Introduction
Glioblastoma multiforme (GBM) is the most common and
lethal primary malignant brain tumor. Even with intensive
multimodality treatment, including surgical resection combined with
radiation and chemotherapy, the prognosis of GBM patients remains
very poor, with a median survival of less than 15 months (1). Chemoresistance to alkylating agents
and to temozolomide (TMZ) in particular has been identified as a
major cause of treatment failure (2). Understanding the mechanisms of TMZ
resistance in GBM may contribute to improving the efficacy of the
conventional chemotherapeutic agents. Researchers have conducted
multiple investigations into the mechanisms of TMZ resistance.
Among these investigations, most have focused on
O6-methylguanine-DNA methyltransferase (MGMT), primarily because
MGMT directly mediates TMZ-induced cytotoxicity. Glioblastoma cells
with a high level of MGMT are resistant to TMZ, whereas a low level
or the absence of MGMT sensitizes glioblastoma cells to TMZ
(3–5). However, MGMT alone does not fully
account for the chemoresistance of GBM to TMZ, as >40% of
glioblastomas with a low level of MGMT remain resistant to TMZ.
These lines of evidence suggest that a high MGMT level is merely
one of the possible mechanisms of TMZ resistance (6–8). In
addition to MGMT, several genes have been reported to be involved
in TMZ resistance. Nevertheless, these data combined are still
unable to fully elucidate the mechanisms of TMZ resistance, which
prompted us to search for other undefined mechanisms that may
contribute to TMZ resistance in GBM.
β-catenin, a subunit of the cadherin protein
complex, serves as a fundamental mediator in the Wnt signaling
pathway. In particular, nuclear β-catenin is the hallmark of active
Wnt/β-catenin signaling (9–11). Multiple studies have shown that
β-catenin contributes to glioblastoma inception and progression.
For instance, β-catenin is positively correlated with the grade of
glial neoplasms (12,13) and has been identified as a marker of
poor prognosis in glial neoplasms (14). In addition, compared to
membrane-localized β-catenin, nuclear β-catenin is associated with
a worse prognosis in cancer (15),
indicating that β-catenin subcellular localization is linked to
prognostic differences (16–18).
Not only does β-catenin play a part in cancer development and
progression but it also contributes to chemoresistance. Nuclear
β-catenin mediates the activation of the Wnt/β-catenin pathway and
confers doxorubicin (DoxR)-resistance in neuroblastoma (NB)
(19). β-catenin activation by the
glycogen synthesis kinase-3 inhibitor induces chemoresistance to
the interferon (IFN)-α/5-fluorouracil (5-FU) combination therapy
for hepatocellular carcinoma (HCC) (20). Importantly, the active β-catenin
(nuclear β-catenin) significantly abrogated the efficacy of three
chemotherapeutic agents, TMZ, cisplatin and doxorubicin (21). By contrast, β-catenin depletion
sensitized resistant glioblastoma cells to chemotherapy (22).
FoxO3a, a Forkhead box O (FoxO) protein member of
the Forkhead family, plays an important role in the regulation of
cell differentiation, proliferation, and survival (23). Although FoxO3a has been defined as a
ubiquitous tumor suppressor, as it induces cell apoptosis (24,25),
emerging evidence indicates that FoxO3a is associated with poor
clinical outcomes in specific cancer types (26–28).
Consistent with this evidence, our previous work (unpublished data)
has shown that in GBM cells, FoxO3a leads to slow cell
proliferation, possibly by regulating its target genes such as
cyclin D1 and p21. It is noteworthy that the dysregulation of genes
involved in survival signal, such as p21, and cell cycle
progression, such as cyclin D1 (2,29),
results in the development of chemoresistance in malignant
glioblastoma. Additionally, Tenbaum et al showed that in
colon cancer, FoxO3a acting in concert with β-catenin conferred
drug resistance in cancer cells (28). Although there is accumulating
evidence on the critical role played by FoxO3a in tumorigenesis and
cancer progression and on the FoxO3a potential connection with
β-catenin and chemoresistance, to the best of our knowledge,
whether FoxO3a is involved in conferring TMZ chemoresistance in GBM
is still unknown. Therefore, the present study was designed to
investigate whether FoxO3a contributes to TMZ resistance in GBM
cells and to understand its molecular mechanism.
Materials and methods
Cell lines and cell culture
The glioma cell line U251-MG was obtained from the
Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and
U87-MG was obtained from the American Type Culture Collection. The
two human glioma cell lines were cultured in Dulbecco's modified
Eagle's medium (DMEM, Gibco, Carlsbad, CA, USA) containing 2 mM
glutamine, 10% fetal calf serum (FBS), 100 U/ml penicillin (Sigma,
St. Louis, MO, USA), and 100 µg/ml streptomycin (Sigma). Cells were
maintained at 37°C in a 5% CO2 incubator.
Generation of temozolomide-resistant glioblastoma
cell lines. The parental U251-MG cells and U87-MG cells were
exposed to 400 µM TMZ for 3 weeks to generate TMZ-resistant
colonies. Initially, we cultured the two cell lines in 6-well
plates separately and allowed them to adhere during overnight
incubation at 37°C. TMZ treatment was repeated every 24 h for 5
consecutive days, and the cells were then exposed to fresh TMZ
every 3 days for a total of 3 weeks. The majority of the cells
died, but a small population survived and propagated. The surviving
colonies were selected and established as TMZ-resistant U251
(U251-TR) and U87 (U87-TR) cell lines.
Lentivirus production and
transduction
The following short hairpin (sh) RNA against FoxO3a
(FoxO3a-knockdown) was used: 5′-GCATGTTCAATGGGAGCTTGGA-3′. Another
construct expressing shRNA against irrelevant gene luciferase
(shRNA-NC) was used as a negative control. These constructs were
co-transfected with packaging plasmids into HEK-293T cells using
Lipofectamine 2000 (Invitrogen) according to the manufacturer's
instructions; the viral particles were harvested 48 h later. U87-TR
and U251-TR cells were infected with the lentivirus with 6 µg/ml
polybrene (Sigma).
Cell viability assay
Cell viability was determined using the Cell
Counting Kit (CCK)-8 assay. Cells were seeded in 96-well plates at
a density of 3×103 cells/well. After overnight
incubation, the cells were transduced with lentivirus and then
treated with various concentrations of TMZ (ranging from 200 to
2000 µM) for 1–5 days. After a 2 h incubation with 10 µl of CCK-8
solution (Dojindo Laboratories, Kumamato, Japan), cell viability
was detected at 490 nm using a microplate reader (BioTek, Winooski,
VT, USA). The survival rate of untreated cells was set at 100% and
used to calculate the half-maximal inhibitory concentration
(IC50). Each experiment was conducted in triplicate.
Real-time PCR
Total RNA was extracted from the TMZ resistant cell
lines and the GBM parental cell lines. cDNA was prepared using 1 µg
of total RNA from each sample, using specific primers (Applied
Biosystems). Six nanograms of cDNA were then used for real-time PCR
analysis in a final reaction volume of 20 µl. Samples were analyzed
in triplicate, and statistical analysis was performed using the
t-test.
Western blotting
Total protein was extracted and separated by gel
electrophoresis. Protein was then transferred to nitrocellulose
membranes and probed overnight using the appropriate primary
antibodies. The antibodies used were against N-cadherin (Cell
Signaling Technology, 4061), β-catenin (Cell Signaling Technology,
8480), and Histone H3 (Abcam, ab1791). Nuclear and cytoplasmic
fractions of total protein were separated using a NE-PER nuclear
and cytoplasmic extraction reagent (Thermo Scientific, Rockford,
IL, USA) and then subjected to western blotting.
Statistical analysis
Data shown in the graphs represent the mean values ±
SDs of three independent experiments. The difference among groups
was determined by ANOVA analysis, and the difference between two
groups was analyzed by Student's t-test. A P-value of <0.05 was
considered to indicate a statistically significant difference.
Results
FoxO3a and β-catenin protein levels
are enhanced in U87-TR and U251-TR cells, compared with their
parental GBM cells U87-MG and U251-MG
To investigate the cytotoxic impact of TMZ on GBM
cells, we analyzed the response of the parental cell lines U87-MG
and U251-MG (also designated as sensitive cell lines) and their
corresponding TMZ-induced drug-resistant cell lines (U87-TR and
U251-TR) to TMZ treatment. Dose titrations of TMZ in the sensitive
and resistant cell lines were performed in parallel to show the
effects of TMZ on cell viability. The survival of the sensitive
cells was significantly reduced within a relatively low TMZ
concentration gradient (50–500 µM). By contrast, this concentration
gradient did not affect the survival of the resistant cells (data
not shown); however, a higher TMZ concentration gradient resulted
in the reduction of their cell viability. To determine whether
FoxO3a and β-catenin were differentially expressed between the
resistant cells and their sensitive counterparts, we measured the
FoxO3a and β-catenin expression levels in the sensitive cells and
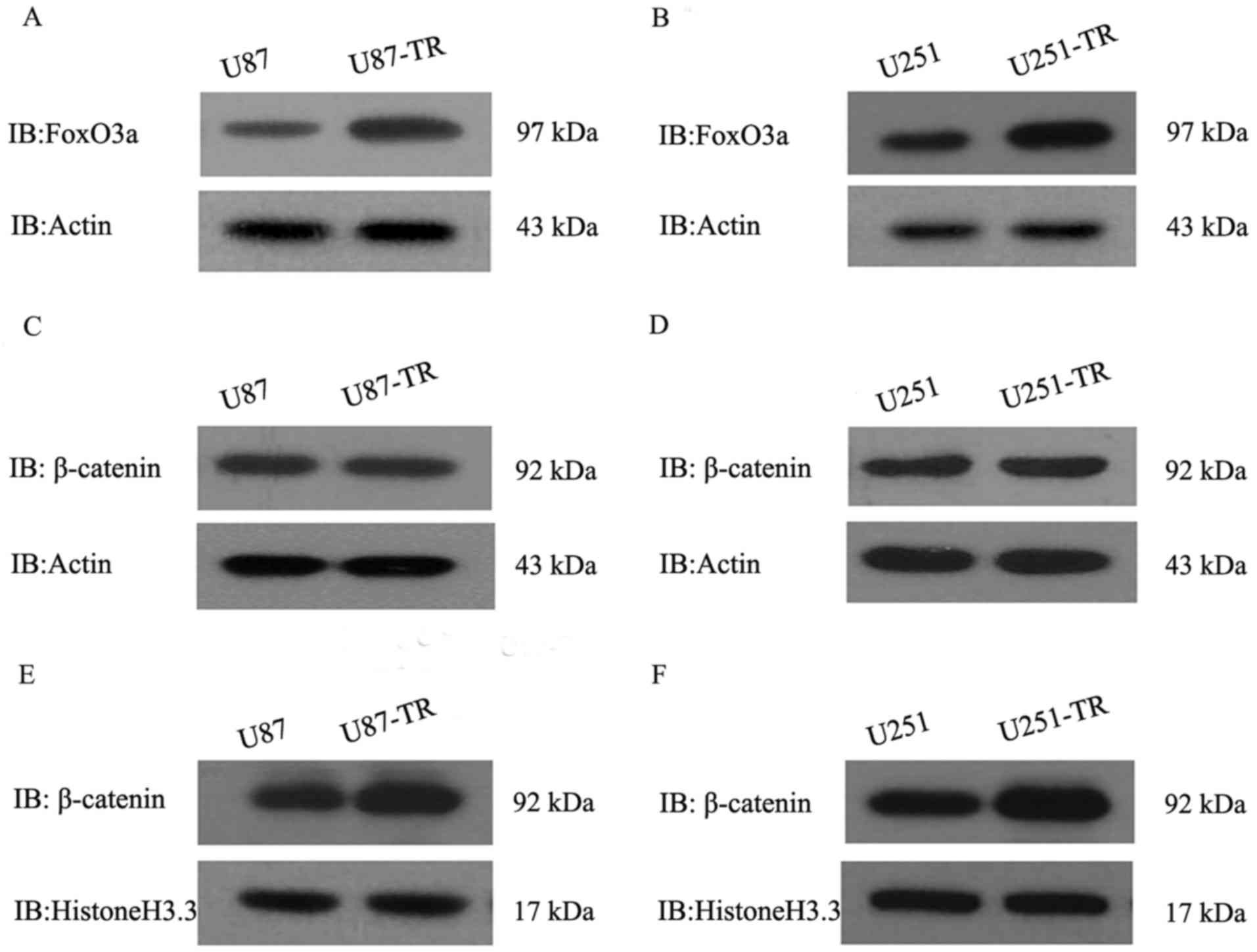
their corresponding resistant counterparts. Fig. 1 shows elevated protein levels of
FoxO3a and β-catenin in U87-TR cells compared with those in U87
cells. Consistently, the levels of these two proteins were higher
in U251-TR cells than in U251 cells, suggesting that the protein
levels of FoxO3a and β-catenin were indeed enhanced in the
resistant cells generated by repeated exposure to TMZ.
FoxO3a depletion increases the
chemosensitivity of U87-TR and U251-TR cells to TMZ
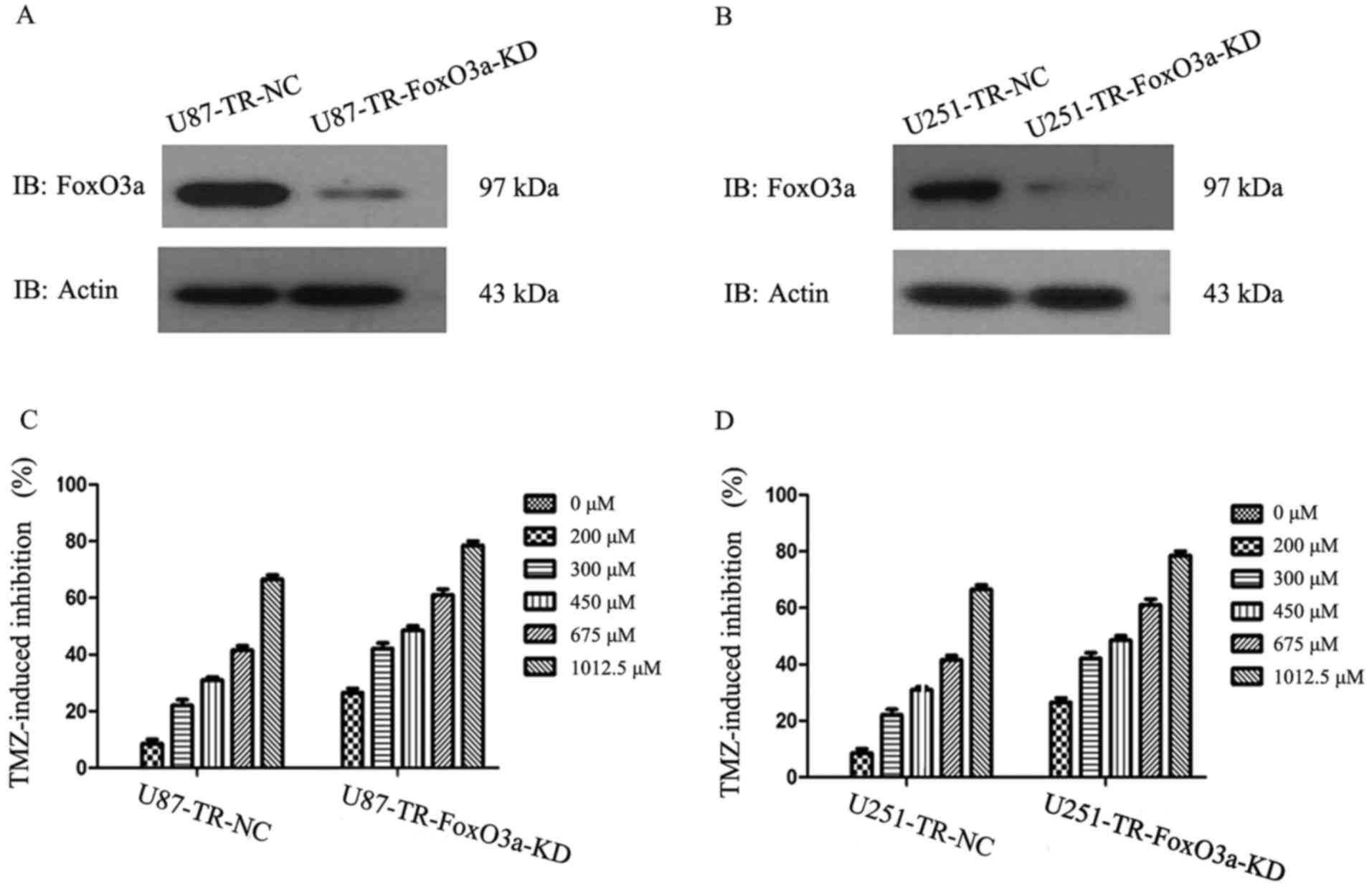
To show that FoxO3a has a critical role in TMZ
resistance in glioma cells, we transduced the resistant cells
expressing a relatively high level of FoxO3a with either lentivirus
FoxO3a-specific shRNA or non-specific control shRNA and studied the
response of these cells to TMZ treatment. As shown in Fig. 2, FoxO3a depletion by
lentivirus-mediated FoxO3a shRNA treatment in U87-TR cells resulted
in a marked reduction of cell viability following TMZ treatment,
and a similar result was observed in U251-TR cells. Moreover, the
dosage of TMZ that could cause a 50% inhibition of cell growth
(IC50) in U87-TR was reduced to 432 µM (nearly 41%)
after FoxO3a depletion. Similar to this finding, the depletion of
FoxO3a in U251-TR cells also led to a significant reduction in the
IC50 of TMZ (817.6 µM vs. 449.1 µM).
Considering the fact that β-catenin has been
implicated in TMZ resistance, coupled with the potential
connections between FoxO3a and β-catenin as revealed by several
studies (28,30–32),
we speculated that the functional contribution of FoxO3a to the
glioma cell resistance to TMZ may be due to its modulating effect
on the expression or the subcelluar distribution of β-catenin. We
found that the depletion of FoxO3a in U87-TR cells did not lead to
any change in β-catenin expression, nor did FoxO3a depletion in
U251-TR cells (data no shown), indicating that the part FoxO3a
plays in GBM cell resistance is not by regulating β-catenin
expression.
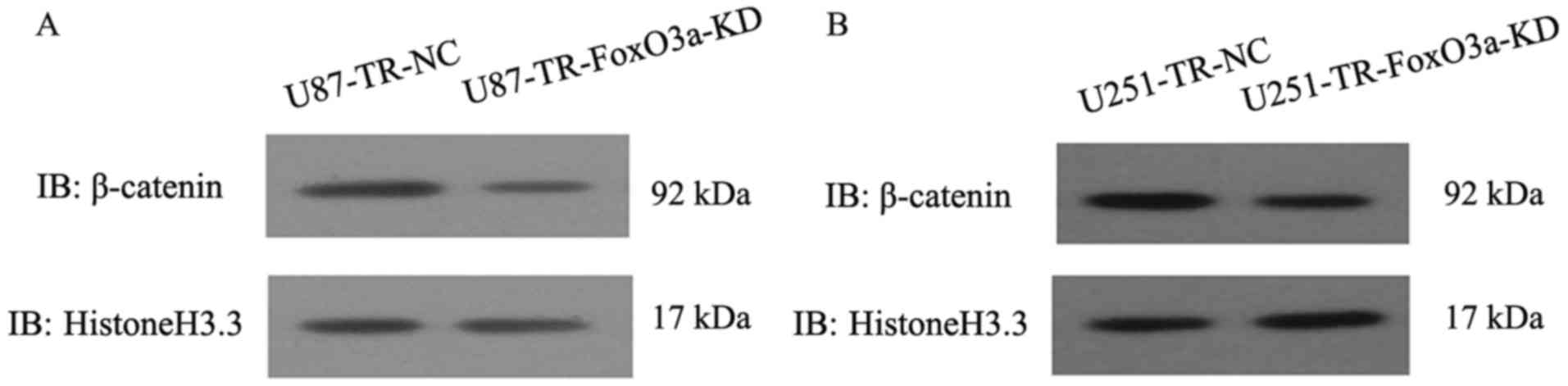
β-catenin nuclear accumulation is the hallmark of
active β-catenin, which has a role in chemotherapeutic resistance
(31,33), we therefore investigated whether
FoxO3a had the ability to affect the subcellular distribution of
β-catenin. The depletion of FoxO3a led to a significantly reduced
nuclear level of β-catenin but no change in its total level,
suggesting that the functional contribution of FoxO3a to TMZ
resistance in GBM cells may depend on it regulating the nuclear
accumulation of β-catenin (Fig. 3),
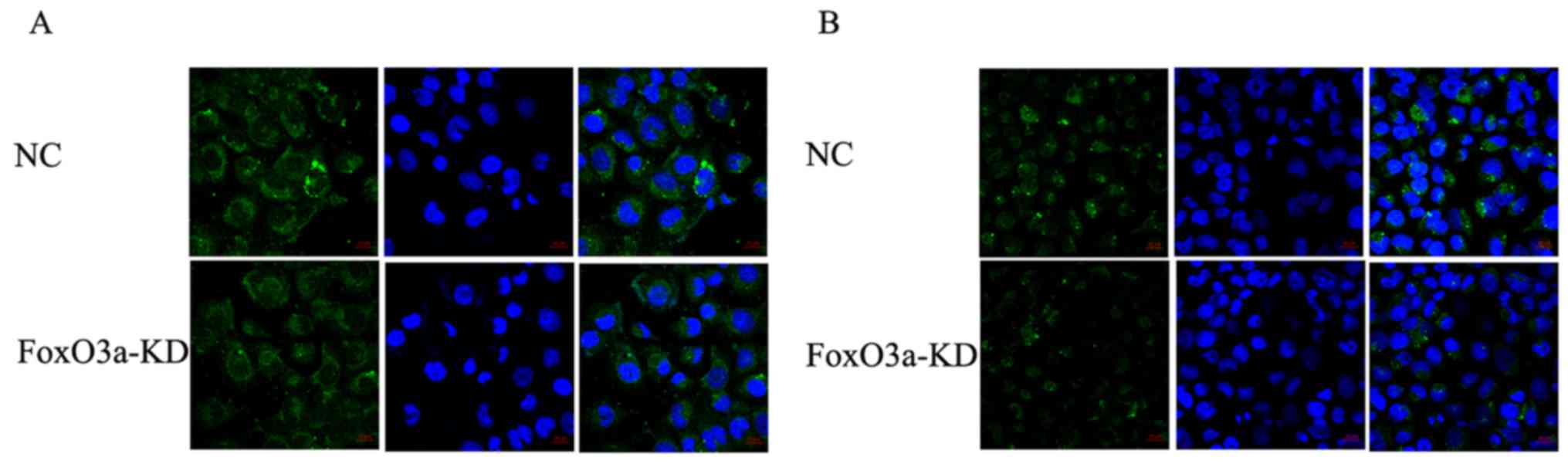
but not its expression. Since β-catenin activation could induce
chemoresistant characteristics by cleaving N-cadherin, with a
consequential effect on β-catenin nuclear accumulation per
se, we tested whether the resistant GBM cells depleted of
FoxO3a could display differential localization of N-cadherin
relative to their untreated counterparts. N-cadherin localization
was affected by FoxO3a depletion, further supporting that
FoxO3a-mediated chemoresistance is dependent on the nuclear level
of β-catenin (Fig. 4).
Discussion
Drug resistance is a crucial clinical feature that
determines the rate of tumor relapse and patient survival. TMZ is a
widely used GBM chemotherapeutic agent, and TMZ chemoresistance has
been identified as a main cause of treatment failure (34,35).
To address this challenge, researchers have studied the molecular
basis of this chemoresistance in GBM. Since the DNA repair system
has been associated with the chemoresistance, there has been
intensive investigation into genes involved in the DNA repair
system, particularly MGMT (5,7).
However, these published findings combined are still unable to
fully explain how GBM cells gain resistance to TMZ.
Herein, we found that FoxO3a, a novel factor for the
known mechanisms of TMZ resistance, may be required for the
TMZ-resistant phenotype in glioblastoma cells. Using the
TMZ-sensitive glioma cell lines U87 and U251, as well as the
TMZ-resistant cell lines U87-TR and U251-TR, we showed that
TMZ-resistant cells exhibited higher expression levels of FoxO3a
and β-catenin than their corresponding parental cells (also
designated as sensitive cells). The depletion of FoxO3a in U87-TR
cells resulted in significantly reduced growth upon TMZ treatment,
relative to the U87-TR control (treated with irrelevant shRNA).
Parallel studies performed using U251-TR cells and U251-TR cells
depleted of FoxO3a showed a similar result. These results indicated
that FoxO3a depletion increased the sensitivity of the resistant
glioma cells to the TMZ treatment. In addition, FoxO3a depletion
reduced the abundance of nuclear β-catenin in the resistant cell
lines. Taken together, these findings suggest that the regulation
of nuclear β-catenin accumulation by FoxO3a could be a novel
mechanism by which glioma cells gain resistance to TMZ
treatment.
Although a number of studies have shown that FoxO3a
functions as a tumor suppressor, there is emerging evidence that
correlates FoxO3a function with metastasis or a poor prognosis of
cancer, suggesting that FoxO3a can be either oncogenic or tumor
suppressive, presumably depending on the cell types and the
specific context. In glioma cells, we have previously demonstrated
that FoxO3a leads to slow cell proliferation and promotes cell
invasion. A study by Osuka et al showed that the nuclear
level of FoxO3a was greater in the radiological-resistant glioma
cells (generated by repeated radiation treatments) than their
sensitive counterparts, indicating that FoxO3a promotes
radioresistance in glioma (36).
Moreover, Tenbaum et al found that FoxO3a,
together with β-catenin, confers chemoresistance in colon cancer
(28). This background and, in
particular, the evidence for the important role of FoxO3a in
chemoresistance in colon cancer cells led us to hypothesize that
FoxO3a contributes to TMZ chemoresistance. The depletion of FoxO3a
significantly reduced the chemoresistance of U87-TR to TMZ
treatment as revealed by a combination of gradually reduced cell
growth upon increasing doses of TMZ treatment and a reduction in
the IC50 dosage of TMZ in U87-TR cells depleted of
FoxO3a relative to the U87-TR control cells. A similar result was
observed in the U251-TR cells and the FoxO3a-depleted U251-TR cells
(Fig. 2). These findings indicate
that FoxO3a contributes to cancer cell survival under TMZ-driven
chemotherapeutic stress, which is consistent with our hypothesis.
Given the critical role of FoxO3a in TMZ resistance in glioma
cells, the combined use of synthetic FoxO3a-siRNA and TMZ may
potentially in part abrogate the TMZ dose-limiting side effect,
which is a main disadvantage of TMZ administration, and improve the
efficacy of TMZ. Notably, the resistant cells that had FoxO3a shRNA
treatment (more than 90% FoxO3a had been depleted) were not as
susceptible to TMZ as their sensitive parental cells (U87 and
U251), suggesting that other mechanisms may contribute to this
pathological process. As U87 and U251 are known to be MGMT-negative
cell lines (37), it is unlikely
that MGMT is involved in the chemoresistance. However, FoxO3a
depletion led to a 41% reduction in U87-TR cell growth upon TMZ
treatment as evaluated by IC50 analysis and a 45%
reduction in U251-TR cell growth, suggesting that some other
mechanisms might exist in both of these glioma cell lines. In our
future investigations, we will attempt to identify these other
mechanisms contributing to the TMZ resistance in the glioma
cells.
β-catenin is a fundamental canonical Wnt/β-catenin
signaling effector, and its preferential nuclear accumulation is
the hallmark of the activation of this pathway. Notably, the
importance of β-catenin has been well established in glioma
tumorigenesis. Studies from independent research groups have shown
that β-catenin is highly expressed in gliomas and is associated
with the poor prognosis and short survival of GBM patients
(13,15). The depletion of β-catenin using the
RNA interference approach resulted in a significant inhibition of
glioma cell proliferation. Moreover, cytoplasmic ATRA (all-trans
retinoic acid)-induced β-catenin retention led to a reduced growth
of glioma cells (30), indicating
that the pharmacological or genetic inhibition of the expression or
nuclear accumulation of β-catenin impairs the glioma cell
growth.
Additionally, the repression of β-catenin by
sulforaphane promoted TMZ-induced cell apoptosis (33), and the blockade of nuclear β-catenin
translocation by FH535, a β-catenin inhibitor, enhanced the
antitumor function of TMZ, establishing a critical role for
β-catenin in TMZ chemoresistance in glioma. Prompted by these
published data, we tested whether TMZ sensitivity induced by FoxO3a
depletion is associated with the level of expression or the nuclear
accumulation of β-catenin. Of note, we only observed a reduced
nuclear β-catenin level and concomitant increased sensitivity to
TMZ in U87-TR and U251-TR cells, whereas the depletion of FoxO3a
did not affect β-catenin expression at the total protein level
(Figs. 2 and 3). As β-catenin functions are dependent on
its nuclear accumulation and transcriptional activity, it is
plausible that β-catenin nuclear accumulation alone may be
sufficient for conferring the chemoresistance. Our findings,
together with the previously published studies, corroborate that
nuclear β-catenin sufficiently causes the chemoresistance of GBM
cells.
Although it is known that β-catenin nuclear
accumulation is important for glioma tumorigenesis, the mechanism
of this molecular event has been surprisingly difficult to define.
Nevertheless, an important mechanism for β-catenin nuclear
translocation via binding to FoxM1, a forkhead box (Fox)
transcription factor, has been discovered (38). Given the antagonistic action between
FoxO3a and FoxM1 and the observation that FoxO3a activation and the
concomitant FoxM1 down-regulation increased chemosensitivity, it is
unlikely that FoxO3a mediates β-catenin nuclear accumulation and
the resulting chemoresistance in glioma cells by regulating FoxM1.
It is important to note that increased cellular oxidative stress
promotes FoxO3a nuclear accumulation and facilitates the binding of
FoxO3a and β-catenin (32). This
finding suggests that FoxO3a could directly bind to β-catenin and
mediate its nuclear import. Taken together, although we observed
FoxO3a depletion resulted in enhanced chemosensitivity and a
reduction in β-catenin nuclear abundance, experimentally
comprehending the molecular basis of FoxO3a-mediated
chemoresistance in glioma cells is still challenging. In our future
studies, we will perform intensive investigation into this
mechanism.
In conclusion, we provide direct evidence of the
critical role of FoxO3a in TMZ resistance in glioma cells. We show
that the repression of FoxO3a using lentivirus-mediated RNA
interference renders glioma cells more susceptible to TMZ treatment
and reduces the nuclear β-catenin level (without affecting its
total level). Our findings reveal a previously unknown role of
FoxO3a as an inducer of TMZ resistance in glioma cells and provide
a novel potential target for chemotherapeutic drug development, as
well as new diagnostic and predictive biomarkers.
Acknowledgements
We thank the National Natural Science Foundation of
China (grant no. 81660502) and the Start-up Foundation for Young
Scientists of Hainan Medical University (2015012) for funding this
study.
References
|
1
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: European Organisation for Research and Treatment of
Cancer Brain Tumor and Radiotherapy Groups; National Cancer
Institute of Canada Clinical Trials Group: Radiotherapy plus
concomitant and adjuvant temozolomide for glioblastoma. N Engl J
Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sarkaria JN, Kitange GJ, James CD, Plummer
R, Calvert H, Weller M and Wick W: Mechanisms of chemoresistance to
alkylating agents in malignant glioma. Clin Cancer Res.
14:2900–2908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang JY and Edelmann W: Mismatch repair
proteins as sensors of alkylation DNA damage. Cancer Cell.
9:417–418. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caporali S, Falcinelli S, Starace G, Russo
MT, Bonmassar E, Jiricny J and D'Atri S: DNA damage induced by
temozolomide signals to both ATM and ATR: Role of the mismatch
repair system. Mol Pharmacol. 66:478–491. 2004.PubMed/NCBI
|
|
5
|
Hegi ME, Liu L, Herman JG, Stupp R, Wick
W, Weller M, Mehta MP and Gilbert MR: Correlation of
O6-methylguanine methyltransferase (MGMT) promoter methylation with
clinical outcomes in glioblastoma and clinical strategies to
modulate MGMT activity. J Clin Oncol. 26:4189–4199. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cahill DP, Levine KK, Betensky RA, Codd
PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR,
Curry WT, et al: Loss of the mismatch repair protein MSH6 in human
glioblastomas is associated with tumor progression during
temozolomide treatment. Clin Cancer Res. 13:2038–2045. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yip S, Miao J, Cahill DP, Iafrate AJ,
Aldape K, Nutt CL and Louis DN: MSH6 mutations arise in
glioblastomas during temozolomide therapy and mediate temozolomide
resistance. Clin Cancer Res. 15:4622–4629. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sathornsumetee S and Rich JN: New
treatment strategies for malignant gliomas. Expert Rev Anticancer
Ther. 6:1087–1104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peifer M, Rauskolb C, Williams M,
Riggleman B and Wieschaus E: The segment polarity gene armadillo
interacts with the wingless signaling pathway in both embryonic and
adult pattern formation. Development. 111:1029–1043.
1991.PubMed/NCBI
|
|
10
|
Noordermeer J, Klingensmith J, Perrimon N
and Nusse R: dishevelled and armadillo act in the wingless
signalling pathway in Drosophila. Nature. 367:80–83. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peifer M, Berg S and Reynolds AB: A
repeating amino acid motif shared by proteins with diverse cellular
roles. Cell. 76:789–791. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang ZQ, Chen HQ, Chen YH and Cheng XF:
Significance of beta-catenin and Cyclin D1 express in glioma. Xi
Bao Yu Fen Zi Mian Yi Xue Za Zhi. 25:1010–1012. 2009.(In Chinese).
PubMed/NCBI
|
|
13
|
Liu X, Wang L, Zhao S, Ji X, Luo Y and
Ling F: β-Catenin overexpression in malignant glioma and its role
in proliferation and apoptosis in glioblastma cells. Med Oncol.
28:608–614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu C, Tu Y, Sun X, Jiang J, Jin X, Bo X,
Li Z, Bian A, Wang X, Liu D, et al: Wnt/beta-Catenin pathway in
human glioma: Expression pattern and clinical/prognostic
correlations. Clin Exp Med. 11:105–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rossi M, Magnoni L, Miracco C, Mori E,
Tosi P, Pirtoli L, Tini P, Oliveri G, Cosci E and Bakker A:
β-catenin and Gli1 are prognostic markers in glioblastoma. Cancer
Biol Ther. 11:753–761. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pukkila MJ, Virtaniemi JA, Kumpulainen EJ,
Pirinen RT, Johansson RT, Valtonen HJ, Juhola MT and Kosma VM:
Nuclear beta catenin expression is related to unfavourable outcome
in oropharyngeal and hypopharyngeal squamous cell carcinoma. J Clin
Pathol. 54:42–47. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Elzagheid A, Buhmeida A, Korkeila E,
Collan Y, Syrjanen K and Pyrhonen S: Nuclear beta-catenin
expression as a prognostic factor in advanced colorectal carcinoma.
World J Gastroenterol. 14:3866–3871. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang CL, Liu D, Ishikawa S, Nakashima T,
Nakashima N, Yokomise H, Kadota K and Ueno M: Wnt1 overexpression
promotes tumour progression in non-small cell lung cancer. Eur J
Cancer. 44:2680–2688. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lentsch AB, Kato A, Yoshidome H, McMasters
KM and Edwards MJ: Inflammatory mechanisms and therapeutic
strategies for warm hepatic ischemia/reperfusion injury.
Hepatology. 32:169–173. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sinnberg T, Menzel M, Ewerth D, Sauer B,
Schwarz M, Schaller M, Garbe C and Schittek B: β-Catenin signaling
increases during melanoma progression and promotes tumor cell
survival and chemoresistance. PLoS One. 6:e234292011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu G, Wu F and Wang E: KLF8 promotes
temozolomide resistance in glioma cells via β-catenin activation.
Cell Physiol Biochem. 38:1596–1604. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Accili D and Arden KC: FoxOs at the
crossroads of cellular metabolism, differentiation, and
transformation. Cell. 117:421–426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Calnan DR and Brunet A: The FoxO code.
Oncogene. 27:2276–2288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Myatt SS and Lam EW: The emerging roles of
forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 7:847–859.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen J, Gomes AR, Monteiro LJ, Wong SY, Wu
LH, Ng TT, Karadedou CT, Millour J, Ip YC, Cheung YN, et al:
Constitutively nuclear FOXO3a localization predicts poor survival
and promotes Akt phosphorylation in breast cancer. PLoS One.
5:e122932010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Storz P, Döppler H, Copland JA, Simpson KJ
and Toker A: FOXO3a promotes tumor cell invasion through the
induction of matrix metalloproteinases. Mol Cell Biol.
29:4906–4917. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tenbaum SP, Ordóñez-Morán P, Puig I,
Chicote I, Arqués O, Landolfi S, Fernández Y, Herance JR, Gispert
JD, Mendizabal L, et al: β-catenin confers resistance to PI3K and
AKT inhibitors and subverts FOXO3a to promote metastasis in colon
cancer. Nat Med. 18:892–901. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shah MA and Schwartz GK: Cell
cycle-mediated drug resistance: an emerging concept in cancer
therapy. Clin Cancer Res. 7:2168–2181. 2001.PubMed/NCBI
|
|
30
|
Lu J, Zhang F, Zhao D, Hong L, Min J,
Zhang L, Li F, Yan Y, Li H, Ma Y, et al: ATRA-inhibited
proliferation in glioma cells is associated with subcellular
redistribution of beta-catenin via up-regulation of Axin. J
Neurooncol. 87:271–277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi ZD, Qian XM, Liu CY, Han L, Zhang KL,
Chen LY, Zhang JX, Pu PY, Yuan XB and Kang CS: Chinese Glioma
Cooperative Group (CGCG): Aspirin-/TMZ-coloaded microspheres exert
synergistic antiglioma efficacy via inhibition of β-catenin
transactivation. CNS Neurosci Ther. 19:98–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Essers MA, de Vries-Smits LM, Barker N,
Polderman PE, Burgering BM and Korswagen HC: Functional interaction
between beta-catenin and FOXO in oxidative stress signaling.
Science. 308:1181–1184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lan F, Pan Q, Yu H and Yue X: Sulforaphane
enhances temozolomide-induced apoptosis because of down-regulation
of miR-21 via Wnt/β-catenin signaling in glioblastoma. J Neurochem.
134:811–818. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mrugala MM and Chamberlain MC: Mechanisms
of disease: Temozolomide and glioblastoma - look to the future. Nat
Clin Pract Oncol. 5:476–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Agnihotri S, Gajadhar AS, Ternamian C,
Gorlia T, Diefes KL, Mischel PS, Kelly J, McGown G, Thorncroft M,
Carlson BL, et al: Alkylpurine-DNA-N-glycosylase confers resistance
to temozolomide in xenograft models of glioblastoma multiforme and
is associated with poor survival in patients. J Clin Invest.
122:253–266. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Osuka S, Sampetrean O, Shimizu T, Saga I,
Onishi N, Sugihara E, Okubo J, Fujita S, Takano S, Matsumura A, et
al: IGF1 receptor signaling regulates adaptive radioprotection in
glioma stem cells. Stem Cells. 31:627–640. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ryu CH, Yoon WS, Park KY, Kim SM, Lim JY,
Woo JS, Jeong CH, Hou Y and Jeun SS: Valproic acid downregulates
the expression of MGMT and sensitizes temozolomide-resistant glioma
cells. J Biomed Biotechnol. 2012:9874952012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bowman A and Nusse R: Location, location,
location: FoxM1 mediates β-catenin nuclear translocation and
promotes glioma tumorigenesis. Cancer Cell. 20:415–416. 2011.
View Article : Google Scholar : PubMed/NCBI
|