Introduction
Oxidative stress in cancer is one of the most
important metabolic stresses caused by both extrinsic (reperfusion
of a hypoxic microenvironment) and intrinsic (uncoupled
mitochondrial activity) factors (1,2). From
the viewpoint of carcinogenesis, oxidative stress is a double-edged
sword, since it promotes genetic alterations through DNA damage to
drive malignant progression while also causing cell damage and
inducing apoptosis (3,4). Under physiological conditions, cells
possess molecular mechanisms that enable them to adapt to these
stresses. In multicellular organisms, adaptive responses to
oxidative stresses are regulated by nuclear factor erythroid
2-related factor 2 (Nrf2), a master transcription factor of many
antioxidant genes and phase II detoxifying enzymes (5). Moreover, recent studies have
demonstrated that cancer cells can also use Nrf2 and its target
genes to adapt to multiple stresses, which is beneficial for cancer
cell survival, leading to malignant progression and lethal
characteristics such as invasion, metastasis and chemotherapy
resistance (6). Therefore, some
researchers have pointed out that under these conditions, Nrf2 can
be defined as a proto-oncogene.
Transcription factor Nrf2, a critical element in the
survival of healthy cells in response to oxidative stress, belongs
to the Keap1-Nrf2-antioxidant response element (ARE) signaling
pathway and upregulates many ARE-containing genes such as
NAD(P)H:quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1),
aldo-keto reductase 1C1 (AKR1C1) and thioredoxin (Trx) (7). In addition to these enzymes, these
proteins also include, for example, those involved in the
biosynthesis and regeneration of glutathione (GSH), a very
effective scavenger of reactive oxygen species (ROS) and
electrophiles, such as heavy and light chains of γ-glutamyl
cysteinyl ligase (8), the x-CT
(subunit of the cystine/glutamate transporter) component of the
cystine/glutamate exchange transport system (9), the glutathione reductase (GR) and the
GSH synthetase (10,11).
In the present study, we first assessed the
expression patterns of Nrf2 in normal human astrocytes and 3 GBM
cell lines, and subsequently silenced Nrf2 expression in the GBM
cell line U251 using RNA interference technology. Functional
analysis demonstrated that Nrf2 knockdown led to a decrease in U251
cell proliferation, as well as resulted in intracellular redox
imbalance (decreased levels of GSH and increased levels of ROS). In
addition, suppression of Nrf2 also resulted in the impairment of
the AKT and ERK1/2 signaling pathways. Finally, we ascertained that
exogenous supplementation of Nrf2-knockdown cells with glutathione
monoethyl ester (GMEE; a membrane-permeable derivative of GSH that
can supplement cellular GSH levels) restored their intracellular
GSH levels, cell proliferation defects and AKT inhibition,
confirming that GSH depletion and AKT pathway inhibition are
critical for GBM cell proliferation. Our findings may shed light on
the role and underlying mechanism of Nrf2 in the regulation of
glioma proliferation via GSH depletion and consequent AKT
inhibition, a new viewpoint by which to comprehend the role and
mechanism of Nrf2 in glioma pathoetiology.
Materials and methods
Reagents and antibodies
N-acetylcysteine (NAC) was obtained from
Beyotime (Shanghai, China). GMEE (sc-203974), AKT inhibitor IV
(sc-203809) and AG490 (sc-202046) were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). U0126 (cat. #9903) was
purchased from Cell Signaling Technology (CST; Danvers, MA, USA).
Antibodies against phospho-AKT (Ser473), AKT, phospho-ERK1/2
(Thr202/Tyr204), ERK1/2, phospho-STAT3 (Tyr705), STAT3, histone H3
and β-actin were purchased from CST. Antibodies against GR, GCLC
and Nrf2 were obtained from Santa Cruz Biotechnology.
Cell culture
Human GBM cell lines A172, U87 and U251 were
obtained from the Cell Bank of Shanghai, Institute of Biochemistry
and Cell Biology, Chinese Academy of Sciences (Shanghai, China).
The cells were cultured at 37°C in 5% CO2/95% air in
Dulbecco's modified Eagle's medium (DMEM) containing 10%
heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin and
100 mg/ml streptomycin (all from Gibco, Los Angeles, CA, USA).
Normal human astrocytes (NHAs) were obtained from
ScienCell (San Diego, CA, USA) (cat. 1800). NHAs were cultured in
astrocyte medium (cat. 1801; ScienCell) and cultured following the
manufacturer's instructions.
Lentivirus-mediated RNA
interference
For lentivirus-mediated silencing of Nrf2, the
lentiviral particles and Polybrene were purchased from GenePharma
(Shanghai, China). The short hairpin RNA (shRNA) sequence that
effectively targets human Nrf2 was: 5′-GCAGTTCAATGAAGCTCAACT-3′. A
non-silencing shRNA, with the scrambled sequence
5′-TTCTCCGAACGTGTCACGT-3′, was used as the negative control. The
lentiviral particles containing Nrf2-specific shRNA or scrambled
shRNA were named Nrf2i or Sc, respectively. Cell transfection was
performed as per the manufacturer's instructions. Briefly, the
cells were plated in a 6-well plate to reach 30–50% confluence on
the day of transfection. After the addition of Polybrene at 4.5
µg/ml, the lentiviral particles were used to transfect the cells.
Transfection was continued for 24 h followed by a 24-h recovery
period in complete medium. For the selection of cells with stable
transfection, cells were grown under selective pressure by
puromycin (2.0 µg/ml; Sigma-Aldrich, St. Louis, MO, USA) for up to
2 weeks.
Quantitative real-time PCR
Total RNA was extracted using a modified TRIzol
one-step extraction method (Tiangen, Beijing, China), and then the
quality of total RNA was assessed by spectrophotometer analysis
(OD260/280:1.8–2.2). An amount 400 ng RNA of total RNA was
reverse-transcribed using First-Strand cDNA Synthesis SuperMix
(TransGen Biotech, Beijing, China). The cDNA obtained was amplified
immediately using the following primers: for Nrf2,
5′-TCAGCGACGGAAAGAGTATGA-3′ and 5′-CCACTGGTTTCTGACTGGATGT-3′; for
NQO1, 5′-ATGGTCGGCAGAAGAGC-3′ and 5′-GGAAATGATGGGATTGAAGT-3′; for
HO-1, 5′-TCTCCGATGGGTCCTTACACTC-3′ and
5′-GGCATAAAGCCCTACAGCAACT-3′; for GCLC, 5′-AGACATTGATTGTCGCTG-3′
and 5′-TGGTCAGACTCATTAGCA-3′; for GR, 5′-TTTGTCTCGGTCTTTCGGGG-3′
and 5′-CCAGGATCTATGGCACCGTC-3′; and for GAPDH,
5′-GAAATCCCATCACCATCTTC-3′ and 5′-GGACTCCACGACGTACTCA-3′. The
amplification and data acquisition were carried out on a Real-Time
PCR System (Agilent Technologies, Santa Clara, CA, USA) using
FastStart Universal SYBR-Green Master (Roche, Mannheim, Germany).
The conditions consisted of pre-denaturation at 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min. The
PCR amplification efficiency of each gene was established by means
of calibration curves. All samples were analyzed in triplicate. The
results were analyzed using the 2−ΔΔCq method. The data
are expressed as the relative mRNA levels and were normalized to
GAPDH, a commonly used control.
Western blot analysis
To prepare the total proteins, cells were lysed in
cold RIPA lysis buffer containing a 0.5% phosphatase inhibitor and
1% phenylmethylsulphonyl fluoride (both from Beyotime, Jiangsu,
China). To prepare the nuclear and cytoplasmic proteins, cells were
lysed using a nuclear and cytoplasmic protein extraction kit
(Beyotime) as per the manufacturer's instructions. Protein content
was quantified with the Braford reagent (Coomassie Plus Protein
Assay Reagent; cat. #23238) (Pierce, Rockford, IL, USA). Equal
amounts of proteins were separated by 8–15% sodium dodecyl sulfate
polyacrylamide gel electrophoresis and subsequently transferred
onto nitrocellulose membranes (Millipore, Billerica, MA, USA).
Following blocking with 5% non-fat milk in TBS-1% Tween-20 (TBST)
for 2 h, the membranes were immunoblotted with primary antibodies:
1:500 anti-Nrf2 (cat. #sc-13032; 100 kDa), 1:500 anti-GCLC (cat.
#sc-22755; 73 kDa), 1:500 anti-GR (cat. #sc-32886; 50–65 kDa) (all
from Santa Cruz Biotechnology) 1:1,000 anti-phospho-AKT (cat.
#4060; 60 kDa), 1:1,000 anti-AKT (cat. #4691; 60 kDa), 1:1,000
anti-phospho-ERK1/2 (cat. #4370; 42.44 kDa), 1:1,000 anti-ERK1/2
(cat. #4695; 42.44 kDa), 1:1,000 anti-phospho-STAT3 (cat. #9145;
79.86 kDa), 1:1,000 anti-STA3 (cat. #12640; 79.86 kDa), 1:2,000
anti-β-actin (cat. #4970; 45 kDa) and 1:1,000 anti-histone H3 (cat.
#4499; 17 kDa) (all from CST). After removal of the primary
antibodies, the membranes were incubated with the horseradish
peroxidase-conjugated secondary antibodies for 2 h at room
temperature and visualized using the enhanced chemiluminescent
(ECL) detection reagent from Pierce. Immunoreactive protein bands
were detected with Tanon 5200 chemiluminescence imaging system
(Shanghai, China).
Doubling time
Aliquots of 5×104 cells were seeded in
35-mm plastic dishes in culture medium. The number of cells was
counted in triplicate at 24-h intervals for 4 days using a particle
distribution counter CDA-500 (Sysmex, Kobe, Japan). The doubling
time of the cell population was estimated from the exponential
growth phase.
Cell proliferation analysis
Cell proliferation was measured by Cell Counting
Kit-8 (CCK-8) (Dojindo, Kumamoto, Japan) as per the manufacturer's
protocol. One day prior to treatment, 1,000 cells/well were plated
in a 96-well tissue culture plate in a total volume of 200 µl/well
and were cultured for 1–4 days, respectively. Subsequently, 10 µl
of CCK-8 solution was added and the cells were further incubated
for 1 h at 37°C in 5% CO2/95% air. Then, the absorbance
was measured at 450 nm using an ELISA microplate reader (Bio-Rad,
Hercules, CA, USA). At least 6 technical replicates were used for
each condition.
Colony formation analysis
Colony formation analysis was carried out 9 days
after lentivirus transfection. U251 cells were plated into 6-well
plates (300 cells/well) and cultured for 9 days. The medium was
updated every 3 days. After 9 days of culture, the cells were
washed with phosphate-buffered saline (PBS), fixed in 4%
paraformaldehyde for 10 min, and were stained with freshly prepared
crystal violet staining solution for 20 min. Colony formation was
observed through a light microscope and a colony count was
performed.
Assessment of total GSH content
Cells were collected and lysed with protein
detergent S solvent. The total GSH was determined by commercially
available Total Glutathione Assay kit (Beyotime Institute of
Biotechnology, Shanghai, China). All procedures completely complied
to the manufacturer's instructions. The protein concentration was
estimated by Coomassie Plus Protein Assay Reagent (Pierce).
Assessment of intracellular ROS
The level of intracellular ROS was quantified using
the Reactive Oxygen Species Assay kit (Beyotime Institute of
Biotechnology). 2′,7′-Dichlorodihydrofluorescein diacetate
(DCFH-DA) is oxidized by ROS in viable cells to dichlorofluorescein
(DCF) which is highly fluorescent at 530 nm. The cells were washed
3 times with PBS. DCFH-DA, diluted to a final concentration of 10
mM, was added and incubated for 30 min at 37°C in the dark. After
being washed 3 times with PBS, the relative levels of fluorescence
were quantified by a multidetection microplate reader (485 nm
excitation and 535 nm emission).
Statistical analysis
Data bars correspond to the mean values ± standard
deviations (SD) of minimum triplicate experiments. The statistical
significance of the differences was assessed using one-way analysis
of variance (ANOVA) or unpaired two-tailed t-tests; the probability
values of <0.05 and <0.01 were deemed statistically
significant and very significant, respectively, for all
analyses.
Results
Nrf2 is overexpressed in GBM cell
lines
In a previous study, we found that the expression
level of Nrf2 was positively correlated with the tumor grade in
glioma tissues (23). In the
present study, we used western blot analyses to examine Nrf2 levels
in various GBM cell lines and normal human astrocyte (NHA) cell
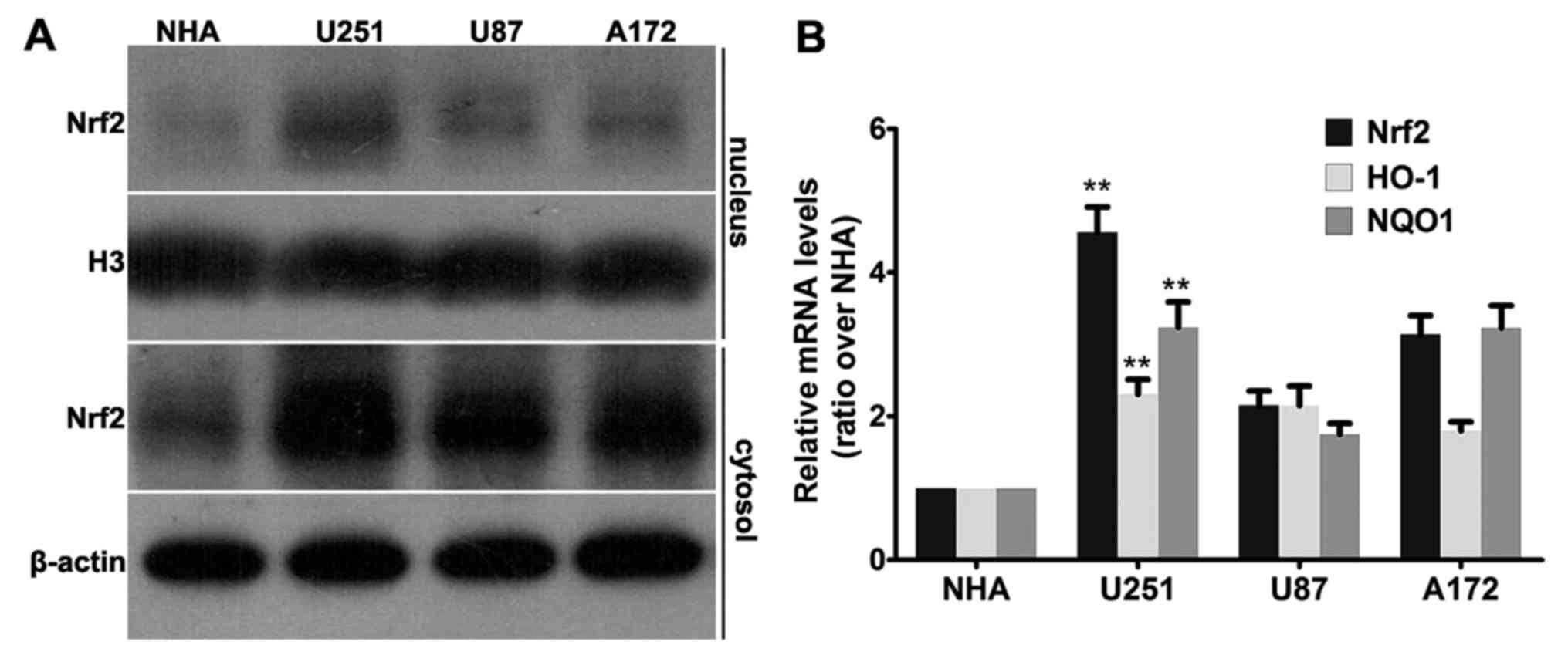
line. As shown in Fig. 1A, western
blot analysis revealed that all 3 cell lines, U251, U87 and A172,
had a higher Nrf2 level than that of the NHA cell line both in the
nuclear fraction and in the cytosol fraction. To be noted, there
was no detectable band in the nuclear fraction of the NHA cell
line, while Nrf2 was accumulated most abundantly in the nuclear
fraction of the U251 cells. Consistent with the aforementioned
results, similar patterns were observed in the mRNA levels of Nrf2.
All three human GBM cell lines (U251, U87 and A172) had increased
relative mRNA levels of Nrf2 compared with the NHA cell line
(Fig. 1B).
As a transcription factor, overexpression of Nrf2
may cause expression of downstream genes; thus, we tested the mRNA
levels of NQO1 and HO-1 using real-time PCR. As shown in Fig. 1B, the highest Nrf2 level was
observed in the U251 cells, as well as the mRNA levels of HO-1 and
NQO1. Therefore, Nrf2 is overexpressed and transcriptionally
active, which may cause overexpression of downstream genes in
multiple GBM cell lines.
We also tested the doubling times for U251, U87 and
A172 cells (~8, 24 and 48 h, respectively). It was determined that
U251, with the highest Nrf2 expression level, also had the fastest
proliferation speed of all 3 cell lines (data not shown). In
accordance with this, we chose the U251 cell line for subsequent
genetic intervention.
Nrf2 deficiency leads to oxidative
stress and redox imbalance in GBM cells
To generate the stable Nrf2-knockdown model, the
U251 cells were transfected with lentivirus-mediated shRNA ecoding
Nrf2-targeting shRNA (Nrf2i) or scrambled control (Sc) shRNA.
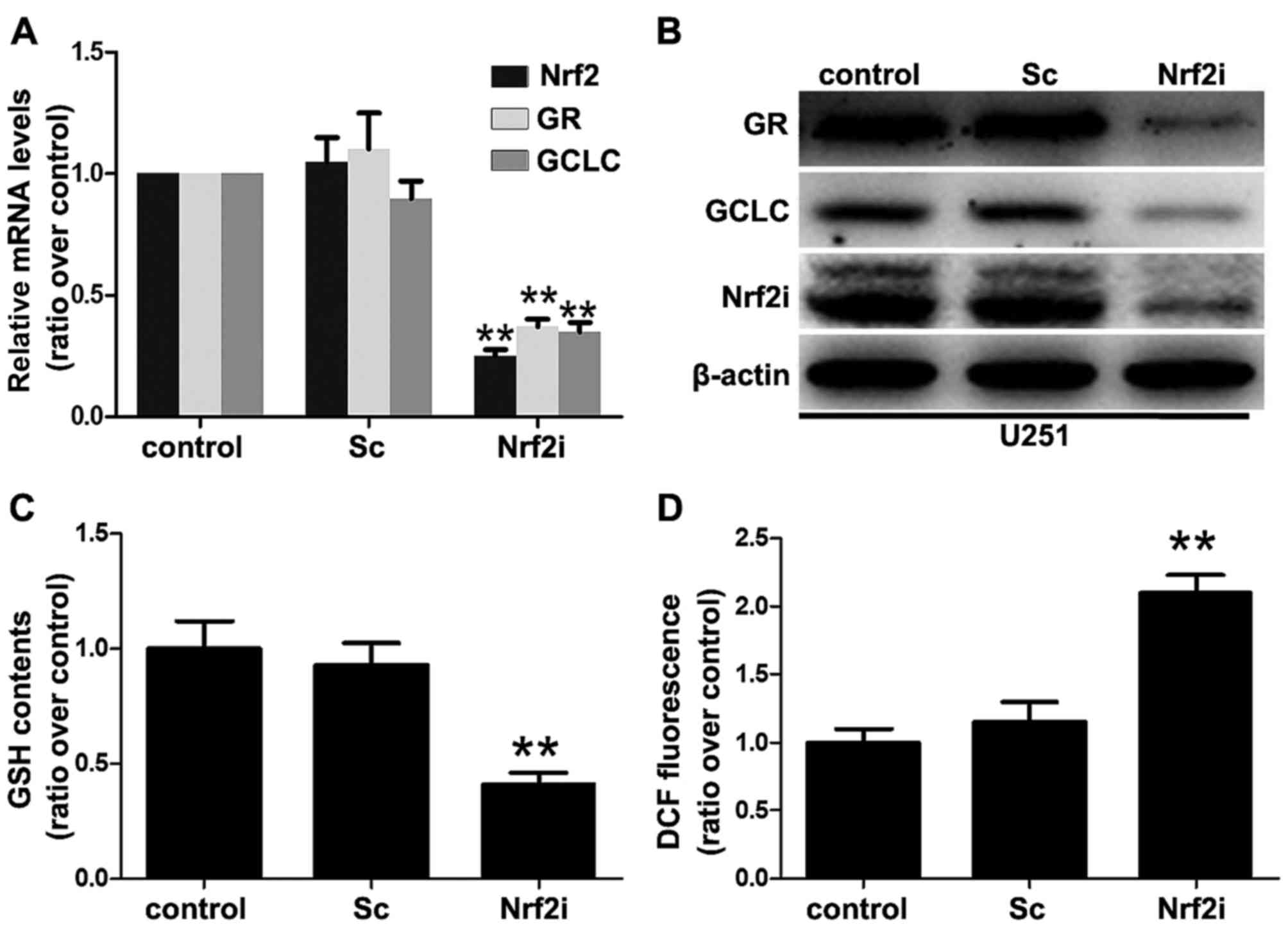
Clearly, transfection with Sc shRNA did not affect the expression
of Nrf2 as the Nrf2 mRNA transcripts and protein expression in the
Sc group were similar to that in the control group (Fig. 2A and B). In contrast, the levels of
Nrf2 mRNA transcripts in the U251-Nrf2i cells were significantly
decreased by ~80% (P<0.01) (as shown in Fig. 2A), compared with the non-treated or
Sc shRNA-treated U251 cells. Additionally, western blot analysis
revealed that the Nrf2 expression was also significantly decreased
in the Nrf2i group (Fig. 2B). These
results indicated that lentivirus-mediated shRNA effectively and
specifically suppressed Nrf2 expression in the U251 cells.
As a transcription factor, knockdown of Nrf2 may
suppress the expression of its regulated genes, for example GCLC
and GR, which are also known as GSH generating enzymes [catalytic
subunit of GCL (GCLC) for de novo GSH biosynthesis and GR
for GSH regeneration]. Thus, we determined the GCLC and GR mRNA
levels using real-time PCR to evaluate the function of Nrf2. As
shown in Fig. 2A, the Nrf2 mRNA
level was suppressed in the U251 cells accompanied by the decreased
mRNA levels of GCLC and GR. Similar patterns were observed in the
immunoblot analysis of the proteins GCLC and GR (Fig. 2B).
Since Nrf2 is the GSH-generation key regulator (as
indicated above), several in vivo studies have shown that an
Nrf2 deficiency results in oxidative stress and redox imbalance
(decreased levels of GSH and increased levels of ROS) (2,3). To
characterize this effect, we determined the GSH levels in the
non-treated, Sc shRNA-treated and Nrf2 shRNA (Nrf2i)-treated cells
(Fig. 2C). Consistent with the
results obtained with decreased GCLC and GR expression, we found
that the GSH levels in the Nrf2-knockdown U251 cells were
significantly decreased (decreased by 70% compared with the Sc and
control groups). To further correlate these results with those
related to cellular redox status, we determined the intracellular
levels of ROS using DCF-DA dye, which emits green fluorescence in
the presence of ROS. As shown in Fig.
2D, U251-Nrf2i cells exhibited a 117.4% increase in ROS
production in comparison to the control group (Fig. 2D). Collectively, these data
indicated that lentivirus-mediated shRNA silencing led to oxidative
stress as a result of decreased GSH levels and redox imbalance in
the GBM cells.
U251 cell proliferation is impaired
with the disruption of Nrf2
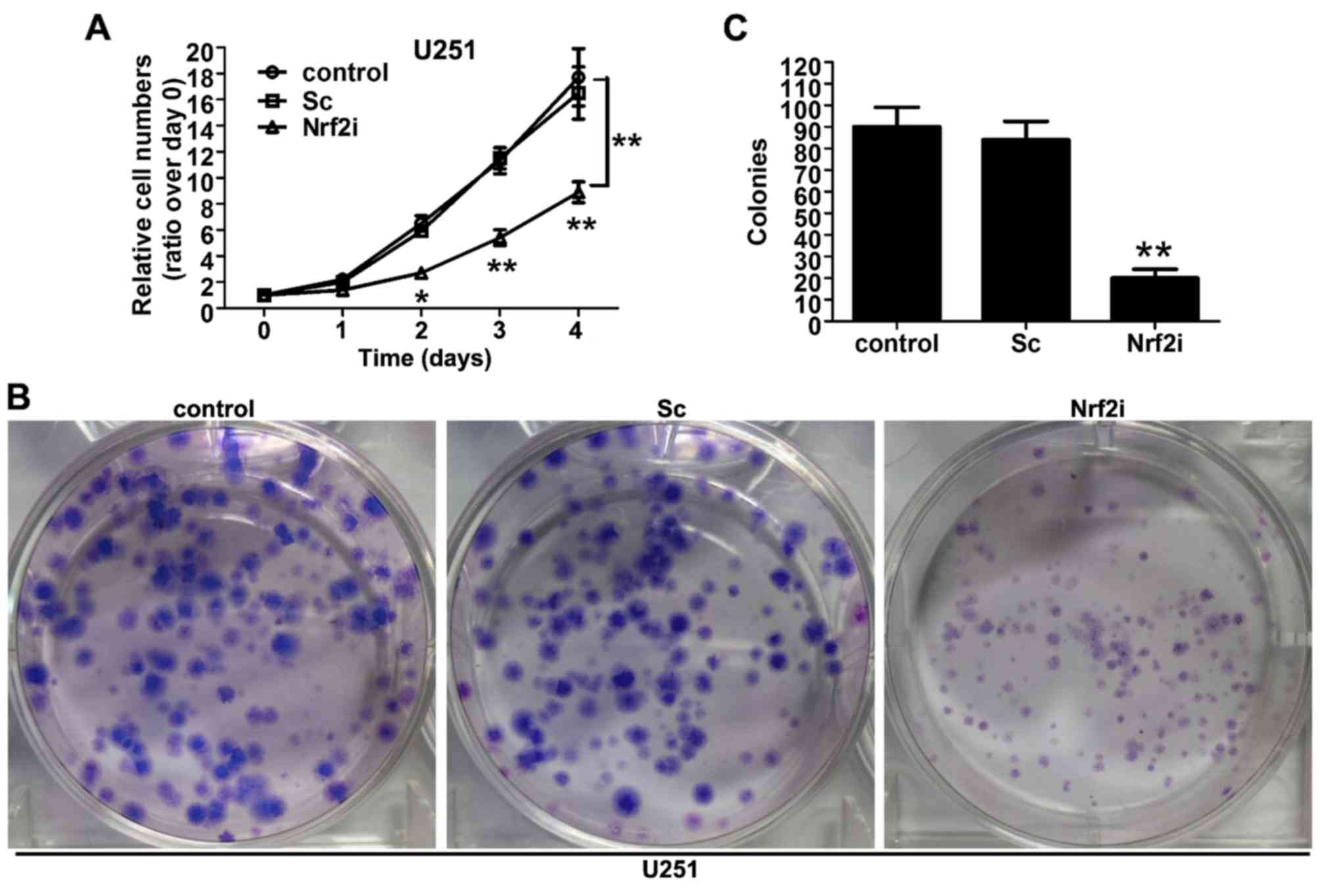
To investigate the effect of Nrf2 silencing on cell
proliferation, the cell viability was observed using CCK-8 assay.
As shown in Fig. 3A, the growth
curve of the Nrf2i group (Nrf2 shRNA-treated U251 cells) began to
decline from the second day, compared with the control group
(non-treated) and Sc group (Sc-treated U251 cells). The decrease
reached 37.1% (P<0.01) and 45.2% (P<0.01) on the third and
fourth day, respectively, compared with the Sc and control groups,
whereas no significant difference relating to cell viability was
observed between the Sc and control groups. To further ascertain
the results of the CCK-8 assay, we conducted the colony formation
assay to evaluate the long-term effect of Nrf2 knockdown on U251
cell proliferation. As shown in Fig.
3B, the size of the independent colonies was much smaller in
the Nrf2i group than that in the control group and Sc group.
Furthermore, the number of colonies that formed in the Nrf2i group
was significantly decreased (P<0.01), compared with the control
and Sc groups (Fig. 3C). The
aforementioned data indicate that Nrf2 knockdown evidently impaired
the proliferation of U251 cells.
Exogenous supplementation with GMEE,
but not NAC, rescues the phenotypic defects associated with Nrf2
deficiency
To further confirm the mechanism of Nrf2
knockdown-induced inhibition of GBM cell proliferation, we wondered
whether exogenous supplementation of the Nrf2-knockdown cells with
NAC or GMEE (a membrane-permeable derivative of GSH that can
supplement cellular GSH levels) may restore their intracellular
redox status and cell proliferation. When Nrf2-knockdown cell
cultures were supplemented on day 1 to day 4 with NAC, GMEE or
vehicle (PBS) at various concentrations (see Fig. 4A and B), we found that
supplementation with GMEE, but not NAC, restored cell proliferation
in the Nrf2-knockdown cells for the most part, although the treated
cells did not reach the same levels in proliferation as was
observed in the control and Sc groups (Fig. 4B). GMEE supplementation also
restored the cellular GSH levels that were decreased in the
Nrf2-deficient U251 cells (Fig.
4C). Notably, we found that supplementation of Nrf2-deficient
cells with NAC, a precursor of GSH, did not improve cell
proliferation (Fig. 4A), even
though it completely eliminated the high levels of ROS that were
otherwise present in the Nrf2-knockdown cells (Fig. 4D). These results collectively
suggest that, in addition to squelching ROS, intracellular GSH
levels are critical for the proliferation of Nrf2-knockdown
cells.
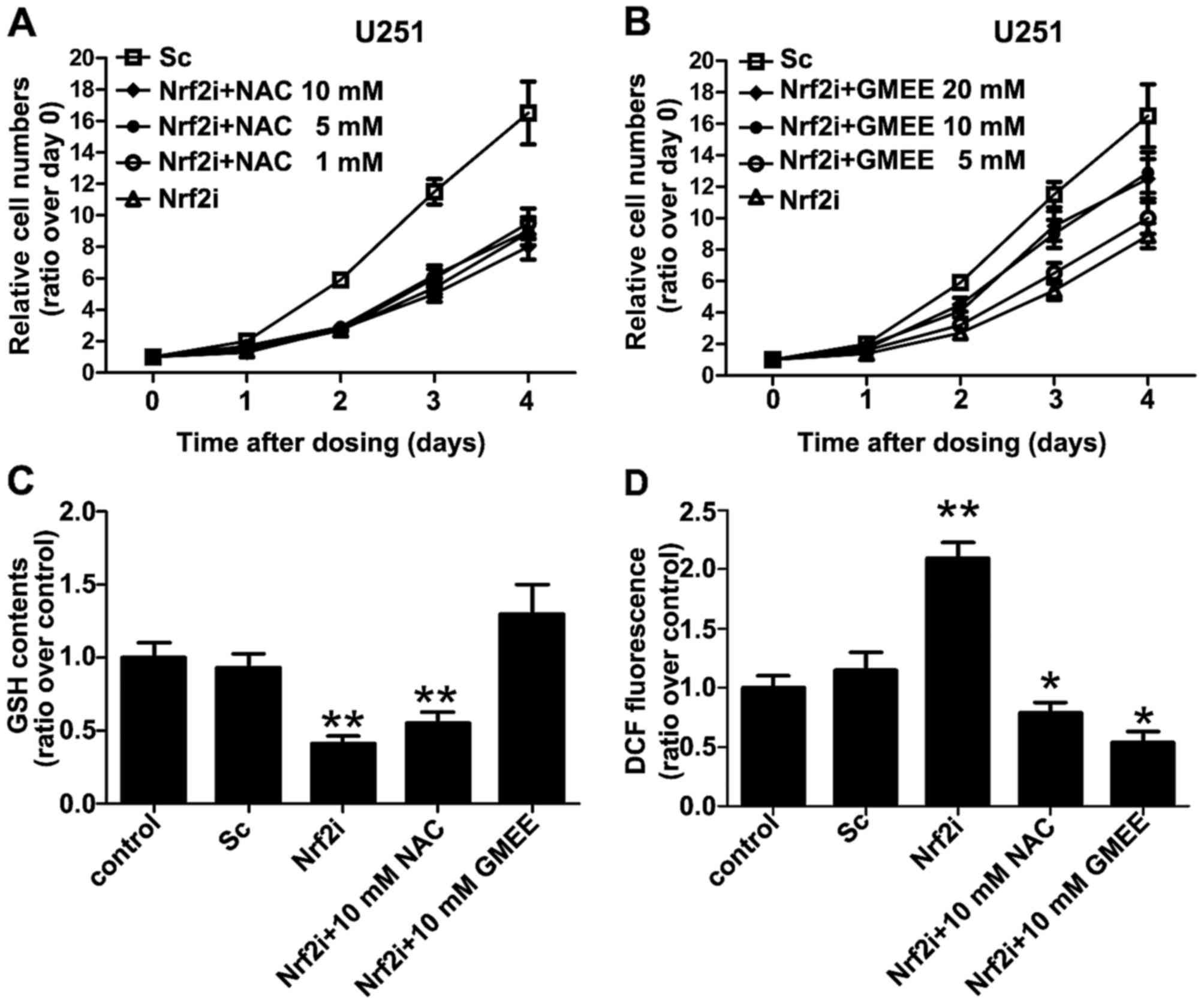 | Figure 4.Exogenous supplementation with
glutathione monoethyl ester (GMEE) restores Nrf2 deficiency-related
phenotypic defects such as cell proliferation. (A and B)
Supplementation with GMEE, but not NAC, mostly restored the arrest
in cell proliferation noted in the Nrf2-knockdown cells, although
the treated cells did not reach the same level as was observed in
the Sc group. Cells were grown in the absence or presence of GMEE
(5, 10 and 20 mM) and NAC (1, 5 and 10 mM) for 4 days and cell
proliferation was assessed every 24 h by CCK-8 assay. (C)
Supplementation of Nrf2-deficient U251 cells with GMEE, but not
NAC, restored normal GSH levels that were decreased in the
Nrf2-knockdown cells. Cells were grown in the absence or presence
of 10 mM GMEE or 10 mM NAC. On day 4 the cells were washed with
PBS, lysed and the GSH levels were determined; **P<0.01 as
compared with PBS (vehicle)-treated control group. (D) Following
supplementation of Nrf2-deficient U251 cells with GMEE or NAC, both
were able to eliminate the increased levels of ROS that were
present in the Nrf2-knockdown cells. Cells were grown in the
absence or presence of 10 mM GMEE or 10 mM NAC. On day 4 the cells
were washed with PBS, lysed and the relative levels of reactive
oxygen species (ROS) generation were determined using the
fluorescence probes DCFH-DA. Values are expressed as the mean ± SD
from 3 experiments; *P<0.05, **P<0.01 as compared with the
PBS (vehicle)-treated control group. |
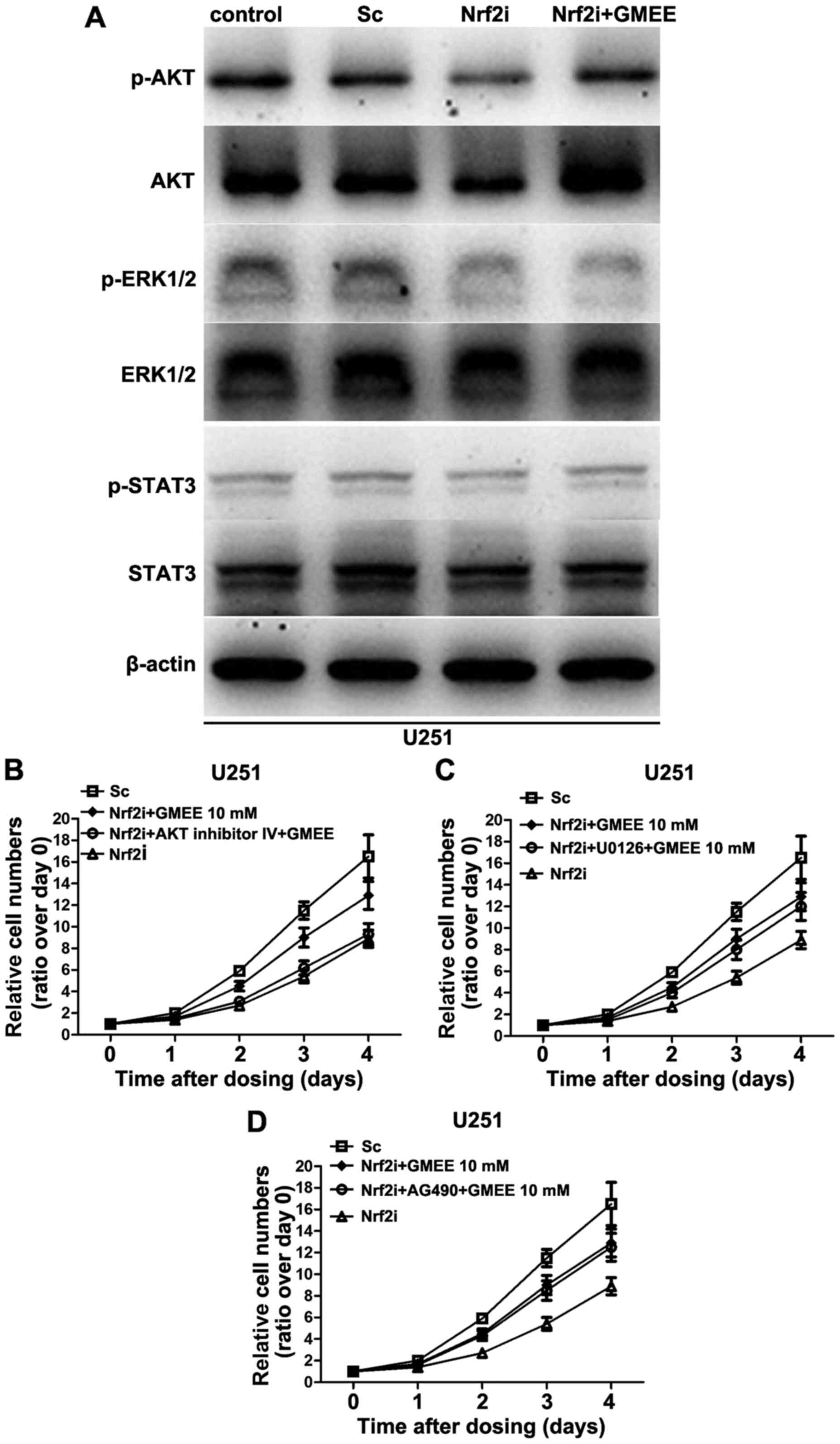
AKT signaling is critical for
GMEE-induced Nrf2-deficient U251 cell proliferation
To further delineate the mechanisms by which Nrf2
regulates cell proliferation, we examined the status of
intracellular signal transduction pathways, such as AKT, ERK1/2 and
STAT3, which are known to be important in cell proliferation. Since
exogenous supplementation of Nrf2-deficient cells with GMEE did not
increase cell proliferation to the same extent as was observed in
the counterpart (as shown in Fig.
4B), we reasoned that knockdown of Nrf2 in U251 cells may cause
the dysregulation of a series of signal transduction pathways, but
GMEE can only restore some, not all of them. In the present study,
we analyzed the phosphorylation status and protein levels of AKT,
ERK1/2 and STAT3 kinases in the Nrf2-deficient cells with or
without GMEE supplementation by immunoblotting. As shown in
Fig. 5A, Nrf2 depletion decreased
the phosphorylation status as well as the protein levels of AKT.
Nrf2 knockdown also significantly decreased the phosphorylation,
but not the level of expression of ERK1/2 kinases (Fig. 5A), whereas the phosphorylation
status and the protein levels of STAT3 exhibited no evident
alteration in the Nrf2-deficient U251 cells. Moreover, the
phosphorylation and expression levels of AKT kinase were restored
when the Nrf2-deficient U251 cell cultures were supplemented with
GMEE. However, the GMEE supplementation failed to restore the
phosphorylation status of ERK1/2 kinases that were also
dysregulated in the Nrf2-deficient cells. Collectively, Nrf2
deficiency caused the inhibition of the AKT and ERK1/2 pathways in
U251 cell lines, but exogenous supplementation of GMEE only
restored AKT but not ERK1/2 kinases.
To further verify the results of western blotting,
we performed a set of experiments with the AKT-specific inhibitor,
AKT inhibitor IV; the ERK inhibitor, U0126; and the STAT3
inhibitor, AG490. AKT inhibitor IV (5 µM), U0126 (10 µM) and AG490
(10 µM) were used to interrupt the signaling transduction of AKT,
ERK1/2 and STAT3 at concentrations that did not affect the cell
viability when applied alone (data not shown). We found that
inhibition of ERK1/2 and STAT3 had no significant effect on
GMEE-induced Nrf2-deficient cell proliferation (Fig. 5C and D). In contrast, blocking AKT
signaling with an AKT-specific inhibitor markedly blocked the
Nrf2-deficient cell proliferation induced by GMEE (Fig. 5B). Collectively, these results
strongly suggest that AKT signaling is important in GMEE-induced
Nrf2-deficient cell proliferation.
Discussion
In the present study, we observed the effects of
Nrf2 knockdown on cell proliferation, intracellular redox status
and the functions of cellular signal transduction pathways. We
found that Nrf2 deficiency led to a decrease in U251 cell
proliferation and caused intracellular redox imbalance (decreased
levels of GSH and increased levels of ROS). Moreover, Nrf2
depletion also resulted in the inhibition of AKT and ERK1/2
signaling. We further found that GMEE supplementation reversed the
Nrf2 deficient-induced cell growth arrest and restored
intracellular GSH levels. In addition, GMEE supplementation also
restored AKT signaling but not ERK1/2 signaling, although they were
both suppressed in the Nrf2-knockdown U251 cells. Notably, we
unexpectedly found that supplementation of cells with antioxidant
NAC appreciably decreased ROS levels, but it failed to counteract
the phenotypic defects associated with cell proliferation. Finally,
we provided evidence that the inhibition of ERK1/2 and STAT3 had no
significant effect on GMEE-induced Nrf2-deficient cell
proliferation. In contrast, blocking AKT signaling with an
AKT-specific inhibitor markedly blocked the Nrf2-deficient cell
proliferation induced by GMEE, confirming that AKT signaling is
important in GMEE-induced Nrf2-deficient cell proliferation. In the
present study, we first revealed that Nrf2 knockdown induced
inhibition of proliferation possibly by GSH depletion and
consequent AKT signaling inhibition, a new perspective by which to
understand the role of Nrf2 activation in cancer pathogenesis.
Of particular note is our observation that
supplementation of Nrf2-knockdown cells with the antioxidant
N-acetylcysteine (NAC), a precursor of GSH, markedly lowered
ROS levels but failed to induce cell proliferation. Several studies
have shown that NAC attenuates or provides protection against
pro-oxidant stimuli both in vitro and in vivo
(24,25). Consistent with these results, our
experiments also demonstrated that NAC could markedly squelch ROS
levels, but it was unable to restore the decreased intracellular
GSH levels that were present in Nrf2-knockdown U251 cells. The
inability of NAC to restore the decreased GSH levels in
Nrf2-knockdown cells is not surprising, since these cells expressed
decreased levels of the γ-glutamyl cysteinyl ligase (GCL)
components GCLC and glutathione reductase (GR), whose activities
are essential for GSH generation from its precursor NAC (26–28).
Although the sources contributing to the high levels of ROS in
Nrf2-knockdown cells remain to be investigated, it is likely that
decreased levels of GSH may lead to an inefficient cellular
detoxification of endogenous ROS levels generated by mitochondria
constitutively in Nrf2-silenced cells. In addition, it is possible
that Nrf2 deficiency may result in dysregulation of ROS generating
enzymes, such as Nox/Duox, Rac or RAS, thereby contributing to
increased levels of ROS. Based on the aforementioned results, we
directly treated the Nrf2-deficient cells with GMEE (a
membrane-permeable derivative of GSH that can supplement cellular
GSH levels) in culture medium. As expected, in addition to
squelching the high levels of ROS, GMEE supplementation reversed
Nrf2 deficiency-induced cell growth arrest and restored the
intracellular GSH levels. Thus, it is likely that Nrf2-knockdown
cells may only inefficiently convert NAC to GSH, a process that may
otherwise cause the intracellular GSH to rise to levels that are
adequate for cell proliferation.
To further define the mechanisms contributing to the
growth arrest caused by Nrf2 deficiency, we assessed the status of
the cellular signaling transduction pathways, since we reasoned
that decreased GSH levels in Nrf2-knockdown cells may cause a
dysregulation of signal transduction pathways that are related to
cell proliferation. This notion is based on recent studies that
have suggested a role for protein glutathionylation, a reversible
post-translational mechanism involved in the modulation of the
activities of the redox-sensitive thiol proteins, particularly
those involved in signal transduction and cell proliferation
(29). In the present study, we
found that suppression of Nrf2 expression could lead to AKT and
ERK1/2 pathway inhibition (the phosphorylation status of STAT3
exhibited no significant alterations), which are both known to be
important in cell proliferation. However, treatment of
Nrf2-knockdown cells with GMEE revealed that exogenous
supplementation with GMEE could only restore AKT signaling but not
ERK1/2 signaling, even though they were both suppressed in the
Nrf2-knockdown U251 cells. Knockdown of Nrf2 in U251 cells caused
the dysfunction of a series of signal transduction pathways, but
GMEE supplementation only restored them in part, not completely.
These observations could be used to explain the results obtained as
indicated in Fig 4 which revealed
that exogenous supplementation of Nrf2-deficient cells with GMEE
did not induce cell proliferation to the same extent as was
observed in the counterparts.
To further confirm these results, we finally
provided evidence that the inhibition of ERK1/2 and STAT3 had no
significant effects on GMEE-induced Nrf2-deficient cell
proliferation, whereas, blocking AKT signaling with an AKT-specific
inhibitor markedly blocked the Nrf2-deficient cell proliferation
induced by GMEE, confirming that AKT signaling is important in
GMEE-induced Nrf2-deficient cell proliferation. Our data are
consistent with a previous study (30) in which Reddy et al found that
Nrf2 deficiency impaired AKT signaling in primary alveolar
epithelial cultures isolated from Nrf2−/− mice. However,
this research group did not point out the precise crosstalk
mechanism between AKT signaling and Nrf2. In the present study, we
provide evidence of the crosstalk between these two pathways
suggesting that protein (AKT kinase) de-glutathionylation (induced
by intracellular decreased GSH levels) may be involved in the AKT
inhibition resulting from Nrf2 disruption.
Emerging studies have pointed out that Nrf2 and its
downstream target genes play a potential role in tumorigenesis, but
the detailed underlying mechanism of the biological effects of Nrf2
activation in cancer remain elusive. In the present study, we
revealed the decrease in Nrf2-impaired GBM cell proliferation via
GSH depletion and consequent AKT signaling inhibition. For the
first time the underlying mechanism of Nrf2 in cancer cell
proliferation from the perspective of cellular metabolism, which
enhances our comprehension of the role of Nrf2 in tumorigenesis was
elucidated. More significantly, knowledge of the association of
Nrf2 and cellular metabolism-related signaling pathways in cancer
cells, may provide new strategies for glioma clinical intervention,
particularly in the absence of effective Nrf2 inhibitors.
Acknowledgements
The present study was supported by grants from the
National Nature Science Foundation of China (nos. 81371357 and
81402072), and the China Postdoctoral Science Foundation Funded
Project (nos. 2014M562665 and 2015M572716).
Glossary
Abbreviations
Abbreviations:
|
AKT
|
protein kinase B
|
|
DCF
|
dichlorofluorescein
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
GBM
|
glioblastoma
|
|
GCLC
|
catalytic subunit of
γ-glutamylcysteinyl ligase
|
|
GMEE
|
glutathione monoethyl ester
|
|
GR
|
glutathione reductase
|
|
GSH
|
glutathione
|
|
HO-1
|
heme oxygenase-1
|
|
NAC
|
N-acetylcysteine
|
|
NHA
|
normal human astrocytes
|
|
NQO1
|
NAD(P)H:quinone oxidoreductase 1
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
ROS
|
reactive oxygen species
|
|
shRNA
|
short hairpin RNA
|
|
STAT3
|
signal transducers and activation of
transcription 3
|
References
|
1
|
Jolly C and Morimoto RI: Role of the heat
shock response and molecular chaperones in oncogenesis and cell
death. J Natl Cancer Inst. 92:1564–1572. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Singh KK: Mitochondrial dysfunction is a
common phenotype in aging and cancer. Ann NY Acad Sci.
1019:260–264. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mena S, Ortega A and Estrela JM: Oxidative
stress in environmental-induced carcinogenesis. Mutat Res.
674:36–44. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gottlieb E and Tomlinson IP: Mitochondrial
tumour suppressors: A genetic and biochemical update. Nat Rev
Cancer. 5:857–866. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Motohashi H and Yamamoto M: Nrf2-Keap1
defines a physiologically important stress response mechanism.
Trends Mol Med. 10:549–557. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gañán-Gómez I, Wei Y, Yang H,
Boyano-Adánez MC and García-Manero G: Oncogenic functions of the
transcription factor Nrf2. Free Radic Biol Med. 65:750–764. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malhotra D, Portales-Casamar E, Singh A,
Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler
TW, Wasserman WW, et al: Global mapping of binding sites for Nrf2
identifies novel targets in cell survival response through ChIP-Seq
profiling and network analysis. Nucleic Acids Res. 38:5718–5734.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wild AC, Moinova HR and Mulcahy RT:
Regulation of gamma-glutamylcysteine synthetase subunit gene
expression by the transcription factor Nrf2. J Biol Chem.
274:33627–33636. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sasaki H, Sato H, Kuriyama-Matsumura K,
Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M and Bannai
S: Electrophile response element-mediated induction of the
cystine/glutamate exchange transporter gene expression. J Biol
Chem. 277:44765–44771. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Itoh K, Chiba T, Takahashi S, Ishii T,
Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et
al: An Nrf2/small Maf heterodimer mediates the induction of phase
II detoxifying enzyme genes through antioxidant response elements.
Biochem Biophys Res Commun. 236:313–322. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishii T, Itoh K, Takahashi S, Sato H,
Yanagawa T, Katoh Y, Bannai S and Yamamoto M: Transcription factor
Nrf2 coordinately regulates a group of oxidative stress-inducible
genes in macrophages. J Biol Chem. 275:16023–16029. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aoki Y, Hashimoto AH, Amanuma K, Matsumoto
M, Hiyoshi K, Takano H, Masumura K, Itoh K, Nohmi T and Yamamoto M:
Enhanced spontaneous and benzo(a)pyrene-induced mutations in
the lung of Nrf2-deficient gpt delta mice. Cancer Res.
67:5643–5648. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K,
Yamamoto M, Talalay P and Kensler TW: Sensitivity to carcinogenesis
is increased and chemoprotective efficacy of enzyme inducers is
lost in nrf2 transcription factor-deficient mice. Proc Natl
Acad Sci USA. 98:3410–3415. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lau A, Villeneuve NF, Sun Z, Wong PK and
Zhang DD: Dual roles of Nrf2 in cancer. Pharmacol Res. 58:262–270.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hayes JD and McMahon M: NRF2 and
KEAP1 mutations: Permanent activation of an adaptive
response in cancer. Trends Biochem Sci. 34:176–188. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hayes JD and McMahon M: The double-edged
sword of Nrf2: Subversion of redox homeostasis during the evolution
of cancer. Mol Cell. 21:732–734. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kensler TW and Wakabayashi N: Nrf2: Friend
or foe for chemoprevention? Carcinogenesis. 31:90–99. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Homma S, Ishii Y, Morishima Y, Yamadori T,
Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N,
et al: Nrf2 enhances cell proliferation and resistance to
anticancer drugs in human lung cancer. Clin Cancer Res.
15:3423–3432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamadori T, Ishii Y, Homma S, Morishima Y,
Kurishima K, Itoh K, Yamamoto M, Minami Y, Noguchi M and Hizawa N:
Molecular mechanisms for the regulation of Nrf2-mediated cell
proliferation in non-small-cell lung cancers. Oncogene.
31:4768–4777. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lister A, Nedjadi T, Kitteringham NR,
Campbell F, Costello E, Lloyd B, Copple IM, Williams S, Owen A,
Neoptolemos JP, et al: Nrf2 is overexpressed in pancreatic cancer:
Implications for cell proliferation and therapy. Mol Cancer.
10:372011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ji XJ, Chen SH, Zhu L, Pan H, Zhou Y, Li
W, You WC, Gao CC, Zhu JH, Jiang K, et al: Knockdown of
NF-E2-related factor 2 inhibits the proliferation and growth of
U251MG human glioma cells in a mouse xenograft model. Oncol Rep.
30:157–164. 2013.PubMed/NCBI
|
|
22
|
Pan H, Wang H, Zhu L, Mao L, Qiao L and Su
X: The role of Nrf2 in migration and invasion of human glioma cell
U251. World Neurosurg. 80:363–370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji X, Wang H, Zhu J, Zhu L, Pan H, Li W,
Zhou Y, Cong Z, Yan F and Chen S: Knockdown of Nrf2 suppresses
glioblastoma angiogenesis by inhibiting hypoxia-induced activation
of HIF-1α. Int J Cancer. 135:574–584. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thimmulappa RK, Lee H, Rangasamy T, Reddy
SP, Yamamoto M, Kensler TW and Biswal S: Nrf2 is a critical
regulator of the innate immune response and survival during
experimental sepsis. J Clin Invest. 116:984–995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rangasamy T, Guo J, Mitzner WA, Roman J,
Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, et
al: Disruption of Nrf2 enhances susceptibility to severe
airway inflammation and asthma in mice. J Exp Med. 202:47–59. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hutter DE, Till BG and Greene JJ: Redox
state changes in density-dependent regulation of proliferation. Exp
Cell Res. 232:435–438. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pani G, Colavitti R, Bedogni B, Anzevino
R, Borrello S and Galeotti T: A redox signaling mechanism for
density-dependent inhibition of cell growth. J Biol Chem.
275:38891–38899. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Menon SG, Sarsour EH, Spitz DR,
Higashikubo R, Sturm M, Zhang H and Goswami PC: Redox regulation of
the G1 to S phase transition in the mouse embryo
fibroblast cell cycle. Cancer Res. 63:2109–2117. 2003.PubMed/NCBI
|
|
29
|
Dalle-Donne I, Carini M, Vistoli G,
Gamberoni L, Giustarini D, Colombo R, Facino Maffei R, Rossi R,
Milzani A and Aldini G: Actin Cys374 as a nucleophilic target of
α,β-unsaturated aldehydes. Free Radic Biol Med. 42:583–598. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reddy NM, Kleeberger SR, Bream JH, Fallon
PG, Kensler TW, Yamamoto M and Reddy SP: Genetic disruption of the
Nrf2 compromises cell-cycle progression by impairing GSH-induced
redox signaling. Oncogene. 27:5821–5832. 2008. View Article : Google Scholar : PubMed/NCBI
|