Introduction
The polycomb group (PcG) of transcription factor
proteins form transcriptional repressor modules that play crucial
roles in many physiological processes, including cell
differentiation, stem cell self-renewal, and gene silencing through
histone modifications (1). Numerous
studies have shown that PcG proteins are involved in malignant
transformation and tumor development in various cancer types
(2). B cell-specific Moloney murine
leukemia virus integration site 1 (BMI1), a member of the PcG
complex, plays an essential role in the maintenance and
self-renewal of hematopoietic and neural stem cells, at least
partly by silencing the Ink4a/Arf locus (3,4). BMI1
has also been linked with a multitude of cellular processes,
including cell cycle progression, apoptosis,
epithelial-to-mesenchymal transition (EMT), senescence,
immortalization and/or induction of telomerase (5–7). BMI1
overexpression is associated with disease progression and poor
clinical outcome in a number of human malignancies (8–11).
Although BMI1 plays a critical role in cancer, the precise
molecular mechanism by which it contributes to cancer development
and therapy failure remains poorly understood.
Several independent studies have demonstrated that
genetic silencing and pharmacologic inhibition of BMI1 suppresses
the growth of various cancers, induces cell cycle arrest, apoptosis
and senescence, and increases susceptibility to chemotherapeutic
agents and ionizing radiation (12–14).
In normal human keratinocytes, BMI1 elicits radioprotective effects
by mitigating the genotoxic effects of ionizing radiation (IR)
(15). In nasopharyngeal carcinoma
cells, targeting BMI1 expression increases their susceptibility to
radiation through the induction of oxidative stress and apoptosis
(13). Elevated expression of BMI1
has been shown to radioprotect CD133-positive cancer-initiating
neural stem cells through recruitment of DNA damage response (DDR)
machinery to DSBs after exposure to radiation (16). Although a role for BMI1 in cancer
progression and its importance as a target for therapy has been
reported, its role in radiosensitization of breast cancer has not
been investigated.
In the present study, we demonstrate that silencing
BMI1 sensitizes MDA-MB-231 and SUM159PT breast cancer cells to
ionizing radiation. We also show that this sensitization occurs
through induction of both the DDR and autophagy pathways. These
results indicate that BMI1 may play an important role in
radioresistance, and that BMI1 suppression may be an important
therapeutic target for breast cancer.
Materials and methods
Cell lines
Human MDA-MB-231 breast cancer cell line obtained
from American Type Culture Collection (ATCC; Manassas, VA, USA) was
maintained in α-MEM (Cellgro, Manassas, VA, USA) containing 10%
fetal bovine serum, 2 mmol/l L-glutamine, and 2 mmol/l
penicillin-streptomycin. SUM159PT cells were obtained from Asterand
Bioscience (Detroit, MI, USA) and maintained in Ham's F-12 media
supplemented with 5% heat-inactivated FBS, 2 mmol/l
penicillin-streptomycin, 10 mM Hepes, and 1 µg/ml insulin. All
cultures were maintained at 37°C in an atmosphere of 5%
CO2 and 95% room air.
Plasmid construction
Sequences (miR shControl: Sense
5′-AGCGATCTCGCTTGGGCGAGAGTAAGTATGAAGCCACAGATGTGACTTACTCTCGCCCAACGAGAG-3′,
Antisense
5′-GGCAACTCTCGCTTGGGCGAGAGTAAGTACATCTGTGGCTTCACTACTTACTCTCGCCCAAGCGAGAT-3′;
miR shBMI1: Sense
5′-AGCGATCCAAGATATTGTATACAAATTAGTGAAGCCACAGATGTAATTTGTATACAATATCTTGGAG-3′,
Antisense
5′-GGCACTCCAAGATATTGTATACAAATTACATCTGTGGCTTCACTAATTTGTATACAATATCTTGGAT-3′)
were cloned into pEN_RmiRc2 (17).
Then, entry vectors containing shRNA sequence were recombined with
the lentiviral destination vector CMV PURO DEST according to
manufacturer's recommendations (Invitrogen, Grand Island, NY,
USA).
siRNA transfection
Transfection of SUM159PT cells with human BMI1 siRNA
(siBMI1) and non-targeting siRNA#3 (siScr) (GE Dharmacon,
Lafayette, CO, USA) was performed in 60-mm dishes using DharmaFECT
2 transfection reagent (GE Dharmacon) according to manufacturer's
instructions. Cells were transfected with siRNA (20 nM) in
serum-free medium. Six hours after transfection, the media was
replaced with fresh medium containing 2% serum. The next day the
cells were irradiated (5 Gy) and harvested after specified
incubation periods for further experiments.
Wound healing assay
MDA-MB-231 shControl and shBMI1 cells were seeded in
60-mm dishes and grown to 80–85% confluence. After 24 h of
incubation the cell monolayers were wounded longitudinally using a
200 µl pipette tip. Cells were washed once to remove detached and
injured cells and were visually assessed immediately, with live
images captured by light microscopy using an attached CCD camera.
After an additional 24 and 48 h incubation, cells were stained with
crystal violet and pictures were taken at the same location as the
initial image to determine any defects in cell migration.
Experiments were done in triplicate. Three random fields of each
well were recorded.
Cell migration assay
Cells were seeded in the upper chamber of the
Transwell (8 µm; BD Biosciences, Bedford, MA, USA) in medium
containing 2% FBS and placed in a 6-well plate filled with 2 ml of
medium containing 20% FBS (lower chamber). After 24, 48, and 72 h
incubation, the inserts were removed, processed, and the number of
migrated cells counted as previously described (18).
Clonogenic survival
The effectiveness of BMI1 knockdown (in both shBMI1
and siBMI1-treated cells) and ionizing radiation was assessed with
clonogenic assays as previously described (19,20).
Cells were irradiated with a high dose-rate 137Cesium
(Cs) unit at room temperature. After treatment, known number of
cells were re-plated in triplicate in 60-mm tissue culture dishes
and returned to the incubator to allow macroscopic colony
development. Ten-to-twelve days after seeding, colonies were
stained with 0.5% gentian violet in methanol. The number of
colonies formed in each treatment group was counted, with a cutoff
of 50 viable cells per colony. The percent plating efficiency (PE)
and fraction surviving a given treatment was calculated based on
the survival of non-irradiated control or BMI1 knockdown cells.
Survival curves were generated after normalizing for the
cytotoxicity generated by control alone.
Western blot analysis
Protein extracts, separated by SDS-PAGE and
transferred onto PVDF membranes, were probed with antibodies
against LC3B, p62 (Santa Cruz Biotechnology, Dallas, TX, USA),
BMI1, p38, phospho-p38, Akt, phospho-Akt, Bcl2, Mcl1, MRE11, Bax,
DNA-PK, Ku70, Ku80 (Cell Signaling Technology, Beverly, MA, USA),
and ATM (Gene Tex, Irvine, CA, USA). β-actin was used as an
internal loading control (Santa Cruz Biotechnology). Proteins were
detected using the appropriate HRP-conjugated secondary antibodies
and visualized by enhanced chemiluminescence with ECL Plus reagent
(Amersham Pharmacia Biotech, Arlington Heights, IL, USA).
Quantitative polymerase chain
reaction
Total RNA was isolated using TRIzol (Life
Technologies, Grand Island, NY, USA) and subjected to reverse
transcription using the Omniscript RT kit (Qiagen Inc., Hercules,
CA, USA). Obtained cDNA was used to perform real-time (RT)-PCR
(Bio-Rad CFX96™ Touch Real-time PCR Detection System) with PerfeCTa
SYBR Green Fast Mix (Quanta BioSciences, Gaithersburg, MD, USA).
The primers for human BMI1, Atg3, Atg5, Atg7, Atg12, LC3A, LC3B,
and GAPDH were obtained from IDT Inc. (Coralville, IA, USA). The
comparative Ct (cycle threshold) method was used to calculate the
relative abundance of mRNA compared with that of GAPDH expression
in triplicate experiments.
Immunofluorescent staining for
γ-H2AX
Cells were grown for 24 h on coverslips. At
specified times, cells were treated with 2 Gray (Gy) radiation,
fixed and stained as previously described (19,20).
Stained slides were imaged using a Nikon fluorescence microscope
with an attached CCD camera using NIS-Elements imaging software
(Nikon Instruments, Melville, NY, USA). For each treatment, γ-H2AX
foci were counted in at least 50 cells from the stored images.
Comet assay
DSBs were detected using a Comet Assay kit
(Trevigen, Gaithersburg, MA, USA) according to the manufacturer's
instructions. Briefly, cells were grown on 35-mm dishes.
Twenty-four hours later, cells were irradiated at 20 Gy, harvested
at specified time points, washed twice, resuspended at
1×105 cells/ml in ice-cold 1X PBS, combined with
low-melting (LM) agarose at a ratio of 1:10 (v/v) and spread on the
Comet slide. The slides were allowed to solidify for 30 min in the
dark at 4°C and then submerged in precooled, neutral lysis buffer
at 4°C overnight. After lysis, slides were washed in 1X TBE buffer
[0.89 mol/l Tris, 0.88 mol/l boric acid, 2 mmol/l EDTA (pH 8.3)]
for 15 min and subjected to electrophoresis at 1.0 V/cm for 45 min.
The slides were then rinsed with distilled water, placed in 70%
ethanol for 5 min, dried at 37°C for 15 min and stained with SYBR
Green for 30 min in the dark. Comet images were obtained using a
Nikon fluorescence microscope with an attached CCD camera and
NIS-Elements imaging software (Nikon Instruments) and analyzed
using Casplab comet assay software. The Olive tail moment was
determined for 50 cells in each sample.
Statistical analysis
Data analysis was performed using the paired t-test.
Data are presented as the mean ± standard deviation at least for
three independent experiments. p<0.05 was considered to indicate
a statistically significant difference.
Results
BMI1 downregulation reduces the
migration potential of MDA-MB-231 cells
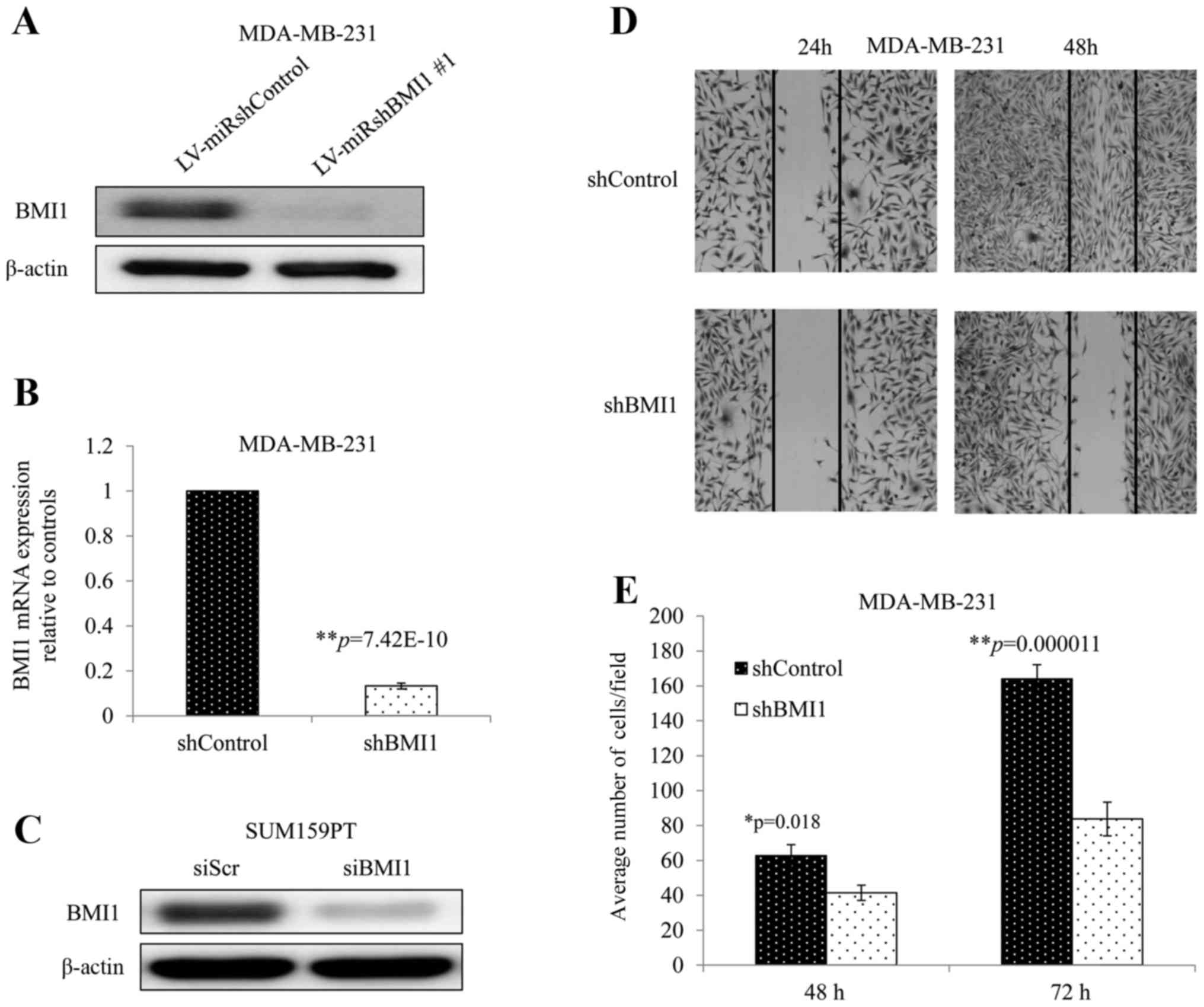
MDA-MB-231 cells transduced with a vector expressing
an shRNA against human BMI1 (shBMI1 cells) demonstrated greatly
reduced BMI1 protein and mRNA expression compared with those
transduced with a vector control (shControl cells) (Fig. 1A and B). Similarly, siBMI1 reduced
BMI1 protein levels significantly in SUM159PT cells when compared
with non-targeted siRNA (siScr) (Fig.
1C). To investigate whether BMI1 knockdown affects cellular
migration, we performed the wound healing and Transwell assays. The
wound healing assay showed that inhibition of BMI1 dramatically
impaired the wound healing rate in MDA-MB-231 cells as observed in
their ability to fill the gap at 24 and 48 h (Fig. 1D). The Transwell assay showed a
significant reduction in the migration potential in shBMI1 cells
with average cells per field decreasing by 34%, and 49% compared
with shControl cells at 48 and 72 h, respectively (Fig. 1E; p<0.02). These two assays
showed that inhibition of BMI1 impaired cell migration in
MDA-MB-231, a very important hallmark in cancer progression.
Knockdown of BMI1 sensitizes breast
cancer cells to radiation and amplifies DNA damage and prolongs
repair
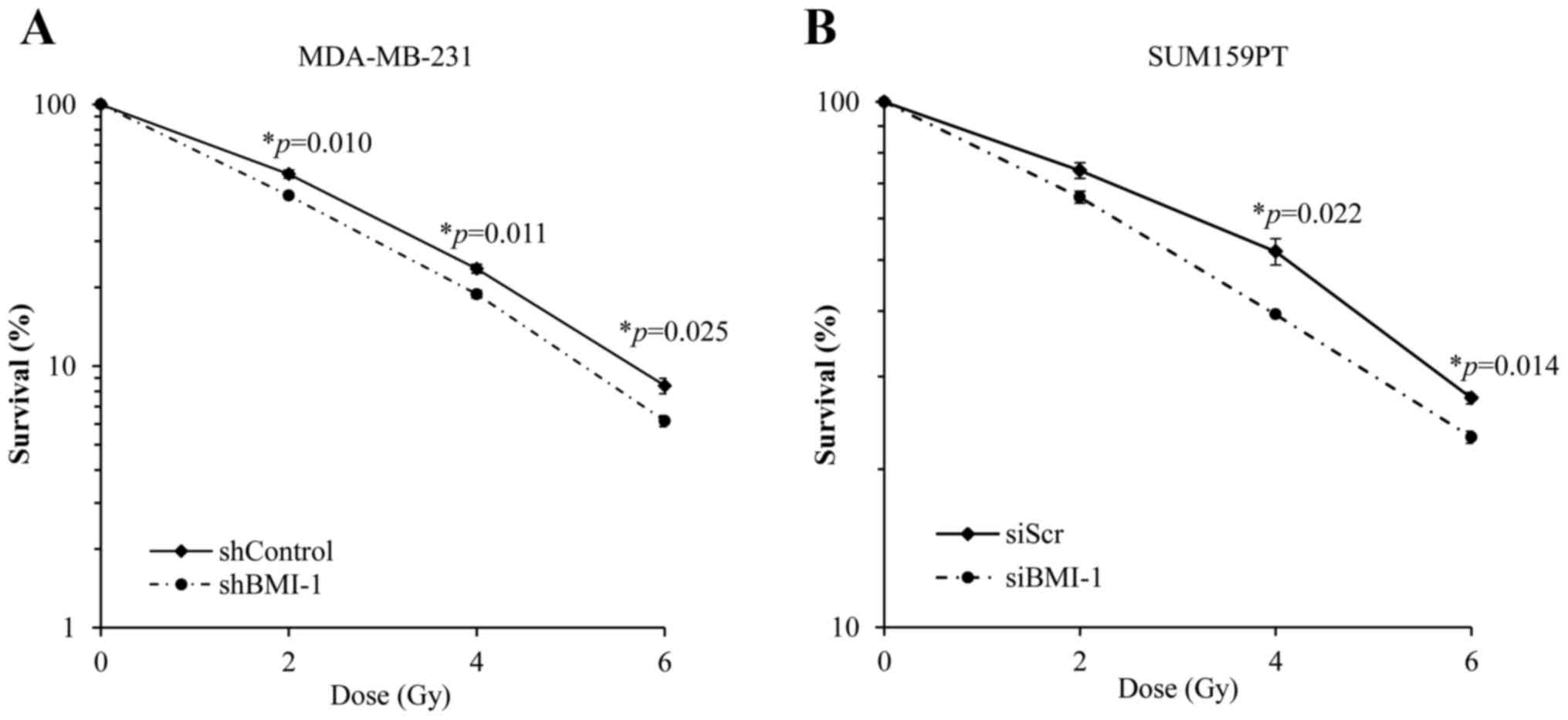
We investigated the effect of silencing BMI1 on the
radiosensitivity of MDA-MB-231 and SUM159PT cells by assessing
their clonogenic potential. BMI1 knockdown was achieved in
MDA-MB-231 cells using an shRNA against BMI1 (as described above)
and in SUM159PT cells using a BMI1-specific siRNA. We found that
silencing BMI1 suppressed the clonogenic survival and
radiosensitized MDA-MB-231 cells (Fig.
2A). Survival at 2 Gy (SF2) was reduced from 54±2% in the
shControl cells to 45±1% in shBMI1 (p=0.01) cells. Dose enhancement
factor was then calculated by dividing the radiation dose that
produced 10% cell survival on the radiation-only survival curve by
that for the corresponding shBMI1 plus ionizing radiation curve.
The dose enhancement factor for MDA-MB-231 cells was 1.12.
Similar results were obtained using the SUM159PT
cell line following treatment with siBMI1. Treatment with siBMI1
resulted in an increase in the radiosensitivity of SUM159PT cell
line as compared with siScr (Fig.
2B). Exposure of SUM159PT cells to siBMI1 reduced the surviving
fraction at 2 Gy from 74±2.5% in the siScr to 65.8±1.7% in
siBMI1-treated cells (p=0.08). The dose enhancement factor for
SUM159PT cells, calculated at 50% survival, was 1.36.
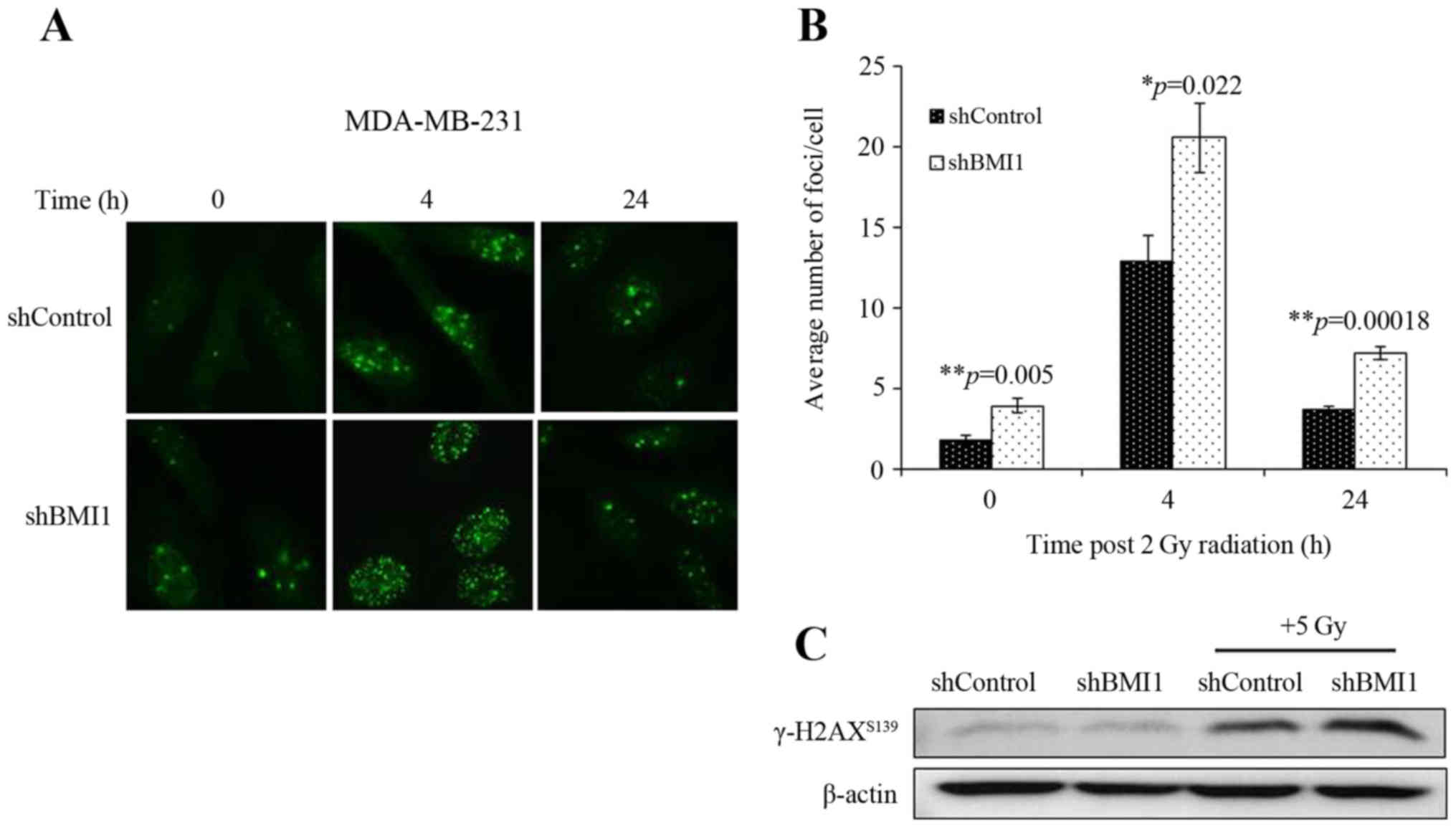
We also assessed the involvement of the DDR in
shBMI1 radiosensitization by determining the number of γ-H2AX foci,
an indicator of radiation-induced DSBs. shControl and shBMI1 cells
were treated with radiation (2Gy) and immunostained at 4 and 24 h
post-radiation. γ-H2AX foci could be clearly distinguished after
irradiation (2 Gy) as shown by the micrographs in Fig. 3A. The average number of γ-H2AX foci
per cell were counted and the results are presented in Fig. 3B. At 0 h (no irradiation), the
average number of γ-H2AX foci per cell was significantly greater in
the BMI1-silenced population than in the shControl (p=0.005;
Fig. 3B). After irradiation with 2
Gy, the average number of γ-H2AX foci in shBMI1 cells significantly
increased compared to the shControl-radiation group at 4 h
(p=0.022) and 24 h (p=0.00018; Fig.
3B). An increase in γ-H2AX levels in the shBMI1 plus radiation
group was also observed by western blot analysis (Fig. 3C). The sustained increase in γ-H2AX
foci after radiation in BMI1-silenced cells suggests that
shBMI1-mediated radiosensitization involves an enhancement of the
DDR and inhibition of DNA repair.
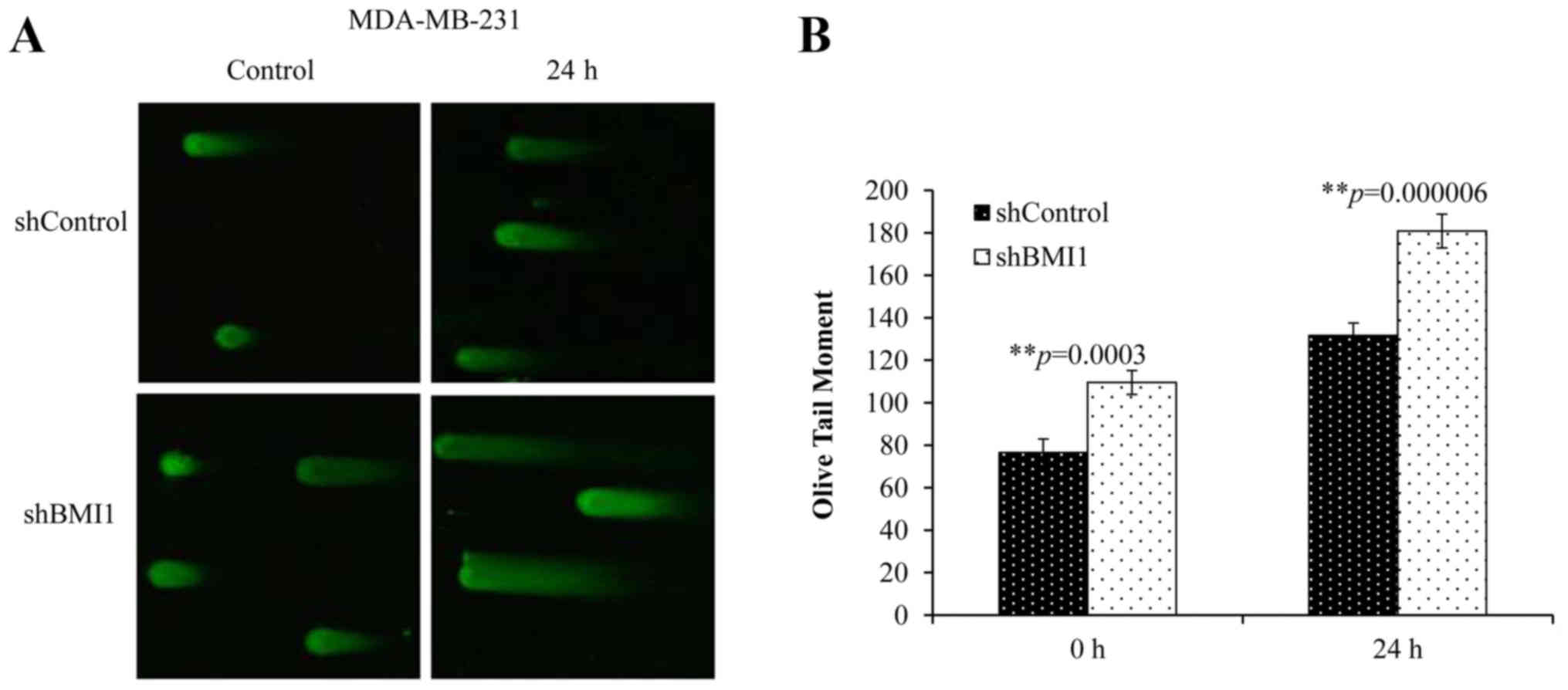
To further understand the extent of BMI1′s influence
on DSBs, we performed the neutral Comet assay. This technique
allows the quantification of DSBs by measuring the length and area
of the ‘comet’ represented by the ‘Olive tail moment’ (OTM). A
greater tail length and a greater tail area indicate that more DNA
breaks have occurred. Elevated basal levels of DNA damage (no
radiation) were observed in shBMI1 cells compared with the
shControl cells (Fig. 4A and B).
Combined shBMI1 plus radiation (20 Gy), however, resulted in
greater DNA damage at 24 h after radiation (Fig. 4B; p<0.001), suggesting that the
repair of radiation-induced DNA damage can be significantly
suppressed upon BMI1 inhibition in MDA-MB-231 cells.
BMI1 modulates expression of DDR and
the Akt and MAPK pathways
In view of the increased DNA damage and modulated
repair processes observed in the neutral Comet assay and γ-H2AX
foci data, we examined the DNA damage response and repair proteins
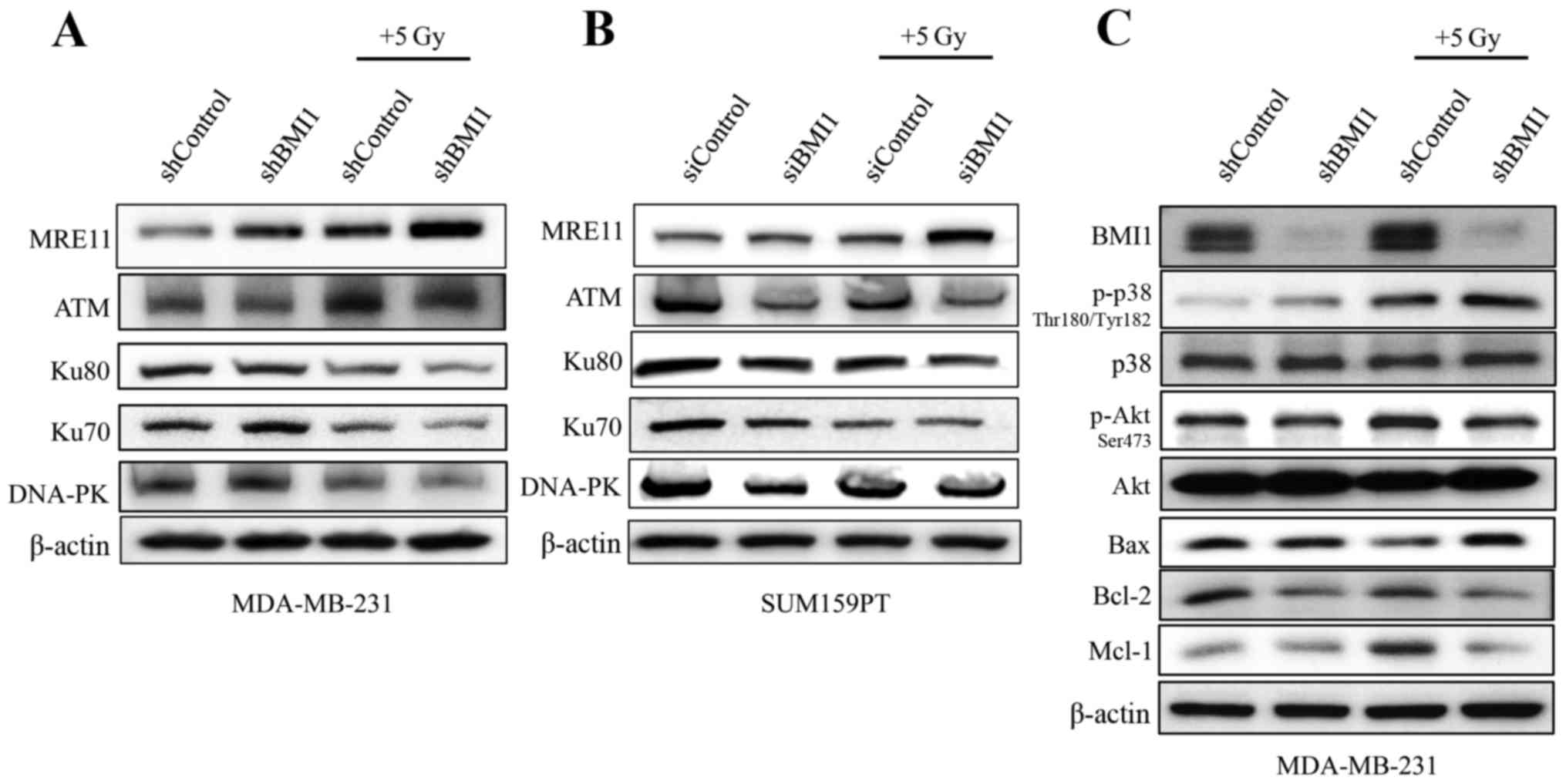
as supportive evidence. Immunoblot analysis showed that BMI1
silencing reduced the levels of ATM which were further reduced in
combination with radiation in both the cell lines (Fig. 5A and B). The expression profiles of
DNA-PK, Ku70 and Ku80, proteins involved in DNA repair, were also
markedly reduced upon BMI1 inhibition and radiation in both
MDA-MB-231 cells and the SUM159PT cells (Fig. 5A and B). However, the reduction in
the expression of the individual proteins varied between the two
cell lines. Expression of MRE11 was higher in shBMI1 cells treated
with radiation, indicating that the maintenance of DNA damage
signaling is likely perturbed.
In addition to increased DNA damage, shBMI1 cells
demonstrated a marked decrease in the expression profiles of
anti-apoptotic proteins Mcl1 and Bcl2, and an increase in
pro-apoptotic Bax when combined with radiation (Fig. 5C). The regulation of apoptotic
proteins can occur through two main pathways: the Akt pathway,
which promotes survival by positively regulating the expression of
anti-apoptotic proteins; and the MAPK pathway, which leads to the
activation of pro-apoptotic proteins. To evaluate the effect of
BMI1 downregulation on these pathways, we analyzed the
phosphorylation levels of Akt and p38MAPK. We found that knockdown
of BMI1 alone increased the activity of pp38Thr180/Tyr182 and these
levels were further increased after radiation (Fig. 5C). In addition, pAktSer473 levels
decreased with BMI1 knockdown and decreased further after exposure
to 5 Gy radiation. These results suggest that perturbation of DNA
damage signaling due to impaired activation of ATM in shBMI1 cells
leads to accumulation of DNA damage and activation of the p38 MAPK
pathway, which in turn modulates the expression of proteins of the
apoptotic machinery. Concomitantly, reduced pAKT, Bcl-2 and Mcl1
protein expression was observed in shBMI1 plus radiation.
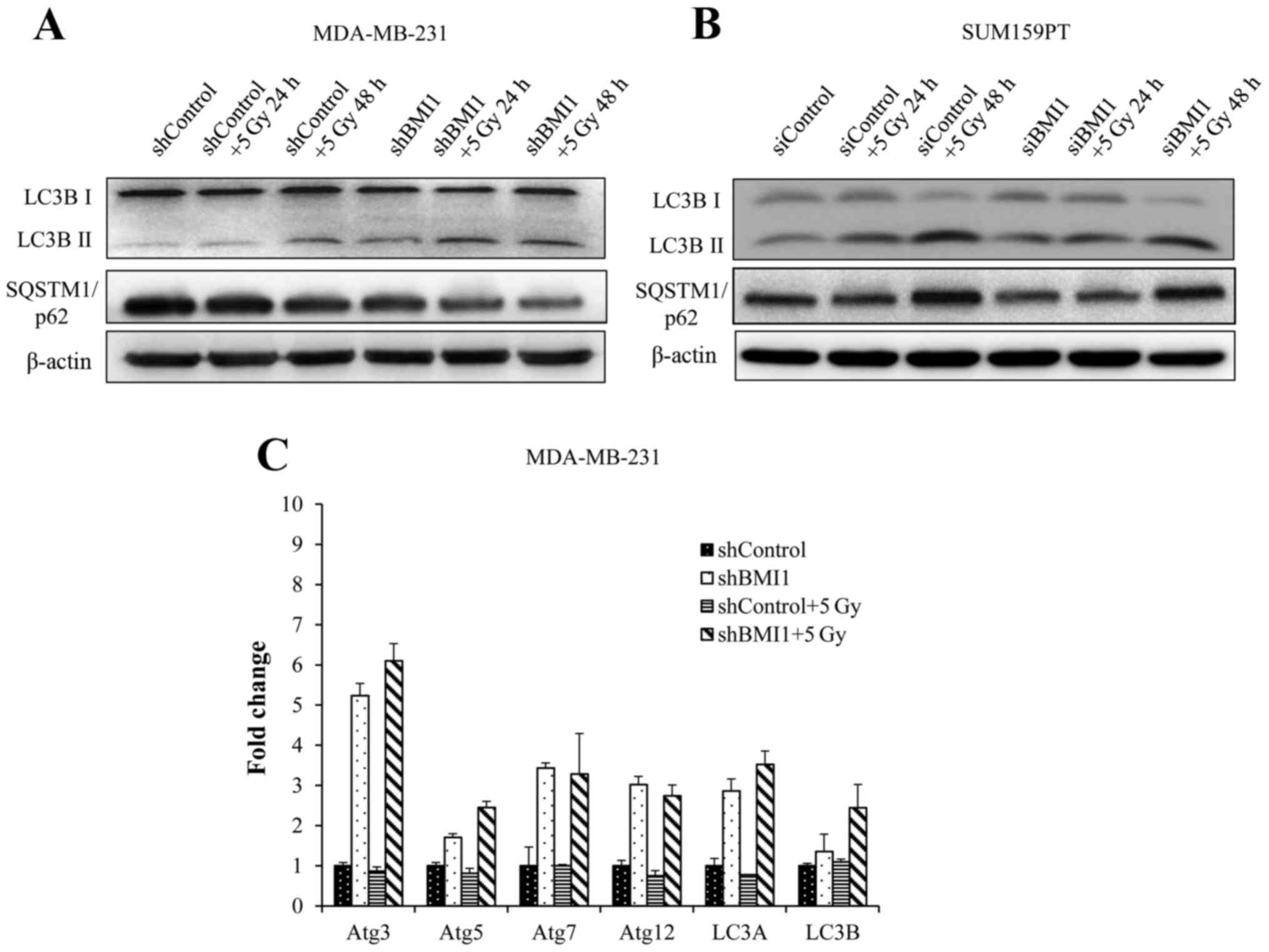
BMI1 influences autophagy
Autophagy is typically associated with survival
after genotoxic stress (21). Our
results showed activation of the autophagic processes in shBMI1 and
siBMI1 treated cells without radiation, as indicated by an increase
in LC3B-II (Fig. 6A and B).
Twenty-four hours after radiation, we observed further enhancement
in LC3B-II expression, continuing through 48 h in the shBMI1 and
the siBMI1 knockdown cells, indicating BMI1′s regulatory role in
autophagy. The shControl cells began to show increase in LC3B-II
above the basal level only after radiation treatment followed by 48
h incubation (Fig. 6A and B).
SQSTM1 (also known as p62) is known to associate with LC3 and
facilitate the delivery of ubiquitinated proteins to autophagic
complexes. As autophagy progresses, p62 and targeted proteins are
degraded. BMI1 knockdown cells were observed to have lower basal
expression of p62 than shControl cells. This expression further
decreases over time after radiation in the shBMI1 as well as the
siBMI1 treated cells (Fig. 6A and
B). We also carried out qRT-PCR to examine the expression of
autophagy-related genes (ATG) in shBMI1 cells, and observed marked
upregulation in ATG3, ATG5, ATG7, ATG12, LC3A, and LC3B mRNAs when
BMI1 was inhibited, further supporting our findings that genetic
inhibition of BMI1 makes breast cancer cells more prone to cell
death by autophagy (Fig. 6C).
Discussion
BMI1 is an important regulator of cancer stem cell
(CSC) phenotype and is often overexpressed in cancer cells leading
to resistance to therapy (10,16,22).
While several studies have shown that BMI1 inhibition increased
susceptibility to cytotoxic agents and radiotherapy, the role of
BMI1 in radiation response, especially in breast cancer, remains
unknown. A number of mechanisms could account for this resistance,
including the tumor microenvironment and altered expression of DDR
and DNA repair pathway components. In order to explore the
mechanisms responsible for radiation resistance in breast cancer,
we first showed that silencing of BMI1 increased the sensitivity of
MDA-MB-231 and SUM159PT cells to radiation in vitro. While
our clonogenic survival data showed that silencing BMI1 resulted in
radiosensitization, the underlying mechanism is still unknown.
Ionizing radiation mainly exerts its therapeutic effect by causing
DNA-DSBs, which are largely responsible for most ionizing
radiation-induced cellular lethality (23,24).
Hence, the ability of cells to recognize and respond to DSBs is
fundamental in determining the sensitivity (or resistance) of cells
to radiation. The non-homologous end-joining (NHEJ) pathway is an
important pathway for repairing radiation-induced DSBs (23–25).
Loss or mutations in any of its subunits (DNA-PK, Ku70 or Ku80) can
lead to extreme radiosensitivity and DSB repair deficiency
(23). We examined the expression
levels of DNA-PK, Ku70, and Ku80, and found them to be markedly
reduced in BMI1 silenced cells after irradiation (Fig. 5A and B). We also assessed the levels
of ATM, key protein in the (HR) pathway in eukaryotes. BMI1
inhibition suppressed ATM levels upon combination with radiation,
indicating that inhibition of the NHEJ and HR pathway, when
targeting BMI1, could be a mechanism underlying the observed
radiosensitization.
Since BMI1 knockdown produced a reduction in the DNA
repair proteins, we hypothesized that the radiosensitizing effect
is due to a delay in the repair of radiation-induced DSBs with
involvement of the DDR pathway. Several published works have used
the phosphorylation of H2AX (γ-H2AX) and its clearance after damage
as a measure of radioresistance (19,20).
To evaluate a potential delay in DNA damage repair, we assessed
γ-H2AX levels and found that BMI1 inhibition greatly impairs the
ability of MDA-MB-231 cells to repair radiation-induced DNA damage
as observed by the persistent levels of γ-H2AX in shBMI1 cells
(Fig. 3). As an additional measure
of the effects of shBMI1 on radiation-induced DSBs, we used the
Comet assay, which under neutral pH conditions selectively detects
DSBs over single-strand breaks. Our data revealed an increased
magnitude of DNA damage in BMI knockdown cells compared with
control cells (Fig. 4). These
results provide additional evidence of defective DSB repair and
offer a partial mechanism for the radiosensitization observed in
clonogenic cell survival assays.
Several researchers have evaluated the relationship
between BMI1, mitochondrial function, and programmed cell death
(PCD) (26). Though the links
between type 1 PCD (apoptosis) and type 2 PCD (autophagy) are well
established, the question of whether autophagy occurs as a final
survival attempt or directly results in cell death is a point of
contention for many experts. For some cancers, the induction of
autophagy results in radioresistance through enhanced clearance of
damaged organelles and protection from cytosolic acidification
(27). Induction of autophagy
through downregulation of Akt/mTOR sensitizes other cancers to
radiation therapy (28). Of note,
p38 MAPK also appears to play a role in the switch between
autophagy and apoptosis, and is capable of acting as both a
positive and negative regulator of autophagy (29). A recent study of T-cell acute
lymphoblastic leukemia cells showed that resveratrol decreased Akt
activation with significant upregulation of autophagy-related genes
in a p38-dependent manner (30). In
our study shBMI1 cells displayed increased phosphorylation of
pp38Thr180/Tyr182, decreased levels of pAktSer473, and significant
evidence supporting increased autophagic activity (Figs. 5 and 6). We found that BMI1 knockdown induced
autophagy that was greatly exacerbated upon radiation as shown by
increased cleavage of LC3B-I to LC3B-II and degradation of p62
(Fig. 6A). Additional evidence
comes from our qRT-PCR analysis that shows a significant
upregulation of the autophagy genes ATG3, ATG5, ATG7, and ATG12
(Fig. 6C), further supporting the
involvement of autophagy in BMI1-mediated radiosensitization.
Altogether, our data present evidence for BMI1 as an
important target for breast cancer therapy and offers a strategy
for incorporating BMI1 targeted therapy with radiation for
achieving enhanced radiosensitivity. Histone deacetylase
inhibitors, trichostatin A and sodium butyrate, have been shown to
reverse acquired radioresistance in esophageal carcinoma cells
through downregulation of BMI1 (31). Similarly, the anti-malarial drug,
artemisinin, inhibits BMI1 at the protein and transcript levels and
radiosensitizes human cervical cancer cells to ionizing radiation
(32,33). While these studies have demonstrated
BMI1 inhibition produces radiosensitization the drugs tested are
not specific BMI1 inhibitors. However, the availability of a novel
small-molecule inhibitor of BMI1, PTC-209, provides an opportunity
to investigate the effects of BMI1 function in various cancers,
including breast. The antitumor effects of PTC-209 have been
demonstrated in colorectal cancer, myeloid leukemia and biliary
cancer (34–36). In particular, PTC-209 has been shown
to target chemotherapy resistant cancer stem cell populations by
reducing the function, activity and amount of BMI1 (34).
Up to now, no data is available describing the
effects of PTC-209 on breast cancer. As more than 50% of breast
cancer patients receive radiotherapy during the treatment of their
disease and breast cancer stem cells are known to be resistant to
ionizing radiation, compounds targeting breast cancer stem cells
may be better therapeutic agents for treating breast cancer and
controlling recurrence and metastasis. Therefore, evaluating
PTC-209 as a potential therapeutic agent to restore the sensitivity
of breast cancer cells to radiation therapy will be of clinical
significance.
Acknowledgements
This study was supported in part by grants received
from an Institutional Development Award (IDeA) from the National
Institute of General Medical Sciences (P20 GM103639) of the
National Institutes of Health (NIH), a National Cancer Institute
(NCI) grant R01 CA167516 received from the NIH, the Stephenson
Cancer Center Seed Grant, and by startup funds received from the
Department of Radiation Oncology, The University of Oklahoma Health
Sciences Center. We thank the Stephenson Cancer Center at the
University of Oklahoma Health Sciences Center, Oklahoma City, OK
for the use of the Functional Genomics Core which provided
molecular analysis service. R.R. is an Oklahoma TSET Research
Scholar and holds the Jim and Christy Everest Endowed Chair in
Cancer Developmental Therapeutics.
References
|
1
|
Simon JA and Kingston RE: Mechanisms of
polycomb gene silencing: Knowns and unknowns. Nat Rev Mol Cell
Biol. 10:697–708. 2009.PubMed/NCBI
|
|
2
|
Wang W, Qin JJ, Voruganti S, Nag S, Zhou J
and Zhang R: Polycomb group (PcG) proteins and human cancers:
Multifaceted functions and therapeutic implications. Med Res Rev.
35:1220–1267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park IK, Qian D, Kiel M, Becker MW,
Pihalja M, Weissman IL, Morrison SJ and Clarke MF: Bmi-1 is
required for maintenance of adult self-renewing haematopoietic stem
cells. Nature. 423:302–305. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bruggeman SW, Valk-Lingbeek ME, van der
Stoop PP, Jacobs JJ, Kieboom K, Tanger E, Hulsman D, Leung C,
Arsenijevic Y, Marino S, et al: Ink4a and Arf differentially affect
cell proliferation and neural stem cell self-renewal in
Bmi1-deficient mice. Genes Dev. 19:1438–1443. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY,
Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, et al: Bmi1 is
essential in Twist1-induced epithelial-mesenchymal transition. Nat
Cell Biol. 12:982–992. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and Polycomb-group gene bmi-1
regulates cell proliferation and senescence through the ink4a
locus. Nature. 397:164–168. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang FB, Sui LH and Xin T: Correlation of
Bmi-1 expression and telomerase activity in human ovarian cancer.
Br J Biomed Sci. 65:172–177. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vonlanthen S, Heighway J, Altermatt HJ,
Gugger M, Kappeler A, Borner MM, van Lohuizen M and Betticher DC:
The bmi-1 oncoprotein is differentially expressed in non-small cell
lung cancer and correlates with INK4A-ARF locus expression. Br J
Cancer. 84:1372–1376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim JH, Yoon SY, Kim CN, Joo JH, Moon SK,
Choe IS, Choe YK and Kim JW: The Bmi-1 oncoprotein is overexpressed
in human colorectal cancer and correlates with the reduced
p16INK4a/p14ARF proteins. Cancer Lett.
203:217–224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim JH, Yoon SY, Jeong SH, Kim SY, Moon
SK, Joo JH, Lee Y, Choe IS and Kim JW: Overexpression of Bmi-1
oncoprotein correlates with axillary lymph node metastases in
invasive ductal breast cancer. Breast. 13:383–388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Silva J, García V, García JM, Peña C,
Domínguez G, Díaz R, Lorenzo Y, Hurtado A, Sánchez A and Bonilla F:
Circulating Bmi-1 mRNA as a possible prognostic factor for advanced
breast cancer patients. Breast Cancer Res. 9:R552007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang R, Xu LB, Yue XJ, Yu XH, Wang J and
Liu C: Bmi1 gene silencing inhibits the proliferation and
invasiveness of human hepatocellular carcinoma cells and increases
their sensitivity to 5-fluorouracil. Oncol Rep. 29:967–974.
2013.PubMed/NCBI
|
|
13
|
Xu XH, Liu XY, Su J, Li DJ, Huang Q, Lu
MQ, Yi F, Ren JH and Chen WH: ShRNA targeting Bmi-1 sensitizes
CD44+ nasopharyngeal cancer stem-like cells to
radiotherapy. Oncol Rep. 32:764–770. 2014.PubMed/NCBI
|
|
14
|
Wang G, Liu L, Sharma S, Liu H, Yang W,
Sun X and Dong Q: Bmi-1 confers adaptive radioresistance to
KYSE-150R esophageal carcinoma cells. Biochem Biophys Res Commun.
425:309–314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dong Q, Oh JE, Chen W, Kim R, Kim RH, Shin
KH, McBride WH, Park NH and Kang MK: Radioprotective effects of
Bmi-1 involve epigenetic silencing of oxidase genes and enhanced
DNA repair in normal human keratinocytes. J Invest Dermatol.
131:1216–1225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Facchino S, Abdouh M, Chatoo W and Bernier
G: BMI1 confers radioresistance to normal and cancerous neural stem
cells through recruitment of the DNA damage response machinery. J
Neurosci. 30:10096–10111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Long CL, Berry WL, Zhao Y, Sun XH and
Humphrey MB: E proteins regulate osteoclast maturation and
survival. J Bone Miner Res. 27:2476–2489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Muralidharan R, Panneerselvam J, Chen A,
Zhao YD, Munshi A and Ramesh R: HuR-targeted nanotherapy in
combination with AMD3100 suppresses CXCR4 expression, cell growth,
migration and invasion in lung cancer. Cancer Gene Ther.
22:581–590. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Munshi A, Tanaka T, Hobbs ML, Tucker SL,
Richon VM and Meyn RE: Vorinostat, a histone deacetylase inhibitor,
enhances the response of human tumor cells to ionizing radiation
through prolongation of gamma-H2AX foci. Mol Cancer Ther.
5:1967–1974. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Munshi A, Kurland JF, Nishikawa T, Tanaka
T, Hobbs ML, Tucker SL, Ismail S, Stevens C and Meyn RE: Histone
deacetylase inhibitors radiosensitize human melanoma cells by
suppressing DNA repair activity. Clin Cancer Res. 11:4912–4922.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Proctor E, Waghray M, Lee CJ, Heidt DG,
Yalamanchili M, Li C, Bednar F and Simeone DM: Bmi1 enhances
tumorigenicity and cancer stem cell function in pancreatic
adenocarcinoma. PLoS One. 8:e558202013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Santivasi WL and Xia F: Ionizing
radiation-induced DNA damage, response, and repair. Antioxid Redox
Signal. 21:251–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Khanna KK and Jackson SP: DNA
double-strand breaks: Signaling, repair and the cancer connection.
Nat Genet. 27:247–254. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olive PL: The role of DNA single- and
double-strand breaks in cell killing by ionizing radiation. Radiat
Res. 150:(Suppl). S42–S51. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu J, Cao L, Chen J, Song S, Lee IH,
Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, et al: Bmi1
regulates mitochondrial function and the DNA damage response
pathway. Nature. 459:387–392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fujiwara K, Iwado E, Mills GB, Sawaya R,
Kondo S and Kondo Y: Akt inhibitor shows anticancer and
radiosensitizing effects in malignant glioma cells by inducing
autophagy. Int J Oncol. 31:753–760. 2007.PubMed/NCBI
|
|
29
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ge J, Liu Y, Li Q, Guo X, Gu L, Ma ZG and
Zhu YP: Resveratrol induces apoptosis and autophagy in T-cell acute
lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating
p38-MAPK. Biomed Environ Sci. 26:902–911. 2013.PubMed/NCBI
|
|
31
|
Dong Q, Sharma S, Liu H, Chen L, Gu B, Sun
X and Wang G: HDAC inhibitors reverse acquired radio resistance of
KYSE-150R esophageal carcinoma cells by modulating Bmi-1
expression. Toxicol Lett. 224:121–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gong XM, Zhang Q, Torossian A, Cao JP and
Fu S: Selective radiosensitization of human cervical cancer cells
and normal cells by artemisinin through the abrogation of
radiation-induced G2 block. Int J Gynecol Cancer. 22:718–724. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu J, Hu D, Yang G, Zhou J, Yang C, Gao Y
and Zhu Z: Down-regulation of BMI-1 cooperates with artemisinin on
growth inhibition of nasopharyngeal carcinoma cells. J Cell
Biochem. 112:1938–1948. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kreso A, van Galen P, Pedley NM,
Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W,
Sydorenko N, et al: Self-renewal as a therapeutic target in human
colorectal cancer. Nat Med. 20:29–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bolomsky A, Schlangen K, Schreiner W,
Zojer N and Ludwig H: Targeting of BMI-1 with PTC-209 shows potent
anti-myeloma activity and impairs the tumour microenvironment. J
Hematol Oncol. 9:172016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mayr C, Wagner A, Loeffelberger M,
Bruckner D, Jakab M, Berr F, Di Fazio P, Ocker M, Neureiter D,
Pichler M, et al: The BMI1 inhibitor PTC-209 is a potential
compound to halt cellular growth in biliary tract cancer cells.
Oncotarget. 7:745–758. 2016.PubMed/NCBI
|