Introduction
Worldwide, and particularly in developing countries,
lung cancer has become one of the leading causes of malignant
cancer-associated death due to its high rates of morbidity and
mortality, and its poor prognosis (1). Non-small cell lung cancer (NSCLC) is
the most common type of lung cancer, accounting for 80% of all lung
cancer cases. The prognosis of NSCLC is poor, with a 5-year overall
survival rate of only ~15% (2).
Surgery, chemotherapy and radiotherapy are the common treatment
options for NSCLC. Surgery is considered the most effective
treatment; however, many patients are not suitable for surgical
intervention because of invasion and metastasis. Additionally, the
adverse effects of radiotherapy and chemotherapy limit their
clinical application (3).
Therefore, searching for novel effective therapeutic agents and
understanding their mechanism of action is of great clinical
significance.
In recent decades, natural products extracted from
herbs, termed phytochemicals, have attracted attention due to their
reliable therapeutic effects and mild side effects (4). Emodin, also referred to as
1,3,8-trihydroxy-6-methyl-anthraquinone, is present in various
Chinese medicinal herbs, including Rheum and Polygonum, which have
been used since ancient times (5).
Modern pharmacological and biomedical analyses have demonstrated
the anticancer effects of emodin on several types of human cancer,
including hepatic cancer (6),
gallbladder cancer (7), cervical
cancer (8), myelocytic leukemia
(9) and lung cancer (10). However, our understanding of the
molecular mechanisms of the effects of emodin as a potent
anticancer agent on NSCLC cells is still inadequate to support the
further clinical application of this phytochemical.
Tribbles homolog 3 (TRIB3) was initially identified
as a pseudokinase that inhibits mitosis in the embryo and germ
cells of Drosophila (11).
Further studies detected the expression of TRIB3 in many human cell
types and suggested that TRIB3 has important regulatory roles in
cell apoptosis, autophagy and migration (12). It was reported that the expression
of TRIB3 is induced when cells are under conditions of stress,
including hypoxia and nutrient starvation (13). Activated endoplasmic reticulum (ER)
stress in particular has been shown to trigger TRIB3 signaling
activation, as transcription of TRIB3 was identified to be
regulated by activating transcriptional factor 4 (ATF4) and C/BEP
homologous protein (CHOP) (14). A
study in pancreatic β cells showed that TRIB3 induced cell
apoptosis via activation of nuclear factor-κB (NF-κB) signaling
(15).
A recent study revealed that TRIB3 expression level
was closely associated with the prognosis of malignant cancer,
suggesting that TRIB3 is involved in cancer development and/or
treatment resistance (16).
Previous studies have confirmed that ER stress-induced cell death
is one of the mechanisms underlying the anti-proliferative effects
of emodin in malignant cancers (8).
Thus, it is reasonable to hypothesize that TRIB3 may be a mediator
of the ER stress-induced apoptosis caused by emodin in NSCLC
cells.
In this study, the A549 and H1299 human NSCLC cell
line were used to investigate the effects of emodin. After
incubation with emodin, the proliferation and apoptosis of lung
cancer cells were assessed. Markers of ER stress, including
glucose-regulated protein 78 (GRP78) and CHOP were also detected.
In addition, ER stress was inhibited using 4-phenylbutyrate (4-PBA)
and RNA interference was used to silence TRIB3 expression. The
results of the current study provide further knowledge of the
anticancer activity of emodin and support for its future clinical
application.
Materials and methods
Cell line and culture
A549 and H1299 human NSCLC cells were purchased from
the China Center for Type Culture Collection (Wuhan, China). A549
cells were cultured in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) while H1299 cells were cultured
in RPMI-1640 (Gibco, Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (FBS; Hyclone; GE Healthcare, Logan,
UT, USA), penicillin (100 U/ml; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany), streptomycin (100 mg/ml; Sigma-Aldrich; Merck
Millipore) and L-glutamine (2 mmol/l; Sigma-Aldrich; Merck
Millipore). Cells were cultured in an incubator with a humidified
condition of 5% CO2 and 95% air at 37°C.
Small interfering RNA (siRNA)
knockdown
siRNA was used to knockdown the expression of TRIB3
in A549 and H1299 cells, respectively. Cells were seeded in 6-well
plates (5×104 cell/well) in 2 ml media 24 h before
transfection; cells were 80–90% confluent. The specific siRNA
against TRIB3 (100 pmol/well, GenePharma, China) was transfected
into A549 and H1299 cells with Lipofectamine 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. After 48 h of transfection, cells were
used for qRT-PCR or apopsis assay. The siRNA sequence used was as
indicated in a previous study (15): 5′-ATCTCTGGCTGCTTCTGCCCATGTT-3′.
Cell grouping and treatment
Lung cancer cells were harvested in the logarithmic
growth phase. Cells were incubated with serially diluted emodin
(Selleck Chemicals, Shanghai, China) solutions (0, 20, 40, 60 and
80 µmol/l) for 48 h. Equal numbers of cells were collected and
divided into 6 different treatment groups as follows: Ctrl (control
group); Ctrl + 4-PBA [treatment with 4-PBA (500 µmol/l)]; Ctrl +
siRNA (treatment with TRIB3 siRNA); emodin [treatment with emodin
(80 µmol/l)]; emodin + 4-PBA [co-treatment with emodin (80 µmol/l)
and 4-PBA (500 µmol/l)]; and emodin + siRNA [co-treatment with
emodin (80 µmol/l) and TRIB3 siRNA].
Cell viability assessment
Colorimetric 3-(4,5-dimethylthiazol-2-yl)
2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich; Merck
Millipore) was used to assess the viability of lung cancer cells in
the afore-mentioned treatment groups. A549 and H1299 cells were
seeded into 96-well plates and incubated with MTT (5 mg/ml) for 4 h
at 37°C. Subsequently, 150 µl dimethylsulfoxide (Sigma-Aldrich;
Merck Millipore) was added to the wells after washing with PBS. A
plate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was
used to detect the absorbance at 540 nm (A540). The cell
viability inhibition rate was calculated using the following
formula: Viability inhibition = [1-A540 (experimental
well) / A540 (control well)] ×100%.
Cell apoptosis analysis
Hoechst fluorescent staining was used to indicate
apoptotic cells. Briefly, cells were harvested and fixed with 4%
paraformaldehyde at 37°C for 1 h. Subsequently, the cells were
washed with PBS and stained with 5 µmol/l Hoechst 33342
(Sigma-Aldrich; Merck Millipore) at 37°C for 30 min in a humidified
dark chamber. The fluorescent images of Hoechst staining were
captured using a fluorescence microscope (Cannon, Japan).
Hoechst-positive cells were considered apoptotic.
Western blotting
Cell lysates of A549 and H1299 cells were collected
after lysis in RIPA buffer (Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) according to the manufacturer's instructions. A protein
extraction kit (Beyotime Institute of Biotechnology, Haimen, China)
was used to extract the total protein, and a R0050 nuclear protein
extraction kit (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) was used to extract the nuclear proteins of
lung cancer cells. Protein concentrations were assessed using a
bicinchoninic acid kit (Thermo Fisher Scientific, Inc.). Vertical
electrophoresis was applied to separate the proteins after loading
in SDS gels, and the proteins were then electrotransferred onto
nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). After
blocking with 5% non-fat milk, the membranes were incubated with
specific antibodies against GRP78 (1:2,500; Abcam, Cambridge, MA,
USA), CHOP (1:3,000; Abcam), TRIB3 (1:3,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), NF-κB p65 (1:1,000; Abcam),
phospho-NF-κB p65 (1:1,000; Cell Signaling Technology, Inc.),
cleaved caspase-3 (1:2000, Abcam), pro-caspase3 (1:2000, Abcam),
GAPDH (1:20,000; Santa Cruz Biotechnology, Inc.) and lamin B
(1:250; Santa Cruz Biotechnology, Inc.). GAPDH was used as the
internal control for total protein and lamin B as the internal
control for nuclear proteins.
Tumor xenograft experiments
All in vivo experiments were approved by the
Institutional Research Committee of Xi'an Jiaotong University
(Xi'an, China). The mice received humane care in compliance with
the Guide for the Care and Use of Laboratory Animals published by
the National Institutes of Health. Cancer cells
(2.5×106) were mixed in a 1:1 (v:v) ratio with growth
factor-reduced Matrigel (BD Biosciences, Franklin Lakes, NJ, USA),
and the mixture was injected subcutaneously into the right flanks
of 6- to 7-week-old BALB/c nu/nu nude mice. At 24 h after
implantation, mice bearing A549 cells were randomly assigned to 4
groups (n=8 per group) as follows: i) Control; ii) 20 mg/kg/day
4-PBA alone; iii) 50 mg/kg/day emodin; and iv) 50 mg/kg/day emodin
combined with 20 mg/kg/day 4-PBA. Emodin and 4-PBA, suspended in
saline, were intraperitoneally injected once per day. Controls
received the vehicle alone (20 ml/kg). Tumor volume (measured in
mm3) was determined using calipers every 2 days and
calculated using the modified ellipse formula: Volume = length ×
width2/2 (17). Mice
were sacrificed by cervical dislocation on day 40. Implanted tumors
were extracted and weighed.
Statistical analysis
Data acquired in this study is presented as the mean
± standard deviation. Differences between groups were compared by
analysis of variance and followed by Tukey's post-hoc tests.
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were processed by SPSS
software (version 16.0; SPSS, Inc., Chicago, IL, USA).
Results
Emodin reduces viability and induces
apoptosis of A549 cells in a concentration-dependent manner
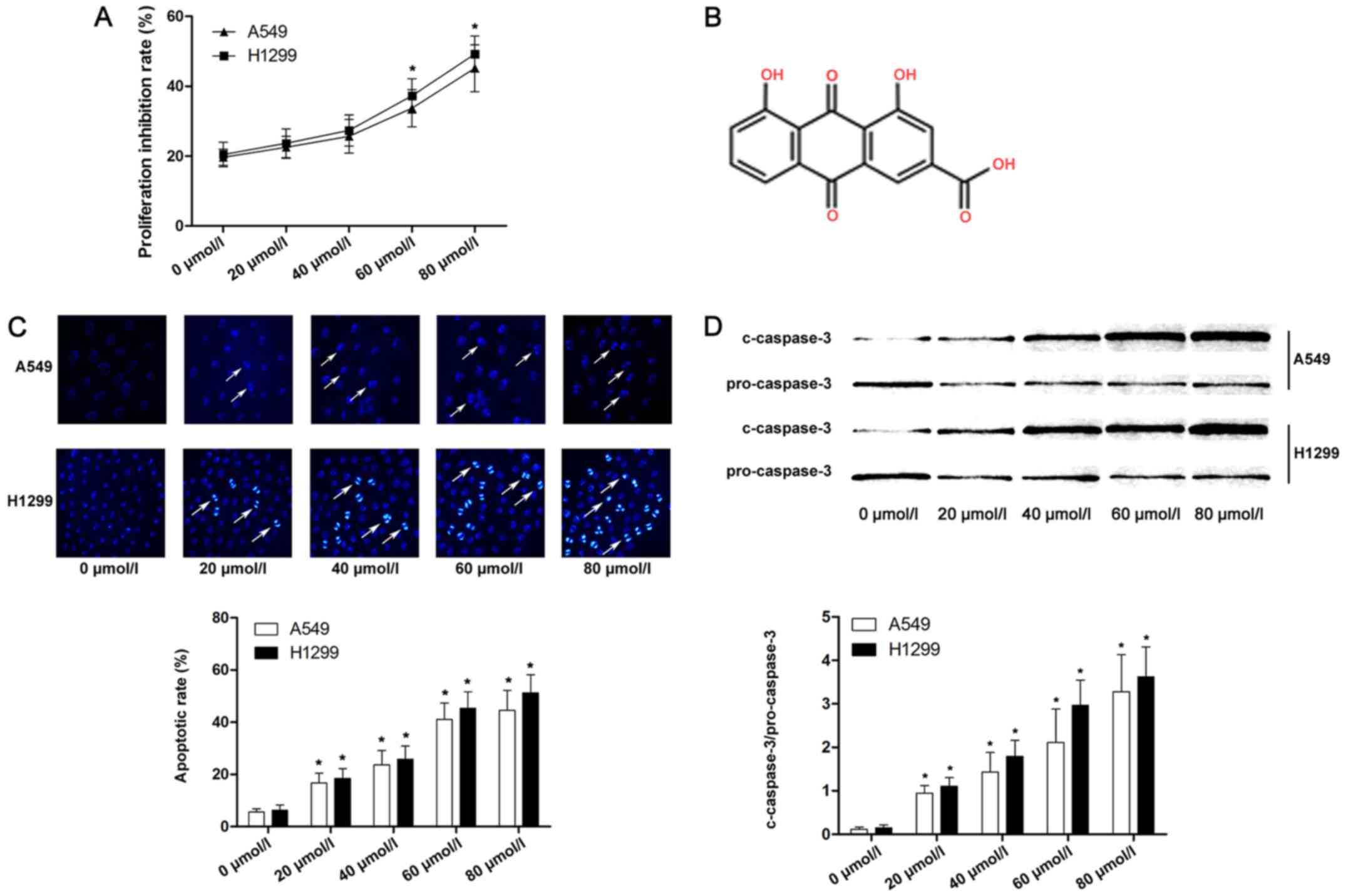
We measured the viability of A549 and H1299 cells
after incubation with emodin at 20, 40, 60 and 80 µmol/l. The
viability was significantly reduced at 60 and 80 µmol/l compared
with the lower concentrations, as shown in Fig. 1A. Cell apoptosis is considered one
of the most important mechanisms that mediates the
proliferation-inhibitory effects of anticancer agents. The
apoptosis of A549 and H1299 cells was assessed by Hoechst staining
and caspase-3 activation. As shown in Fig. 1C and D, Hoechst-positive lung cancer
cells were marked as apoptotic. Furthermore, as the concentration
of emodin increased, the apoptotic rate and expression of cleaved
caspase-3 of lung cancer cells were significantly increased
compared with lower concentrations.
Apoptosis of lung cancer cells induced
by emodin is mediated by ER stress
When exposed to harmful stimuli, prolonged ER stress
causes cell death (18). GRP78 and
CHOP were used as markers of ER stress in the current study.
Furthermore, as a pro-apoptotic factor, CHOP is also closely
associated with ER stress-induced cell death (19). 4-PBA has been applied as specific ER
stress inhibitor in many previous studies (20). The current study demonstrated that
the apoptosis induced by emodin was reduced significantly following
4-PBA treatment (Fig. 2A and B).
Additionally, the expression level of the anti-apoptotic factor
GRP78 was increased after 4-PBA treatment compared with
emodin-treated lung cancer cells (Fig.
2C). Furthermore, the level of the pro-apoptotic factor CHOP
was decreased after 4-PBA treatment compared with emodin-treated
A549 and H1299 cells (Fig. 2C).
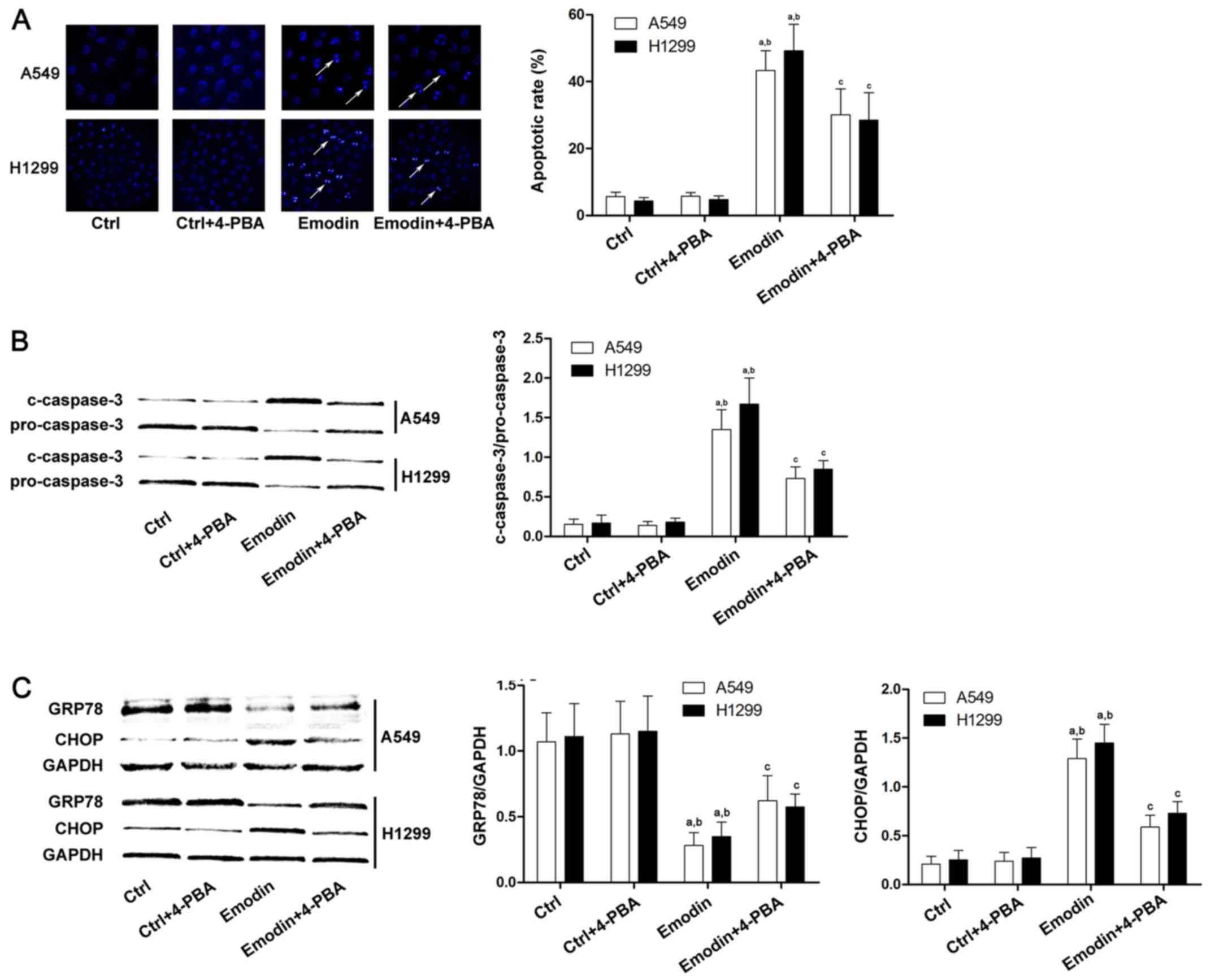 | Figure 2.Apoptosis of A549 and H1299 cells
induced by emodin is ER stress-mediated. Lung cancer cells were
treated with emodin (80 µmol/l) with or without a specific ER
stress inhibitor, 4-PBA (500 µmol/l), for 72 h. (A) The apoptosis
of A549 and H1299 cells was detected by Hoechst staining. The white
arrows indicate the Hoechst-positive cells. Columns demonstrated
the apoptotic rates of A549 and H1299 cells in the Ctrl, Ctrl +
4-PBA, emodin, and emodin + 4-PBA groups. (B) Caspase-3 activation
in different groups was detected by western blots. Columns on the
right panel indicated the ratio of c-caspase-3 over pro-caspase-3
in lung cancer cells. The data are presented as the mean ± standard
deviation from three independent experiments. (C) Western blots of
GRP-78, CHOP and GAPDH in the Ctrl, Ctrl + 4-PBA, emodin, and
emodin + 4-PBA groups. GAPDH was used as the internal reference.
Quantitation of western blots are presented as fold changes of
GRP-78 and CHOP normalized to GAPDH. The data are presented as the
mean ± standard deviation from three independent experiments
(aP<0.05 vs. Ctrl; bP<0.05 vs. Ctrl +
4-PBA; cP<0.05 vs. emodin). ER, endoplasmic
reticulum; Ctrl, control; 4-PBA, 4-phenylbutyrate; GRP78, glucose
regulating protein 78; CHOP, C/BEP homologous protein. |
Emodin-induced apoptosis of lung
cancer cell is TRIB3 dependent
ER stress in particular was previously shown to
trigger TRIB3 signaling activation (14) and TRIB3 has been demonstrated to be
important in the regulation of cell apoptosis (12), the current study aimed to clarify
whether TRIB3 regulated emodin-induced apoptosis of A549 and H1299
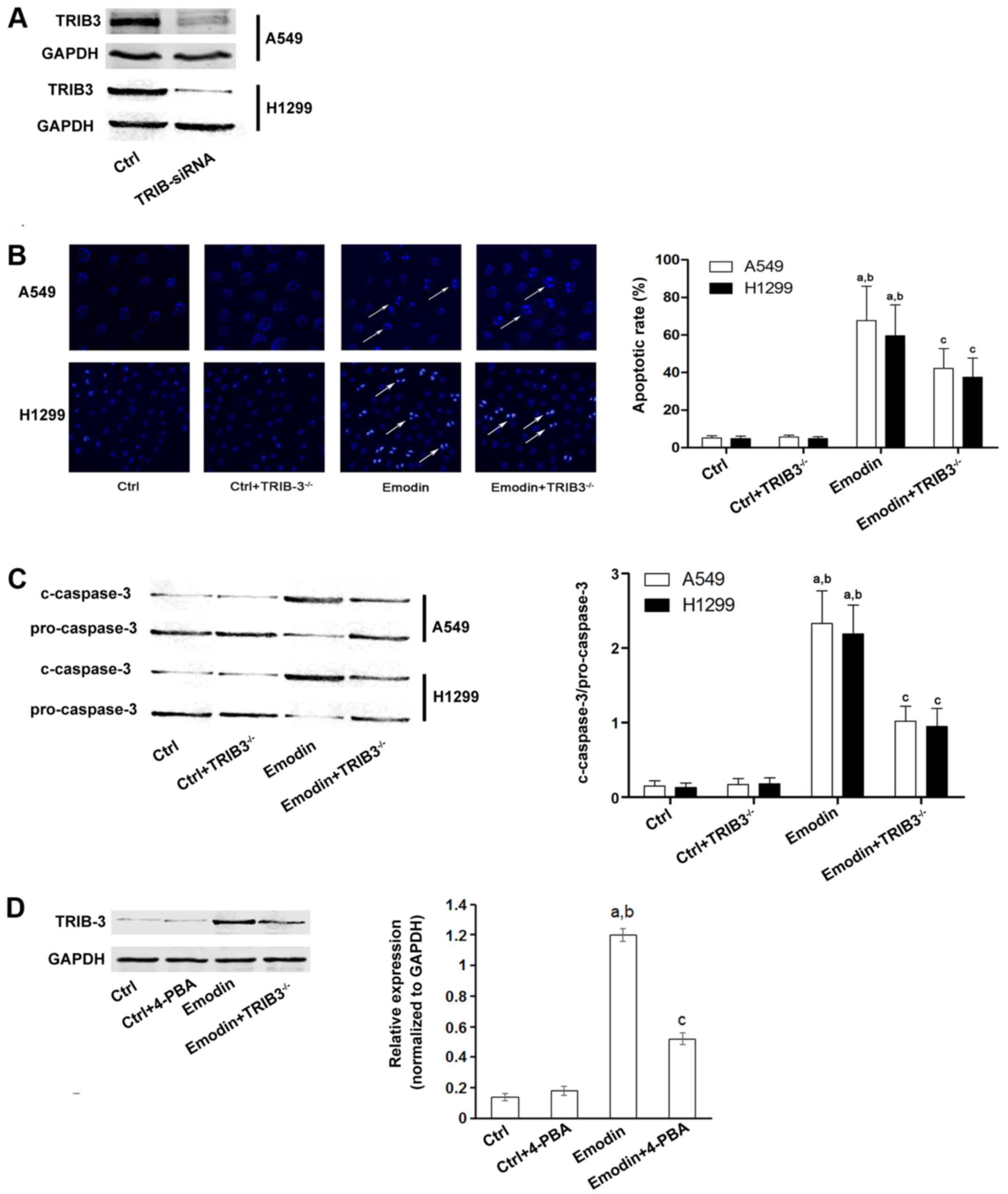
cells. siRNA was used to knock down TRIB3 expression in lung cancer
cells (Fig. 3A) and cell apoptosis
was then evaluated by Hoechst staining. As shown in Fig. 3B and C, downregulation of TRIB3
significantly inhibited cancer cell apoptosis induced by emodin.
Furthermore, emodin also increased the expression of TRIB3; and
this effect was inhibited by pretreatment with 4-PBA, which
inhibits ER stress (Fig. 3D).
Apoptosis of lung cancer cells caused
by emodin-induced ER stress is associated with the TRIB3/NF-κB
pathway
A previous study reported that TRIB3 altered ER
stress-induced cell apoptosis via the NF-κB pathway (15). In order to investigate the potential
mechanism involved in the anticancer effects of emodin, the current
study focused on the ER stress/TRIB3/NF-κB pathway in A549 and
H1299 cells treated with emodin. To determine the effect of NF-κB
inhibition during emodin-induced A549 and H1299 cell apoptosis, a
selective inhibitor of NF-κB, PDTC (Sigma-Aldrich; Merck
Millipore), was used. As shown in Fig.
4A, emodin failed to increase apoptosis in lung cancer cells
after the cells were exposed to PDTC. Furthermore, treatment with
emodin increased NF-κB translocation from the cytoplasm to the
nucleus and downregulation of TRIB3 inhibited this effect (Fig. 4B), indicating that TRIB3/NF-κB
pathway is involved in emodin-induced lung cancer apoptosis.
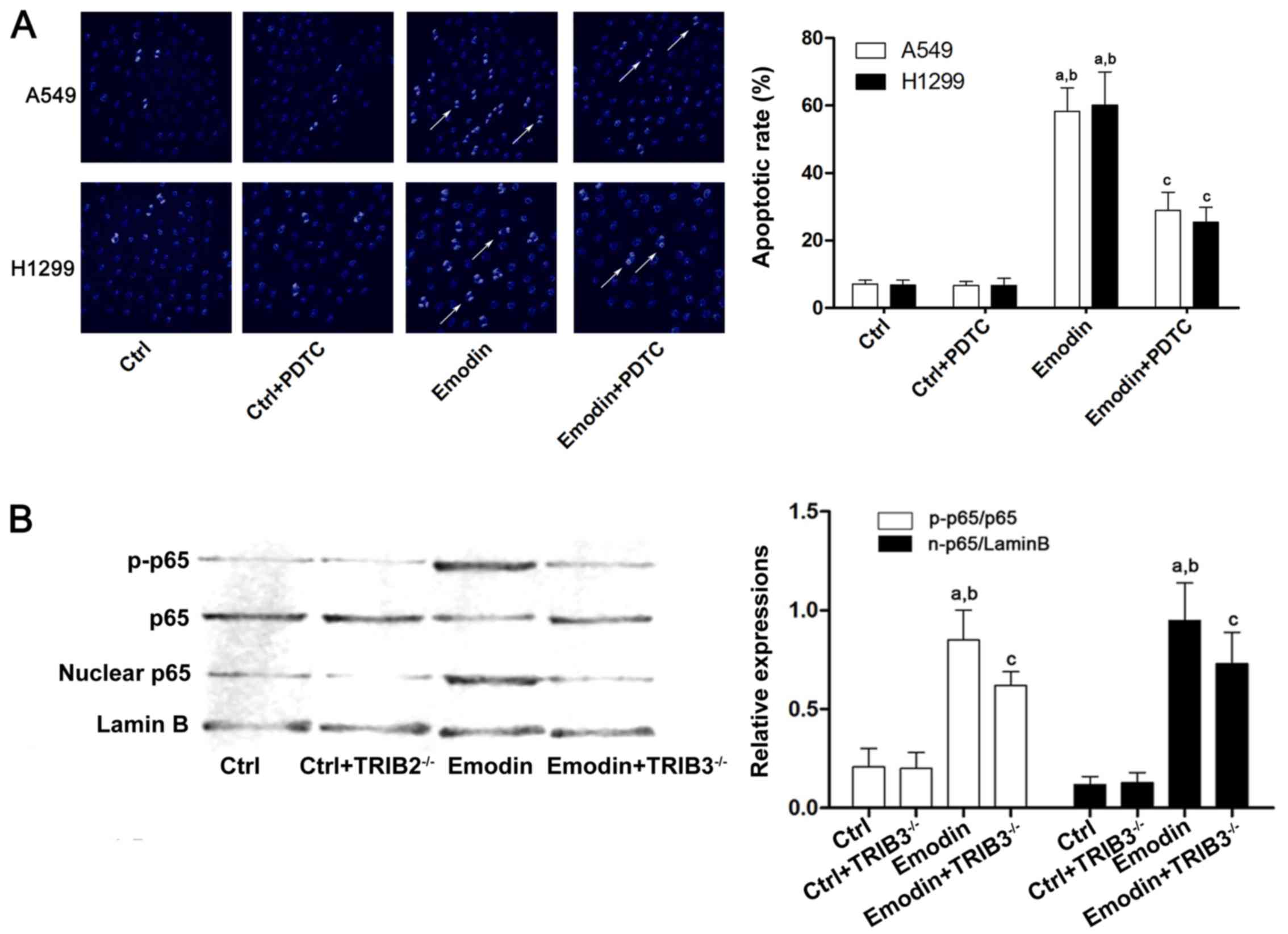 | Figure 4.ER stress-induced apoptosis of A549
cells caused by emodin is associated with TRIB3/NF-κB. A549 and
H1299 cells were treated with emodin (80 µmol/l) with or without a
specific NF-κB inhibitor, PDTC (1 µmol/l), for 72 h. (A) The
apoptosis of A549 cells was detected by Hoechst staining. Columns
demonstrate the apoptotic rates of A549 an H1299 cells in the Ctrl,
Ctrl + PDTC, emodin, and emodin + PTDC groups
(aP<0.05 vs. Ctrl; bP<0.05 vs. Ctrl +
PDTC; cP<0.05 vs. emodin). (B) Western blotting was
used to detect p-p65, p65, nuclear p65 and lamin B in the Ctrl,
Ctrl+TRIB3−/−, emodin, and emodin + TRIB3−/−
groups of A549 cells. The quantitation data of western blots are
presented as the mean ± standard deviation from three independent
experiments (aP<0.05 vs. Ctrl; bP<0.05
vs. Ctrl + TRIB3−/−; cP<0.05 vs. emodin).
ER, endoplasmic reticulum; PDTC, pyrrolidinedithiocarbamate; Ctrl,
control; TRIB, tribbles homolog 3; NF-κB, nuclear factor-κB. |
Emodin inhibits subcutaneous tumors
generated by inducing ER stress-dependent apoptosis in vivo
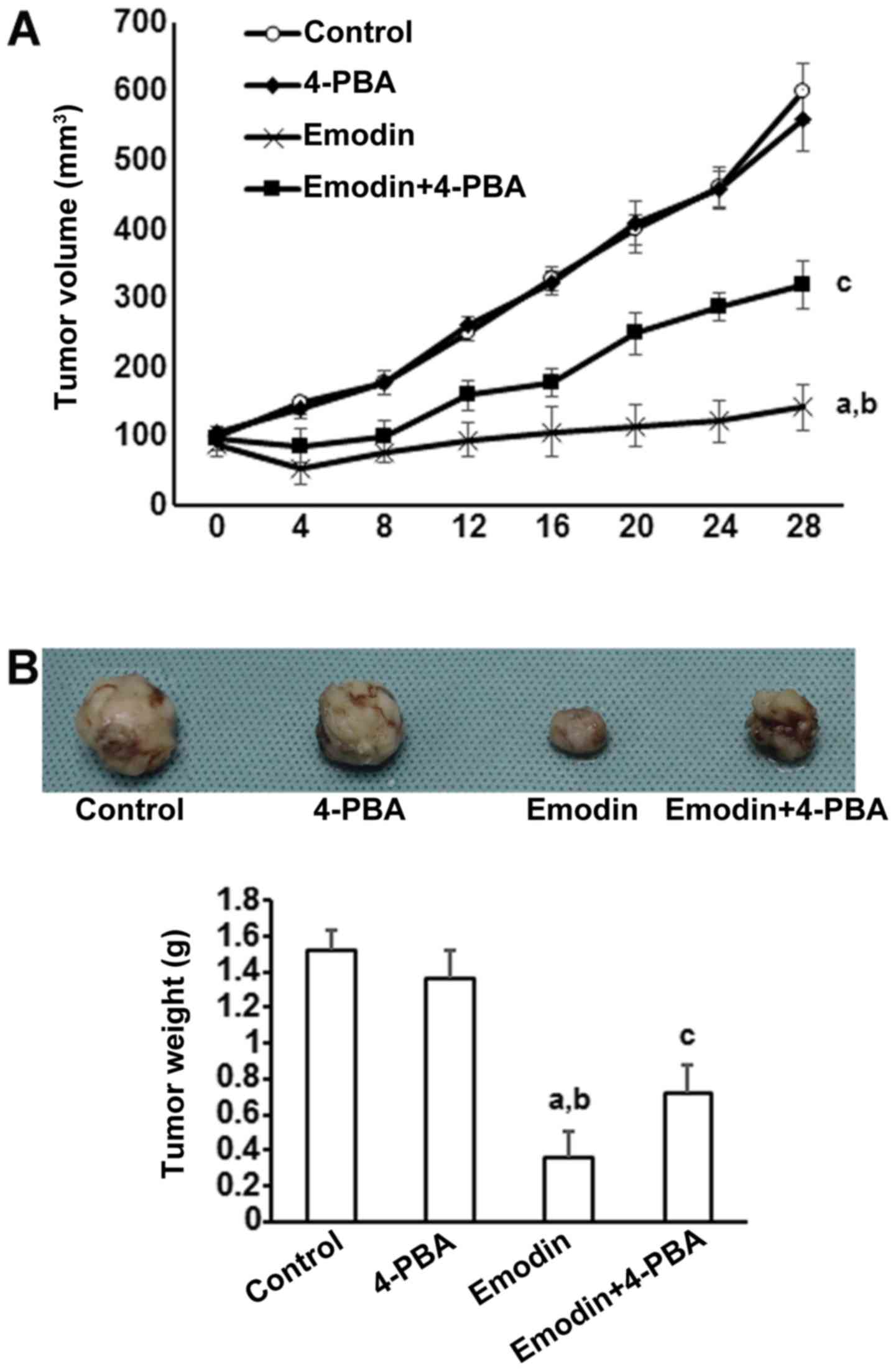
To investigate the in vivo actions of emodin
on tumor development, subcutaneous tumors were generated by
implanting A549 cells in BALB/c nu/nu nude mice. Supporting the
in vitro results, tumors treated with emodin grew more
slowly than control tumors (Fig.
5A). However, the inhibitory effects of emodin on tumor growth
were significantly reduced by co-treatment with 4-PBA (Fig. 5A). At 40 days after tumor
initiation, tumors were removed and macroscopically measured. As
shown in Fig. 5B, emodin
significantly reduced tumor volume and this effect was inhibited by
co-treatment with 4-PBA.
Discussion
The results of the current study, which demonstrated
the inhibitory effects of emodin on NSCLC cell viability, were in
accordance with previous studies (21). The results also confirmed that
emodin reduced lung cancer cell viability by inducing apoptosis. By
detecting an ER stress marker, GRP78, and an ER stress
pro-apoptotic factor, CHOP, we demonstrated that the
apoptosis-inducing effect of emodin on A549 and H1299 cells was
mediated by ER stress. In order to identify a specific molecular
target of emodin, the TRIB3/NF-κB signaling pathway was
investigated. The results revealed that emodin did affect TRIB3
expression, which may induce cell apoptosis in emodin-treated A549
and H1299 cells.
As one of the most basic and important organelles of
mammalian cells, the ER executes multiple vital biological
functions, including protein folding, protein maturation, protein
post-translational modification, calcium signaling and lipid
synthesis to maintain intracellular homeostasis (22). When a cell encounters harmful
stimuli, ER stress is triggered and initially presents as the
unfolded protein response (UPR) (23). UPR eliminates damaged proteins and
initiates global inhibition of protein transcription (24). However, when the stimulus is
prolonged and aggravated, the apoptotic signals are transduced by
ER stress pathways to destroy damaged cells (25). GRP78 and CHOP are generally accepted
as molecular markers of ER stress. Furthermore, CHOP is recognized
as a pro-apoptotic factor during ER stress (26).
In the present study, incubation with emodin reduced
the viability of A549 and H1299 cells in a concentration-dependent
manner. Apoptosis analysis also demonstrated that emodin induced
apoptosis of lung cancer cells in a concentration-dependent manner.
These results were in accordance with previous studies. Our results
suggested that the apoptotic avidity of emodin was mediated by ER
stress, as demonstrated by the observation that an ER stress
inhibitor, 4-PBA, reduced the expression levels of GRP78 and CHOP,
and also impaired emodin-induced apoptosis in lung cancer
cells.
TRIB3 is a mammalian homolog of the protein tribbles
in Drosophila which participates in various cellular
processes, such as migration and mitosis. Several recent studies
reported that cancer prognosis and TRIB3 levels are closely
correlated (16). It was reported
that cancer patients with higher levels of TRIB3 protein had better
prognosis compared with patients with low TRIB3 levels (13). These results indicated that TRIB3
was functionally associated with malignant cancer. ER stress is
activated by stimuli, and apoptotic signaling following ER stress
is transduced through an ATF4/CHOP pathway following
phosphorylation of eukaryotic initiation factor 2α. A previous
study reported that TRIB3 expression was induced by ER stress via
the ATF4/CHOP pathway as the promoter region of TRIB3 is an ER
stress response element with a CHOP binding site (27). In the present study, the TRIB3
expression level was elevated by emodin-induced ER stress.
In this study, emodin treatment of A549 and H1299
cells triggered ER stress-mediated apoptosis. When ER stress was
suppressed by 4-PBA, the expression levels of CHOP/TRIB3 were also
reduced. As a result, emodin-induced lung cancer cell apoptosis was
also inhibited. Furthermore, TRIB3 silencing using siRNA impaired
emodin-induced ER stress-mediated apoptosis, even though ER stress
was not inhibited in lung cancer cells. These results indicated
TRIB3 may be a molecular target of emodin when inducing apoptosis
via ER stress.
Previous studies reported that TRIB3 activates NF-κB
activity by directly binding to p65, promoting its nuclear
translocation and phosphorylation (15), which would promote cell death due to
the increased transcription of target genes, including cytokines
(such as tumor necrosis factor) and c-Jun N-terminal kinase, which
triggers the caspase cascade (28).
Additionally, we found that when ER stress was activated by emodin,
TRIB3/NF-κB signaling was also activated. However, when ER stress
was repressed or TRIB3 was silenced, NF-κB signaling was also
inhibited, reducing the pro-apoptotic effects of emodin.
In conclusion, the results of the current study
suggested that the potent anticancer effects of emodin are caused
by ER stress-mediated apoptosis in lung cancer cells. We also
provided evidence that TRIB3 is one of the important molecules
involved in mediating emodin-induced apoptosis, and that
TRIB3/NF-κB signaling participated in this process. Together, these
findings improve the understanding of the mechanisms of the
anticancer activity of emodin against lung cancer, and provide a
theoretical basis for the clinical application of novel
emodin-based anticancer agents in the future.
Acknowledgements
This study was supported by grant from the National
Natural Science Foundation of China (no. 81502295).
References
|
1
|
Yin QW, Sun XF, Yang GT, Li XB, Wu MS and
Zhao J: Increased expression of microRNA-150 is associated with
poor prognosis in non-small cell lung cancer. Int J Clin Exp
Pathol. 8:842–846. 2015.PubMed/NCBI
|
|
2
|
Wang Z, Fu J, Diao D and Dang C:
Pre-operative plasma D-dimer level may predict the poor prognosis
within one year after the surgery for non-small cell lung cancer.
Zhongguo Fei Ai Za Zhi. 14:534–537. 2011.(In Chinese). PubMed/NCBI
|
|
3
|
Cooper S and Spiro SG: Small cell lung
cancer: Treatment review. Respirology. 11:241–248. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arumuggam N, Bhowmick NA and Rupasinghe
HP: A review: phytochemicals targeting JAK/STAT signaling and IDO
expression in cancer. Phytother Res. 29:805–817. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He L, Bi JJ, Guo Q, Yu Y and Ye XF:
Effects of emodin extracted from Chinese herbs on proliferation of
non-small cell lung cancer and underlying mechanisms. Asian Pac J
Cancer Prev. 13:1505–1510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu JQ, Bao W and Lei JC: Emodin regulates
apoptotic pathway in human liver cancer cells. Phytother Res.
27:251–257. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li XX, Dong Y, Wang W, Wang HL, Chen YY,
Shi GY, Yi J and Wang J: Emodin as an effective agent in targeting
cancer stem-like side population cells of gallbladder carcinoma.
Stem Cells Dev. 22:554–566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yaoxian W, Hui Y, Yunyan Z, Yanqin L, Xin
G and Xiaoke W: Emodin induces apoptosis of human cervical cancer
hela cells via intrinsic mitochondrial and extrinsic death receptor
pathway. Cancer Cell Int. 13:712013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chun-Guang W, Jun-Qing Y, Bei-Zhong L,
Dan-Ting J, Chong W, Liang Z, Dan Z and Yan W: Anti-tumor activity
of emodin against human chronic myelocytic leukemia K562 cell lines
in vitro and in vivo. Eur J Pharmacol. 627:33–41. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ok S, Kim SM, Kim C, Nam D, Shim BS, Kim
SH and Ahn KS, Choi SH and Ahn KS: Emodin inhibits invasion and
migration of prostate and lung cancer cells by downregulating the
expression of chemokine receptor CXCR4. Immunopharmacol
Immunotoxicol. 34:768–778. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Prudente S, Sesti G, Pandolfi A, Andreozzi
F, Consoli A and Trischitta V: The mammalian tribbles homolog
TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr
Rev. 33:526–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fontanesi L, Colombo M, Scotti E,
Buttazzoni L, Bertolini F, Dall'Olio S, Davoli R and Russo V: The
porcine tribbles homolog 3 (TRIB3) gene: Identification of a
missense mutation and association analysis with meat quality and
production traits in Italian heavy pigs. Meat Sci. 86:808–813.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wennemers M, Bussink J, Grebenchtchikov N,
Sweep FC and Span PN: TRIB3 protein denotes a good prognosis in
breast cancer patients and is associated with hypoxia sensitivity.
Radiother Oncol. 101:198–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nicoletti-Carvalho JE, Nogueira TC, Gorjão
R, Bromati CR, Yamanaka TS, Boschero AC, Velloso LA, Curi R, Anhê
GF and Bordin S: UPR-mediated TRIB3 expression correlates with
reduced AKT phosphorylation and inability of interleukin 6 to
overcome palmitate-induced apoptosis in RINm5F cells. J Endocrinol.
206:183–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fang N, Zhang W, Xu S, Lin H, Wang Z, Liu
H, Fang Q, Li C, Peng L and Lou J: TRIB3 alters endoplasmic
reticulum stress-induced β-cell apoptosis via the NF-κB pathway.
Metabolism. 63:822–830. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miyoshi N, Ishii H, Mimori K, Takatsuno Y,
Kim H, Hirose H, Sekimoto M, Doki Y and Mori M: Abnormal expression
of TRIB3 in colorectal cancer: A novel marker for prognosis. Br J
Cancer. 101:1664–1670. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang X, Taeb S, Jahangiri S, Emmenegger
U, Tran E, Bruce J, Mesci A, Korpela E, Vesprini D, Wong CS, et al:
miRNA-95 mediates radioresistance in tumors by targeting the
sphingolipid phosphatase SGPP1. Cancer Res. 73:6972–6986. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Z, Lv Y, Zhao N, Guan G and Wang J:
Protein kinase R-like ER kinase and its role in endoplasmic
reticulum stress-decided cell fate. Cell Death Dis. 6:e18222015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leamy AK, Egnatchik RA, Shiota M, Ivanova
PT, Myers DS, Brown HA and Young JD: Enhanced synthesis of
saturated phospholipids is associated with ER stress and
lipotoxicity in palmitate treated hepatic cells. J Lipid Res.
55:1478–1488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cho JA, Zhang X, Miller GM, Lencer WI and
Nery FC: 4-Phenylbutyrate attenuates the ER stress response and
cyclic AMP accumulation in DYT1 dystonia cell models. PLoS One One.
9:e1100862014. View Article : Google Scholar
|
|
21
|
Wei W-T, Lin S-Z, Liu D-L and Wang Z-H:
The distinct mechanisms of the antitumor activity of emodin in
different types of cancer (Review). Oncol Rep. 30:2555–2562.
2013.PubMed/NCBI
|
|
22
|
Pluquet O, Pourtier A and Abbadie C: The
unfolded protein response and cellular senescence. A review in the
theme: Cellular mechanisms of endoplasmic reticulum stress
signaling in health and disease. Am J Physiol Cell Physiol.
308:C415–C425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lakshmanan AP, Harima M, Suzuki K,
Soetikno V, Nagata M, Nakamura T, Takahashi T, Sone H, Kawachi H
and Watanabe K: The hyperglycemia stimulated myocardial endoplasmic
reticulum (ER) stress contributes to diabetic cardiomyopathy in the
transgenic non-obese type 2 diabetic rats: A differential role of
unfolded protein response (UPR) signaling proteins. Int J Biochem
Cell Biol. 45:438–447. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vidal RL, Figueroa A, Court FA, Thielen P,
Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo
AM, et al: Targeting the UPR transcription factor XBP1 protects
against Huntington's disease through the regulation of FoxO1 and
autophagy. Hum Mol Genet. 21:2245–2262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fribley AM, Miller JR, Reist TE, Callaghan
MU and Kaufman RJ: Large-scale analysis of UPR-mediated apoptosis
in human cells. Methods Enzymol. 491:57–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Guo Y, Tang J, Jiang J and Chen Z:
New insights into the roles of CHOP-induced apoptosis in ER stress.
Acta Biochim Biophys Sin (Shanghai). 47:146–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bromati CR, Lellis-Santos C, Yamanaka TS,
Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anhê GF
and Bordin S: UPR induces transient burst of apoptosis in islets of
early lactating rats through reduced AKT phosphorylation via
ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr
Comp Physiol. 300:R92–R100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang R, Cao X, Wang C, Hou L, Nie J, Zhou
M and Feng Y: An antitumor peptide from Musca domestica pupae
(MATP) induces apoptosis in HepG2 cells through a JNK-mediated and
Akt-mediated NF-κB pathway. Anticancer Drugs. 23:827–835. 2012.
View Article : Google Scholar : PubMed/NCBI
|