petrucelli N, Daly MB and Feldman GL:
BRCA1 and BRCA2 hereditary breast and ovarian
cancerGeneReviews(®) [Internet]. Pagon RA, Adam MP,
Ardinger HH, et al: University of Washington; Seattle, WA: 1993.
2016
|
|
7
|
Thull DL and Vogel VG: Recognition and
management of hereditary breast cancer syndromes. Oncologist.
9:13–24. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gligorov J, Doval D, Bines J, Alba E,
Cortes P, Pierga JY, Gupta V, Costa R, Srock S, De Ducla S, et al:
Maintenance capecitabine and bevacizumab versus bevacizumab alone
after initial first-line bevacizumab and docetaxel for patients
with HER2-negative metastatic breast cancer (IMELDA): A randomised,
open-label, phase 3 trial. Lancet Oncol. 15:1351–1360. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leegte B, van der Hout AH, Deffenbaugh AM,
Bakker MK, Mulder IM, ten Berge A, Leenders EP, Wesseling J, De
Hullu J, Hoogerbrugge N, et al: Phenotypic expression of double
heterozygosity for BRCA1 and BRCA2 germline mutations. J Med Genet.
42:e202005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi DH, Lee MH and Haffty BG: Double
heterozygotes for non-Caucasian families with mutations in BRCA-1
and BRCA-2 genes. Breast J. 12:216–220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fodor FH, Weston A, Bleiweiss IJ, McCurdy
LD, Walsh MM, Tartter PI, Brower ST and Eng CM: Frequency and
carrier risk associated with common BRCA1 and BRCA2 mutations in
Ashkenazi Jewish breast cancer patients. Am J Hum Genet. 63:45–51.
1998. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Satagopan JM, Offit K, Foulkes W, Robson
ME, Wacholder S, Eng CM, Karp SE and Begg CB: The lifetime risks of
breast cancer in Ashkenazi Jewish carriers of BRCA1 and BRCA2
mutations. Cancer Epidemiol Biomarkers Prev. 10:467–473.
2001.PubMed/NCBI
|
|
13
|
Peto J, Collins N, Barfoot R, Seal S,
Warren W, Rahman N, Easton DF, Evans C, Deacon J and Stratton MR:
Prevalence of BRCA1 and BRCA2 gene mutations in patients with
early-onset breast cancer. J Natl Cancer Inst. 91:943–949. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Andersen TI, Børresen AL and Møller P: A
common BRCA1 mutation in Norwegian breast and ovarian cancer
families? Am J Hum Genet. 59:486–487. 1996.PubMed/NCBI
|
|
15
|
Simard J, Tonin P, Durocher F, Morgan K,
Rommens J, Gingras S, Samson C, Leblanc JF, Bélanger C, Dion F, et
al: Common origins of BRCA1 mutations in Canadian breast and
ovarian cancer families. Nat Genet. 8:392–398. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dørum A, Heimdal K, Hovig E, Inganäs M and
Møller P: Penetrances of BRCA1 1675delA and 1135insA with respect
to breast cancer and ovarian cancer. Am J Hum Genet. 65:671–679.
1999. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Novaković S, Milatović M, Cerkovnik P,
Stegel V, Krajc M, Hočevar M, Zgajnar J and Vakselj A: Novel BRCA1
and BRCA2 pathogenic mutations in Slovene hereditary breast and
ovarian cancer families. Int J Oncol. 41:1619–1627. 2012.PubMed/NCBI
|
|
18
|
Tea M-KM, Kroiss R, Muhr D,
Fuerhauser-Rappaport C, Oefner P, Wagner TM and Singer CF: Central
European BRCA2 mutation carriers: Birth cohort status correlates
with onset of breast cancer. Maturitas. 77:68–72. 2014. View Article : Google Scholar : PubMed/NCBI
|