Introduction
Small cell lung cancer (SCLC), accounting for
~15–20% of all lung cancer cases, is characterized by its
neuroendocrine origin, rapid development and early metastasis. SCLC
has the lowest differentiation and highest malignant degree among
lung cancers (1). Approximately 70%
of SCLC patients are diagnosed at a late stage with accompanying
metastasis. Therefore, the 5-year survival rate of patients with
SCLC is only 5–10% (2). The
development of novel targeted therapeutics for SCLC is one of the
promising strategies by which to improve the outcome of SCLC
treatment.
Genomic instability and high frequency of p53
mutations are two common characteristics of most human tumors
(3). Genomic instability is an
important impetus for tumor occurrence and development (4,5).
Genomic instability is principally acquired through reprogramming
the DNA repair pathway by oncogenes (6).
Among all types of DNA damage, DNA double-strand
breaks (DSBs) pose the greatest threat to cells (3). DSBs not timely and properly repaired
lead to chromosomal rearrangements, aneuploidy and other serious
genomic mutations (7). An aberrant
DSB repair pathway diminishes the fidelity and efficiency of DNA
repair; therefore, tumors can significantly gain genomic
instability to accelerate progression (8). Meanwhile, abnormal enhancement of
specific DNA repair pathways can undoubtedly lead to tumor cell
radioresistance and chemoresistance, since radiation and most
first-line chemotherapeutics kill tumor cells mainly by DNA damage
(9).
Research has demonstrated that certain DNA repair
pathways, such as transcription-coupled and expression-linked
repairs (10), are inactivated,
while others are upregulated in various SCLC cell lines. For
example, PARP expression in SCLC is 2.06 times higher than that in
NSCLC (11). Thus, differences in
DNA repair pathways and repair ability between tumor and normal
cells have been increasingly assessed to identify promising targets
(2,12).
Homologous recombination (HR) repairs DNA
double-strand breaks with high fidelity. RAD51, the core protein of
HR, is expressed 4–6 times higher in the majority of tumor cells
both at the gene and protein levels, compared with corresponding
normal cells, with its activity increasing by up to 840-fold
(13). Abnormal expression of RAD51
can lead to diminished fidelity of HR repair, and promote
occurrence of translocation and other chromosome mutations
(14). Due to the close correlation
between the aberrant activity of RAD51 and tumor occurrence,
regulation of the activity of RAD51 has attracted increased
attention from scientists.
Fidgetin-like 1 (FIGNL1) belongs to the AAA-ATPase
protein family (15), and plays an
important role in meiosis and mitosis (16,17).
FIGNL1 is an indispensable component of HR, specifically
interacting with RAD51 via its conserved RAD51 binding domain
(18). The above findings indicate
that FIGNL1 can directly regulate the activity of RAD51 and
indirectly modulate that of HR in DNA DSB repair. The present study
aimed to assess FIGNL1 in SCLC patients and cell lines to
explore the function of HR in SCLC.
Materials and methods
Patients and tissue samples
Specimens were collected from patients who underwent
surgical resection for lung cancer or suspected lung cancer
patients through bronchoscopic biopsy at Shanghai Pulmonary
Hospital from 2013 to 2015, with the approval of the Ethics
Committee of Tong Ji University. All patients involved in the
present study had provided written informed consent for the use of
their tissue samples in the present study. The specimens that were
confirmed to be SCLC or normal (tumor negative) by pathological
examination were used for the following experiments. Detailed
clinical information is shown in Table
I. All specimens were immediately preserved in RNAstore reagent
(Tiangen Biotech, Beijing, China) at 4°C until total RNA
extraction.
 | Table I.Specimens assayed for FINGL1
expression. |
Table I.
Specimens assayed for FINGL1
expression.
| Variables (n) | Normal | SCLC | P-value |
|---|
| Total | 45 | 42 |
|
| Gender |
|
| 0.2853 |
|
Male | 34 | 36 |
|
|
Female | 11 | 6 |
|
| Age (years) |
|
| 0.2005 |
|
≤60 | 26 | 18 |
|
|
>60 | 19 | 24 |
|
| TNM stage |
|
| – |
|
I–IIa | – | 0 |
|
|
IIb-IIIa | – | 13 |
|
|
IIa-IV | – | 29 |
|
Cell culture
The human SCLC NCI-H446 cell line was obtained from
the Cell Bank of the Chinese Academy of Sciences and was cultured
in RPMI-1640 medium (HyClone, Logan, UT, USA) containing 10% fetal
bovine serum (FBS) (Gibco, Grand Island, NY, USA). The human
embryonic lung fibroblast MRC-5 cell line was a kind gift from
Professor Zhiyong Li at the College of Life Science and Technology,
Shanghai Jiao Tong University, and was cultured in Minimum
essential medium (MEM) containing 10% FBS. The Platinum-A
Retroviral Packaging Cell Line was a kind gift from Professor
Songcheng Zhu of Tongji University. Culture medium was Dulbeccos
modified Eagles medium (DMEM) containing 10% FBS, with blasticidin
and puromycin (Sigma-Aldrich, St. Louis, MO, USA) added to final
concentrations of 10 and 1 µg/ml, respectively.
Chemical and reagents
The chemicals used in the present study were
purchased from Sigma-Aldrich.
Total RNA extraction and real-time
PCR
Total RNA extraction kit was used for total RNA
extraction. Approximately 1,000 ng total RNA was reverse
transcribed in 20 µl volume using FastQuant cDNA First Chain
Synthesis kit (both from Tiangen) according to the manufacturer's
instructions. Quantitative real-time PCR (qPCR) was performed using
Super Real PreMix Plus (SYBR-Green) (Tiangen) on Eppendorf
Mastercycler ep realplex4, with GAPDH as an internal
control. The PCR program was as following: 3 min at 95°C, followed
by 40 cycles at 95°C for 30 sec, 60°C for 30 sec and 72°C for 15
sec, and last step was the melting curve analysis program. All
experiments were independently performed three times, and three
replicates each time. The following primers were used:
5′-TCCTGCACCACCAACTGCTT-3′ (forward) and 5′-GGGGCCATCCACAGTCTTCT-3′
(reverse) for FIGNL1; 5′-CTCAGCGTGCATCAGGGTCT-3′ (forward)
and 5′-CTGCTCTCCCCCATCTTGCT-3′ (reverse) for GAPDH;
5′-GTTCCTCCTTGGAAAGCAAACAGTA-3′ (forward) and
5′-CAGGGCATCTTCACGCTCTATTT-3′ (reverse) for cyclin A2;
5′-AGAAATGGCCAAAATCGA CA-3′ (forward) and
5′-CCCGGTCATCATCTTCTTTG-3′ (reverse) for cyclin E1;
5′-TATGCCTGATTACAAGCCAAGTTTC-3′ (forward) and
5′-GATAACAAGCTCCGTCCATCTTCAT-3′ (reverse) for CDK2;
5′-CATTGTTGTGTTTCACTGCGAGTTT-3′ (forward) and
5′-GGACATACAGCTCAGGGTAGTGGAG-3′ (reverse) for cdc25A. Relative gene
expression was calculated by the 2−ΔCt method.
Western blotting
Cells were lysed using RIPA lysis solution (Tiangen
Biotech). After the protein concentrations were determined, the
protein samples were separated by 10% SDS-PAGE, transferred onto
0.45-µm polyvinylidene fluoride (PVDF) membranes (Millipore,
Darmstadt, Germany) and probed with relevant antibodies.
Anti-FIGNL1 (cat. no. 7604-1-APl; 1:1,000) and anti-GAPDH (cat. no.
10494-1-AP; 1:2,000) were obtained from ProteinTech (Chicago, IL,
USA). After incubation with the primary antibody overnight at 4°C,
the membranes were washed with Tris-buffered saline with Tween-20
(TBST) three times, 5 min each. HRP-labeled secondary antibodies
(cat. no. 111-035-003; 1:2,000) were obtained from Jackson
ImmunoResearch (West Grove, PA, USA). After incubation with the
secondary antibody for 2 h at room temperature, the membranes were
washed for three time with TBST for 10 min each, Protein bands were
detected by enhanced chemiluminescence (ECL; Sigma-Aldrich) and
quantitated using Gel-Pro Analyzer 4.0 (Media Cybernetics,
Rockville, MD, USA), and the results were obtained from three
independent experiments.
shRNA and SCLC cell transfection
Short hairpin RNA (shRNA) targeting FIGHL1
and a negative control were designed by Origene Technologies, Inc.
(Rockville, MD, USA). shRNA sequences for FIGHL1 were:
5′-AGCACATCCAGTTGATGAGCGTCTGAAGA-3′ for shFIGNL1-B,
5′-CAGAAGCTTCAGCCAGGAAACAGATAGTA-3′ for shFIGNL-D, and
5′-GCACTACCAGAGCTAACTCAGATAGTACT-3 for sh-control.
H446 cells were transfected using 0.45 µM filtered
and Polybrene supplemented (8 µg/ml final concentration) culture
media of the Platinum-A Retroviral Packaging Cell Line transfected
with the shRNA plasmid for 48 h by Lipofectamine 2000. H446 cells
were screened with complete culture medium containing puromycin at
1.0 µg/ml for 6 days. Afterwards, the cells were cultured with
complete medium with 0.5 µg/ml puromycin.
Cell cycle analysis
The cells were cultured for ~2 days following
synchronization for 12 h. Then, the cells were collected, washed
with PBS and fixed with 75% ethanol at −20°C overnight. After
incubation with 0.1 mg/ml RNase A at 37°C for 30 min and staining
with 40 µg/ml PI (Sigma, St. Louis, MO, USA) in the dark for 30
min, DNA content was detected by flow cytometry (FACSCalibur;
Becton-Dickinson, San Jose, CA, USA). Cell cycle distribution was
analyzed and calculated by ModFit (Verity Software House, Topsham,
ME, USA).
Cell proliferation assay
Cells were plated at 3×103/well in
96-well plates. MTT assay was adopted to assess cell viability.
Absorbance was measured on an EnSpire 2300 microplate reader
(Perkin Elmer, Waltham, MA, USA) at 550 nm. For drug sensitivity
assay, cells were seeded in 96-well plates at a density of
3×103 cells/well and cultured for 24 h. Then, the cells
were exposed to 0, 20, 40, 80 and 160 µM etoposide, respectively,
for 24 h, or 0, 1.25, 2.5, 5, 10 and 20 µM, respectively, for 48 h;
cisplatin was assayed at 0, 2, 4, 8, 16 and 32 µM, respectively,
for 24 h, or 0.5, 1, 2, 4, and 8 µM, respectively, for 48 h. Cell
survival was measured by the MTT assay, and half maximal inhibitory
concentration (IC50) values of etoposide or cisplatin
were calculated by GraphPad Prism 5.0.
Statistical analysis
Statistical analysis was performed using SPSS 17.0
or GraphPad Prism 5.0. Two groups of data, such as FIGNL1
expression between normal and SCLC specimens, was analyzed using a
t-test to determine statistical significance. For three or more
groups of data, such as comparison of FIGNL1 expression
among the different H446 transfected cells, one-way analysis of
variance (ANOVA) with Bonferoni post test was adopted. Results are
presented as mean ± SD. P<0.05 was considered to indicate a
statistically significant result.
Results
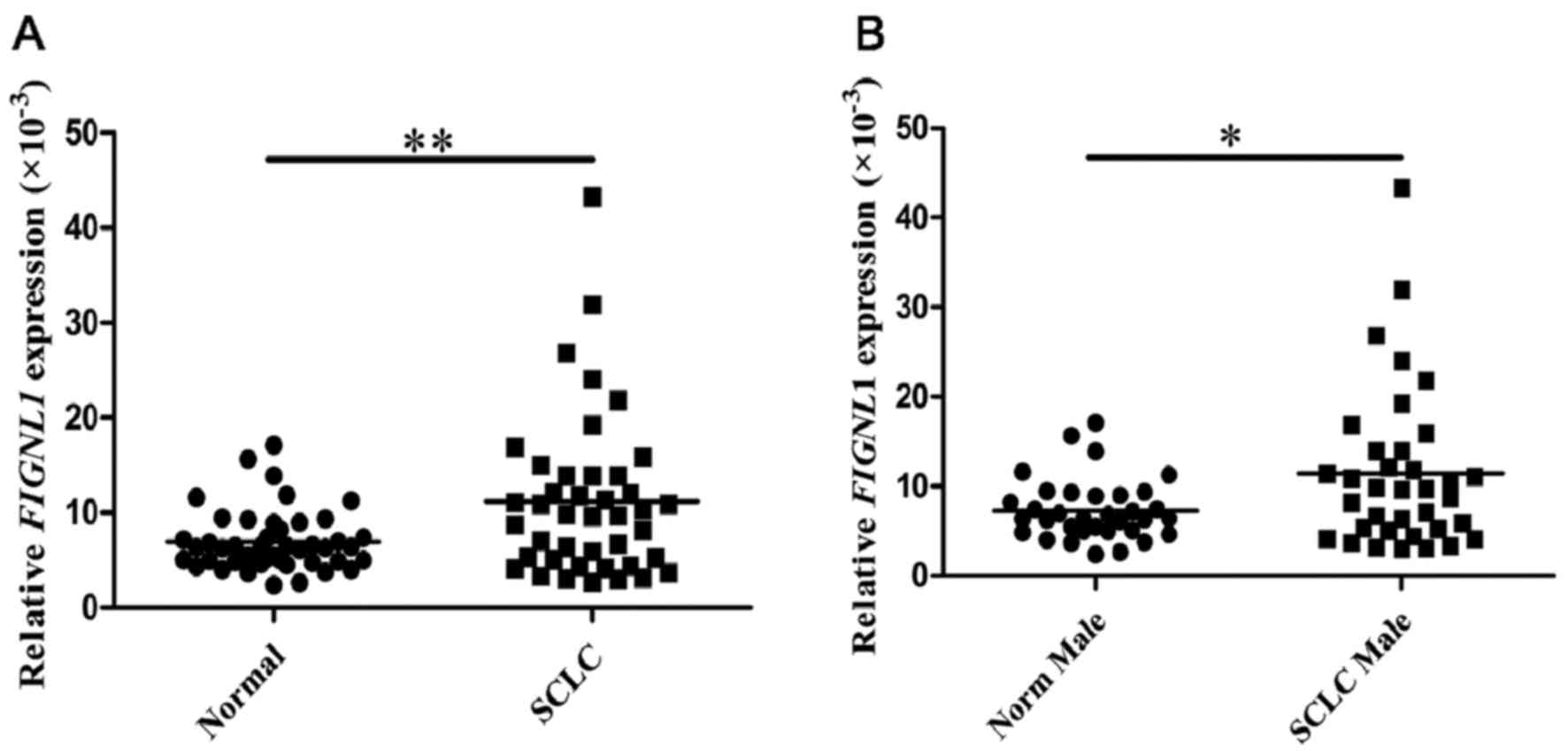
FIGNL1 expression in SCLC
specimens
A total of 45 normal lung samples was assessed, from
34 male and 11 female patients, respectively. In these specimens,
the relative expression level of FIGNL1 was
6.965±3.204×10−3. Meanwhile, 42 SCLC samples from 36
males and 6 females were assessed, and yielded a value of 11.197±
8.466×10−3 (Table I).
Comparison of the FIGNL1 expression levels between normal
and SCLC samples showed a statistical significance (P=0.004;
Fig. 1A). A similar result was
obtained for comparison between normal and SCLC specimens from the
male patients (P=0.014; Fig. 1B).
No significant difference in FIGNL1 expression levels was
observed between age groups below and above 60 years and early and
advanced stage groups (data not shown).
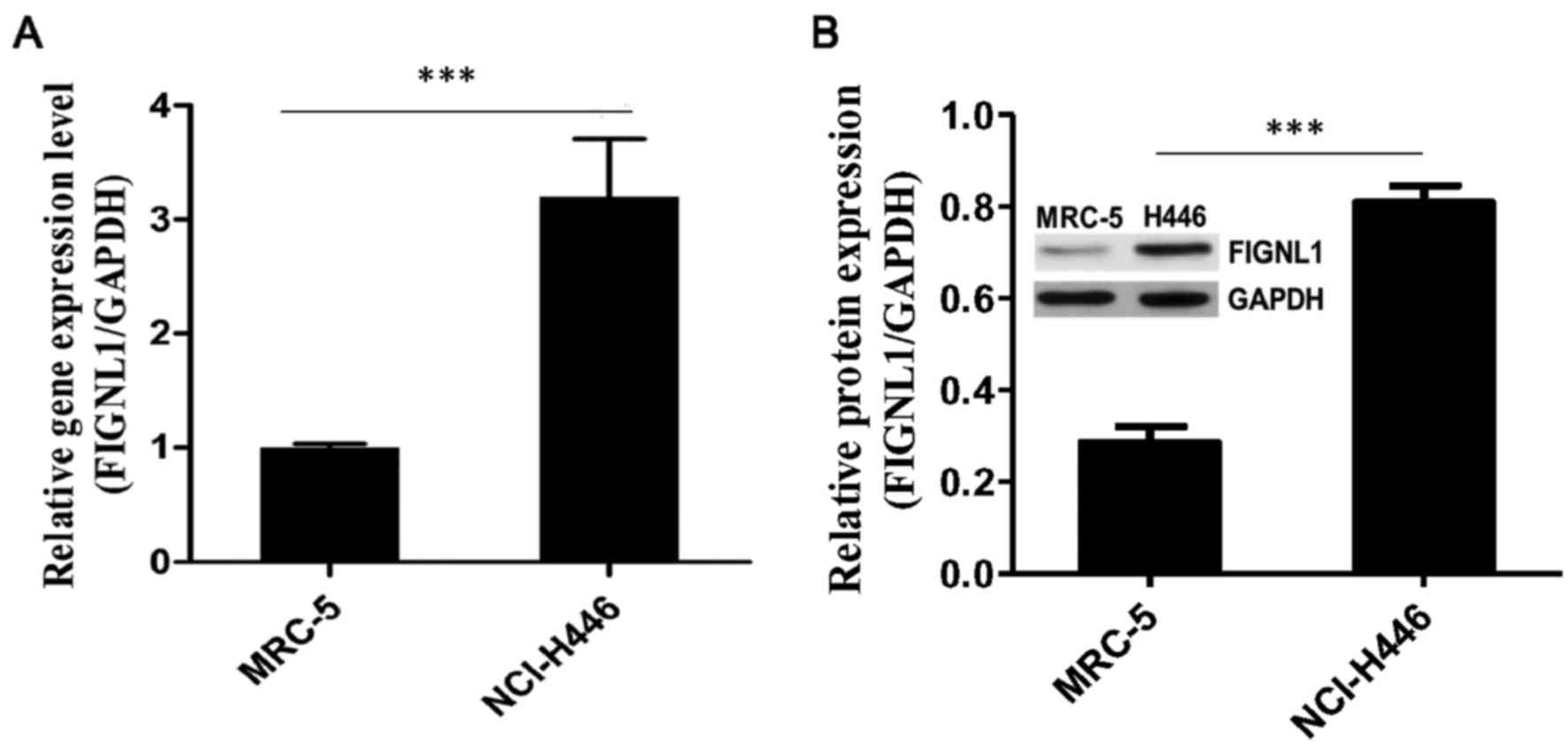
FIGNL1 expression in SCLC and normal
lung fibroblast cell lines
The FIGNL1 transcription level was 3.46-fold
in the SCLC H446 cells [9.83±0.12 (ΔCt)] compared to this level in
the normal lung fibroblast MRC-5 cells [11.13±0.04 (ΔCt)]
(P<0.001; Fig. 2A).
Moreover, FIGNL1 protein expression was assessed in
the different cell lines. Notably, the FIGNL1 protein level
(FIGNL1/GAPDH) in the H446 cells (0.808±0.035) was 2.84 times
higher compared to amounts obtained in the MRC-5 cells
(0.285±0.035) (P<0.001; Fig.
2B).
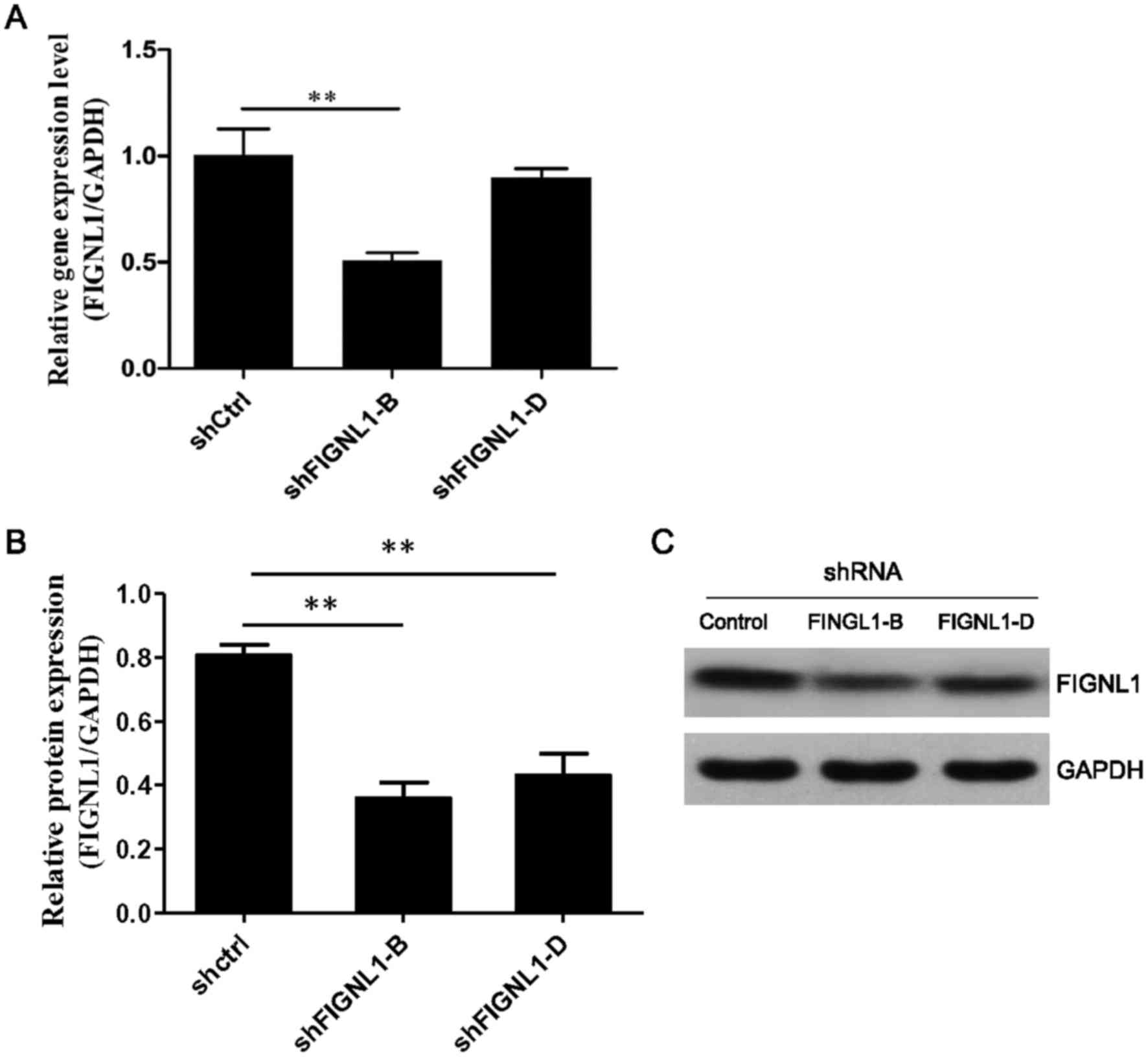
Effect of FIGNL1 knockdown on the
growth and cell cycle distribution of H446 cells
To further explore the biological function of
FIGNL1 overexpression in SCLC cells, shRNA was used to
silence FIGNL1 in the H446 cell line.
ΔCt values for FIGNL1 were 10.81±0.11 and
9.98±0.06 in the H446 cells transfected with shFIGNL1-B and
shFIGNL1-D, respectively. Compared to the control group [9.77±0.10
(shCtrl)], FIGNL1 mRNA amounts were reduced by 48.6%
(P=0.006) and 13.5% (P>0.05) in the shFIGNL1-B and shFIGNL1-D
group cells, respectively (Fig.
3A). Western blot results further confirmed that the FIGNL1
protein level (FIGNL1/GAPDH) was reduced by 55.5% (P=0.002) and
46.5% (P=0.002), respectively, in the H446 cells transfected with
shFIGNL1-B (0.359±0.087) or shFIGNL1-D (0.431±0.117) compared to
the control (0.806±0.059) (Fig. 3B and
C).
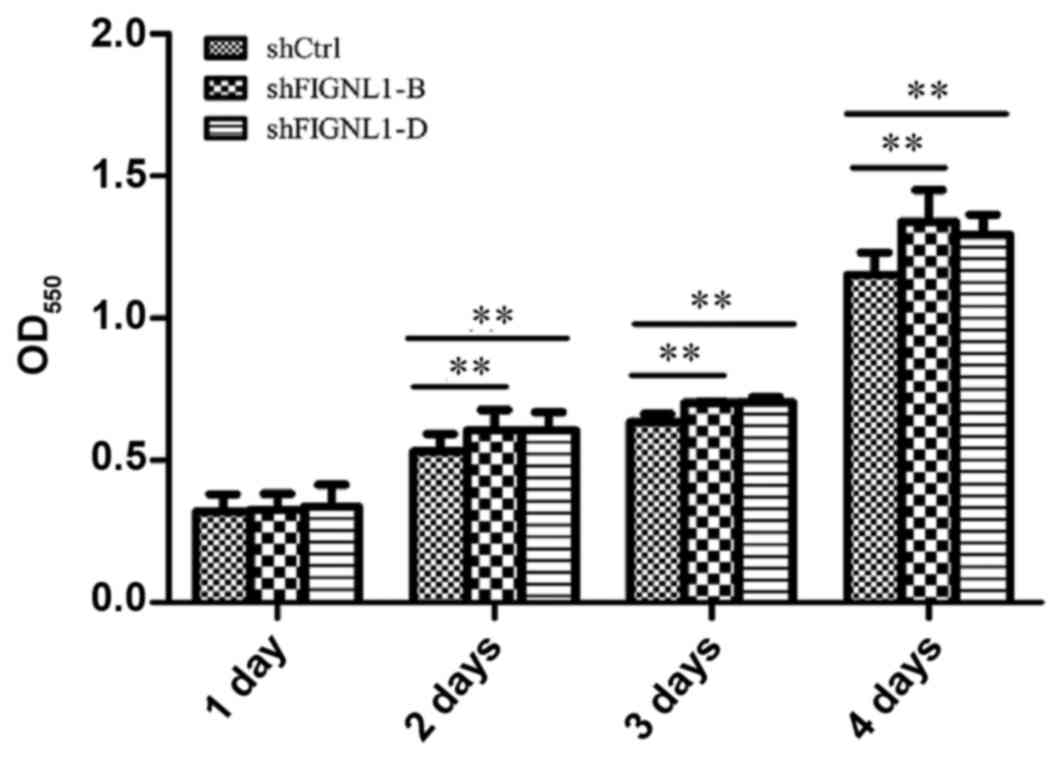
FIGNL1 silencing slightly affects H446
cell growth
Three groups of H446 cells with the same initial
density were cultured for two days, and 13.8 and 12.5% more cells
were transfected with shFIGNL1-B (P=0.0011) or shFIGNL1-D
(P=0.0017), compared with the control group. After three days of
culture, 12.8 and 11.5% more cells were found in the shFIGNL1-B
(P=0.003) and shFIGNL1-D (P=0.002) groups compared with the control
group. After culture for four days, 17.1% (P=0.003) and 15.6%
(P=0.004) more cells, respectively, were obtained in the shFIGNL1-B
and shFIGNL1-D groups compared with the control group. These
findings indicated that FIGNL1 suppression slightly accelerated
growth in the H446 cells (Fig.
4).
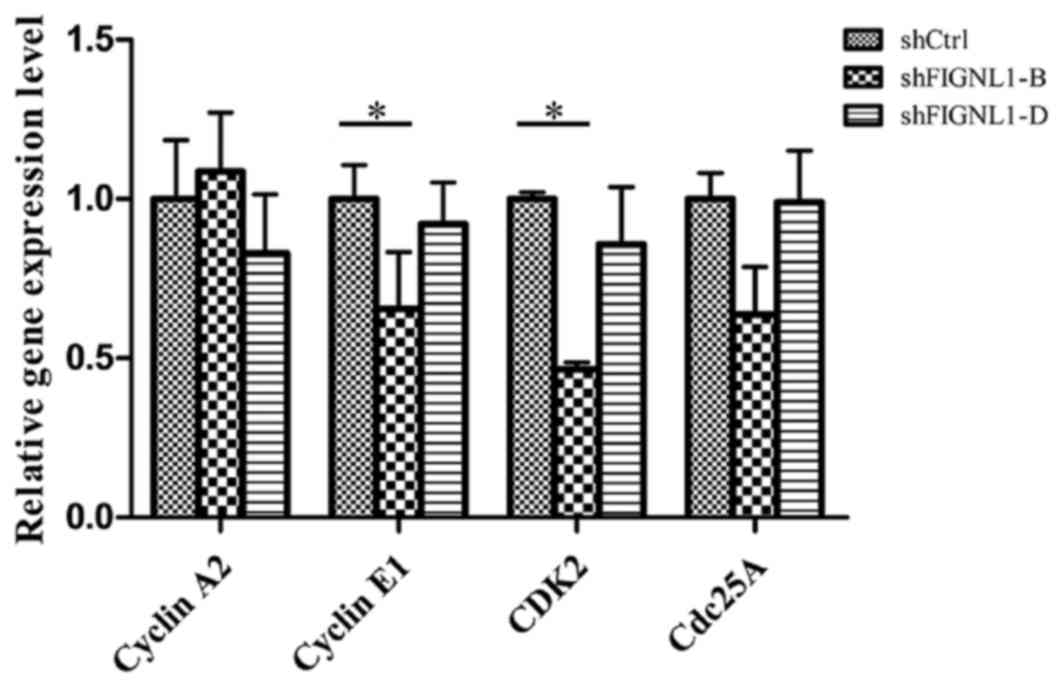
FIGNL1 silencing alters the expression
of key genes involved in cell cycle regulation
To assess whether FIGNL1 expression affects the cell
cycle, cyclin A2, cyclin E1, CDK2 and cdc25A expression levels were
quantified in the H446 cells transfected with the different
shRNAs.
As shown in Fig. 5,
in the H446 cells transfected with shFIGNL1-D, no significant
change in the expression of cyclin A2, cdc25A, cyclin E1 and CDK2
was observed. However, in the shFIGNL1-B-transfected cells, cyclin
E1 and CDK2 mRNA amounts decreased by 34.94% (P=0.016) and 53.03%
(P=0.018), respectively, at same time the expression of cyclin A2
and cdc25A remained unchanged.
Cyclin-dependent kinases (CDKs) function mainly in S
and G1 phases of the cell cycle and most tumors abrogate the cell
cycle regulatory mechanism by directly or indirectly enhancing the
activity of CDKs (19,20). Thus, cell cycle analysis was
conducted to assess whether this downregulation impacted the H446
cell cycle.
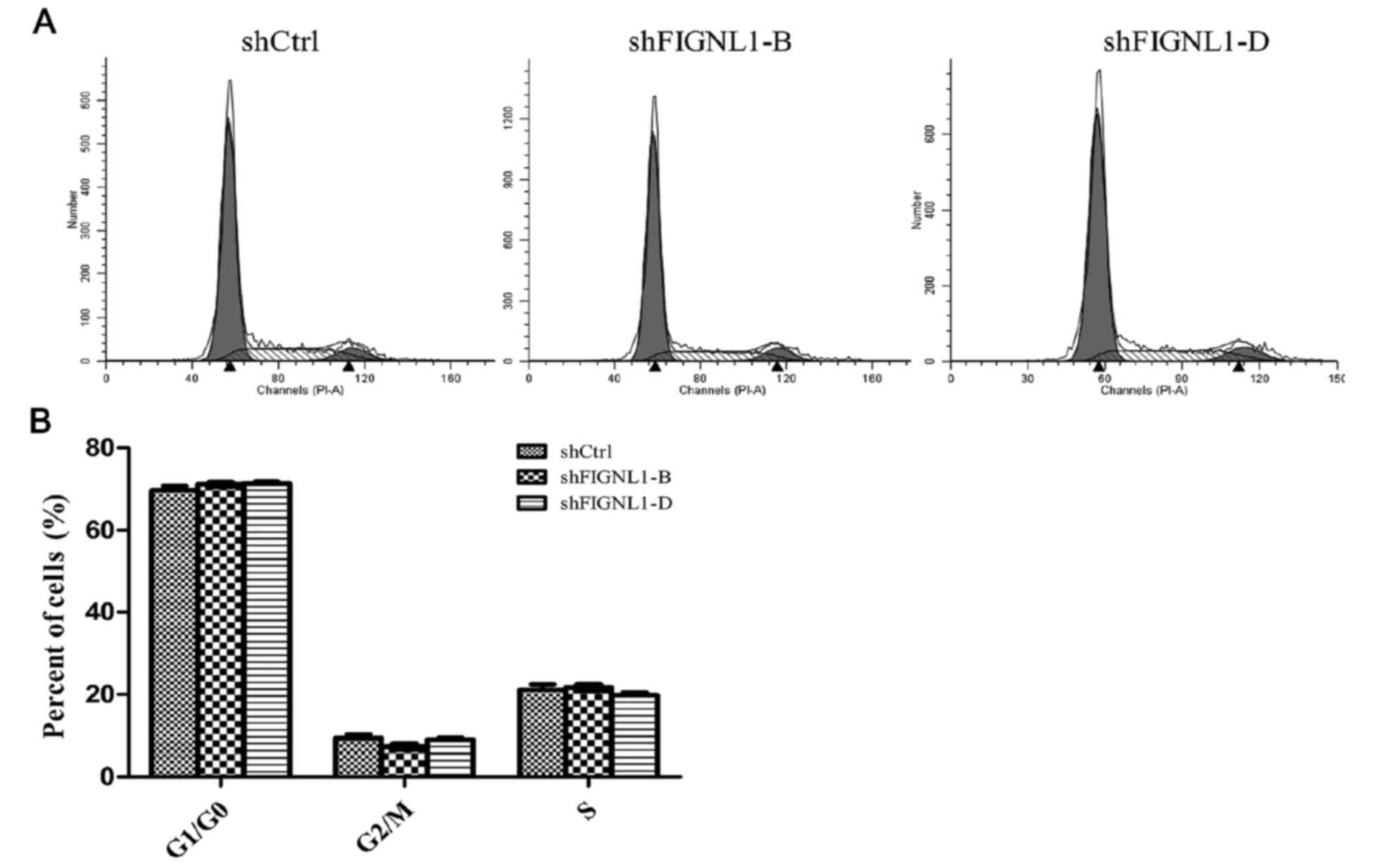
Effect of FIGNL1 silencing on H446
cell cycle distribution
Flow cytometry was used to assess the cell cycle
distribution of H446 cells transfected with the different shRNAs.
As shown in Fig. 6, cell cycle
distribution was not significantly different among the groups.
Although, FIGNL1 knockdown greatly reduced cyclin E1
and CDK2 expression, it did not affect the cell cycle.
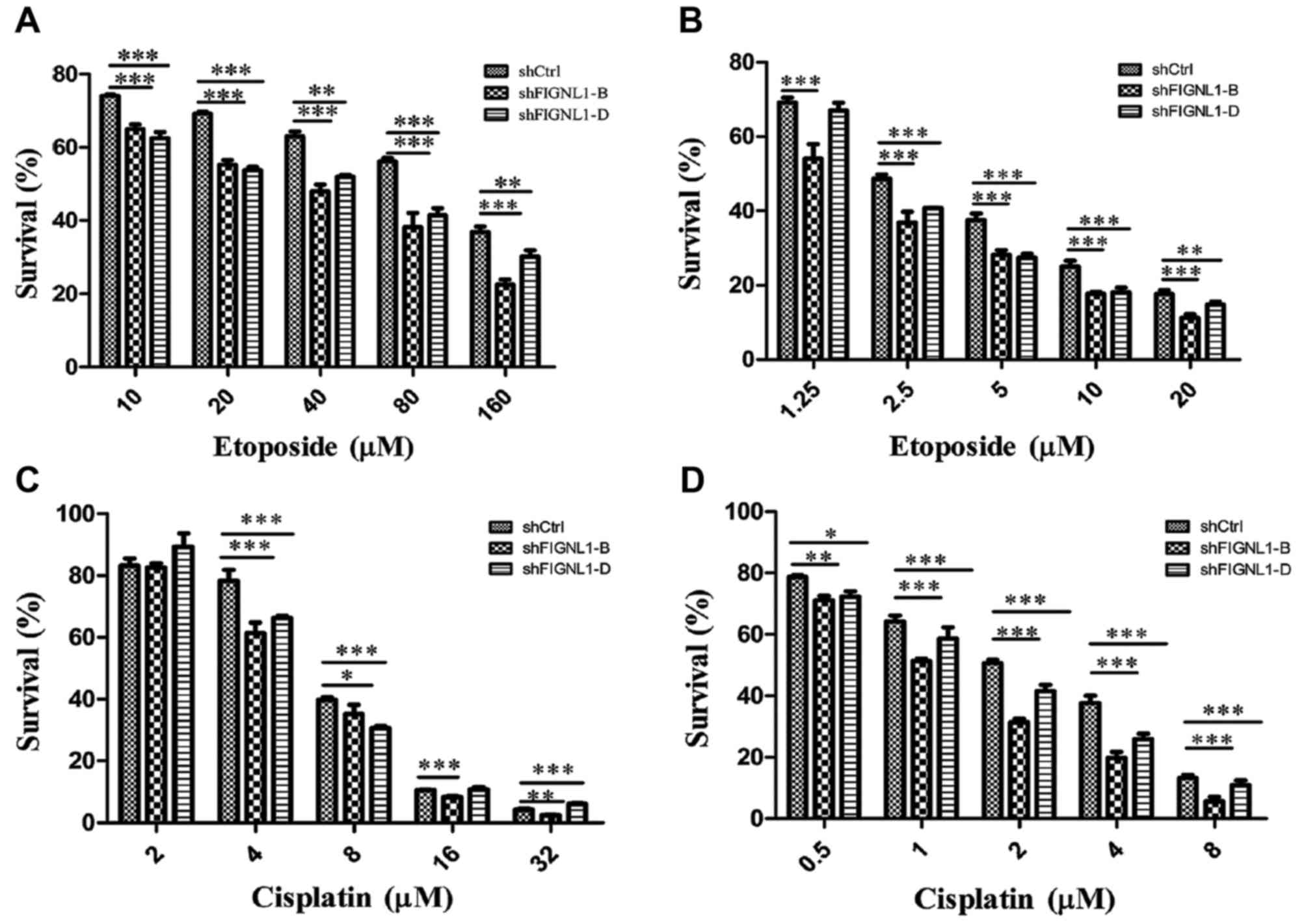
FIGNL1 silencing sensitizes H446 cells
to etoposide and cisplatin
Following treatment with etoposide for 24 h, the
sensitivity to etoposide of H446 cells transfected with shFIGNL1-B
or shFIGNL1-D was increased by 65.6 and 61.8%, respectively,
compared to the control group (Fig.
7A; Table II).
| Table II.IC50 values of etoposide
in the H446 cells transfected with the different shRNAs. |
Table II.
IC50 values of etoposide
in the H446 cells transfected with the different shRNAs.
| Construct
transfected | IC50 (24
h) (µM) | IC50 (48
h) (µM) |
|---|
| shRNA-Ctrl | 87.44 | 2.76 |
| shRNA-FIGNL1-B | 30.10 | 1.44 |
| shRNA-FIGNL1-D | 33.39 | 2.09 |
After treatment with etoposide for 48 h, sensitivity
of the H446 cells transfected with shFIGNL1-B or shFIGNL1-D to
etoposide was increased by 47.8 and 24.3%, respectively, compared
to the control group (Fig. 7B;
Table II).
Changes in the sensitivity to
cisplatin in H446 cells after FIGNL1 silencing
As shown in Fig. 7C
and Table III, H446 cells treated
with cisplatin for 24 h showed IC50 values of 6.463,
5.157 and 5.446 µM in the shCtrl, shFIGNL1-B and shFIGNL1-D groups,
respectively. Compared with the control group, sensitivity of H446
cells to cisplatin was increased by 20.1 and 15.6% after
FIGNL1 suppression by 48.6 or 13.5%, respectively.
| Table III.IC50 value of cisplatin in
the H446 cells transfected with the different shRNAs. |
Table III.
IC50 value of cisplatin in
the H446 cells transfected with the different shRNAs.
| Construct
transfected | IC50 (24
h) (µM) | IC50 (48
h) (µM) |
|---|
| shRNA-Ctrl | 6.46 | 1.89 |
| shRNA-FIGNL1-B | 5.16 | 1.05 |
| shRNA-FIGNL1-D | 5.45 | 1.34 |
H446 cells treated with cisplatin for 48 h showed
IC50 values of 1.889, 1.046 and 1.342 µM in the shCtrl,
shFIGNL1-B and shFIGNL1-D groups, respectively (Fig. 7D; Table III). Compared with the control
group, the sensitivity of shFIGNL1-B- and shFIGNL1-D-transfected
cells to cisplatin was increased by 44.4 and 29.1%,
respectively.
The above results showed a negative correlation
between the FIGNL1 expression level in H446 cells and H446
cell sensitivity to etoposide and cisplatin.
Discussion
Assessment of 45 normal lung and 42 SCLC clinical
specimens revealed that FIGNL1 expression in the SCLC
samples was 1.5 times increased in the SCLC specimens when compared
to that noted in the normal lung tissue specimens.
As an AAA-ATPase protein family member (21), FIGNL1 is involved in various
important cellular activities via regulation of microtubules,
chromosome scaffold and assembly and depolymerization of other
important protein complexes (22–25).
FIGNL1, an indispensable member of the homologous recombination DNA
repair system, plays an important role in DNA double-strand break
repair through interacting with RAD51 (18). The essential function and unique
mechanism of FIGNL1 in DNA repair makes it an attractive target for
modulation of HR pathway activity.
Abnormalities in the DNA double-strand break repair
pathway is a main contributor to cell genomic instability (8), which is an important driving force in
tumorigenesis (26), development
and metastasis (27). Meanwhile,
abnormalities in the DNA repair pathway directly affect the outcome
of radiotherapy and chemotherapy in lung cancer (28–30),
since radiotherapy and the majority of first-line chemotherapeutic
drugs kill tumor cells mainly through DNA damage (12).
SCLC commonly shows sensitivity to initial
chemotherapy and radiotherapy with >50% remission rate, yet
radiation or drug resistance quickly develops, The disease usually
relapses or progresses within 1 year (2). Therefore, abnormal overexpression of
FIGNL1 indicates abnormal enhancement of the DNA double-strand
repair system which may be one of the main mechanisms underlying
the clinical features of SCLC as mentioned above.
As shown above, a significant increase in H446 cell
sensitivity to etoposide and cisplatin was found after
FIGNL1 silencing. Indeed, the lower the FIGNL1
expression, the higher the sensitivity of cells to
etoposide/cisplatin (DNA damaging agents). These results further
support the previous notion that in SCLC, DNA double-strand break
repair pathways are abnormally enhanced.
The mechanism of etoposide and cisplatin involving
the induction of DNA damage is different. Etoposide functions by
inactivating DNA topoisomerase II (31), whereas the action of cisplatin is
due to its ability to promote intra-strand and inter-strand
crosslinking between adjacent purine bases of the DNA strand
(32). Once topoisomerase II in
tumor cells is inhibited by etoposide, DNA damage occurs
immediately. However, for cisplatin, DNA damage in tumor cells may
gradually occur and accumulate in DNA replication and transcription
process after increasing DNA adducts are formed.
In the present study, FIGNL1 knockdown
sensitized H446 cells to etoposide and cisplatin in different
patterns (Tables II and III). For etoposide, the sensitization
reached its highest level at an earlier stage (24 h), while
cisplatin showed an opposite pattern. In addition, there was an
obvious positive correlation between the severity of DNA damage and
sensitization. These results are consistent with the mechanism of
these two chemotherapeutic agents. The results mentioned above were
obtained using NCI-H446 cells, and may be verified in more SCLC
cell lines, i.e. NCI-H1688, in future research.
DNA repair pathway abnormalities contribute directly
to the main causes of the poor outcome of cancer treatment, i.e.
chemoresistance, radioresistance and relapse (28,29).
Therefore, several studies aimed to identify targets of cancer
therapy and biomarkers in the DNA repair pathway (33,34).
Although the cell cycle was not effected, FIGNL1 knockdown
significantly inhibited the expression of cyclin E1 and CDK2. This
implies that FIGNL1-mediated regulation of the expression of cell
cycle genes may exist, and our future research aims to investigate
the underlying mechanisms.
Based on the finding of the present study and
studies from other investigators, we suggest that DNA double-strand
break repair HR pathways are abnormally enhanced in SCLC and the
activity can be markedly suppressed through manipulation of
FIGNL1. Thus, FIGNL1 is a promising target for
SCLC.
Acknowledgements
The present study was supported by grants from the
Ministry of Agriculture in China (2012ZX08011002-004), the National
Natural Science Foundation of China (31121064), and the State Key
Laboratory of Microbial Metabolism (no. 2011DA105494).
References
|
1
|
Chapman CJ, Thorpe AJ, Murray A,
Parsy-Kowalska CB, Allen J, Stafford KM, Chauhan AS, Kite TA,
Maddison P and Robertson JF: Immunobiomarkers in small cell lung
cancer: Potential early cancer signals. Clin Cancer Res.
17:1474–1480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mamdani H, Induru R and Jalal SI: Novel
therapies in small cell lung cancer. Transl Lung Cancer Res.
4:533–544. 2015.PubMed/NCBI
|
|
3
|
Halazonetis TD, Gorgoulis VG and Bartek J:
An oncogene-induced DNA damage model for cancer development.
Science. 319:1352–1355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schvartzman JM, Sotillo R and Benezra R:
Mitotic chromosomal instability and cancer: Mouse modelling of the
human disease. Nat Rev Cancer. 10:102–115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goode EL, Ulrich CM and Potter JD:
Polymorphisms in DNA repair genes and associations with cancer
risk. Cancer Epidemiol Biomarkers Prev. 11:1513–1530.
2002.PubMed/NCBI
|
|
6
|
Yaglom JA, McFarland C, Mirny L and
Sherman MY: Oncogene-triggered suppression of DNA repair leads to
DNA instability in cancer. Oncotarget. 5:8367–8378. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scott SP and Pandita TK: The cellular
control of DNA double-strand breaks. J Cell Biochem. 99:1463–1475.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jeggo PA and Löbrich M: How cancer cells
hijack DNA double-strand break repair pathways to gain genomic
instability. Biochem J. 471:1–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Helleday T, Petermann E, Lundin C, Hodgson
B and Sharma RA: DNA repair pathways as targets for cancer therapy.
Nat Rev Cancer. 8:193–204. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pleasance ED, Stephens PJ, O'Meara S,
McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman
C, et al: A small-cell lung cancer genome with complex signatures
of tobacco exposure. Nature. 463:184–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Byers LA, Wang J, Nilsson MB, Fujimoto J,
Saintigny P, Yordy J, Giri U, Peyton M, Fan YH, Diao L, et al:
Proteomic profiling identifies dysregulated pathways in small cell
lung cancer and novel therapeutic targets including PARP1. Cancer
Discov. 2:798–811. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Damia G and D'Incalci M: Targeting DNA
repair as a promising approach in cancer therapy. Eur J Cancer.
43:1791–1801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hine CM, Seluanov A and Gorbunova V: Use
of the Rad51 promoter for targeted anti-cancer therapy. Proc Natl
Acad Sci USA. 105:20810–20815. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richardson C, Stark JM, Ommundsen M and
Jasin M: Rad51 overexpression promotes alternative double-strand
break repair pathways and genome instability. Oncogene. 23:546–553.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cox GA, Mahaffey CL, Nystuen A, Letts VA
and Frankel WN: The mouse fidgetin gene defines a new role for AAA
family proteins in mammalian development. Nat Genet. 26:198–202.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luke-Glaser S, Pintard L, Tyers M and
Peter M: The AAA-ATPase FIGL-1 controls mitotic progression, and
its levels are regulated by the CUL-3MEL-26 E3 ligase in
the C. elegans germ line. J Cell Sci. 120:3179–3187. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
L'Hôte D, Vatin M, Auer J, Castille J,
Passet B, Montagutelli X, Serres C and Vaiman D: Fidgetin-like1 is
a strong candidate for a dynamic impairment of male meiosis leading
to reduced testis weight in mice. PLoS One. 6:e275822011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan J and Chen J: FIGNL1-containing
protein complex is required for efficient homologous recombination
repair. Proc Natl Acad Sci USA. 110:10640–10645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hartwell LH and Kastan MB: Cell cycle
control and cancer. Science. 266:1821–1828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rajagopalan H and Lengauer C: Aneuploidy
and cancer. Nature. 432:338–341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lupas AN and Martin J: AAA proteins. Curr
Opin Struct Biol. 12:746–753. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hanson PI and Whiteheart SW: AAA+
proteins: Have engine, will work. Nat Rev Mol Cell Biol. 6:519–529.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tucker PA and Sallai L: The AAA+
superfamily - a myriad of motions. Curr Opin Struct Biol.
17:641–652. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Snider J and Houry WA: AAA+ proteins:
Diversity in function, similarity in structure. Biochem Soc Trans.
36:72–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wendler P, Ciniawsky S, Kock M and Kube S:
Structure and function of the AAA+ nucleotide binding pocket.
Biochim Biophys Acta. 1823:2–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yachida S, Jones S, Bozic I, Antal T,
Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al:
Distant metastasis occurs late during the genetic evolution of
pancreatic cancer. Nature. 467:1114–1117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heaphy CM, Bisoffi M, Joste NE,
Baumgartner KB, Baumgartner RN and Griffith JK: Genomic instability
demonstrates similarity between DCIS and invasive carcinomas.
Breast Cancer Res Treat. 117:17–24. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
O'Grady S, Finn SP, Cuffe S, Richard DJ,
O'Byrne KJ and Barr MP: The role of DNA repair pathways in
cisplatin resistant lung cancer. Cancer Treat Rev. 40:1161–1170.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Willers H, Azzoli CG, Santivasi WL and Xia
F: Basic mechanisms of therapeutic resistance to radiation and
chemotherapy in lung cancer. Cancer J. 19:200–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peters GJ, Avan A, Ruiz MG, Orsini V, Avan
A, Giovannetti E and Smit EF: Predictive role of repair enzymes in
the efficacy of Cisplatin combinations in pancreatic and lung
cancer. Anticancer Res. 34:435–442. 2014.PubMed/NCBI
|
|
31
|
Eastman A: The formation, isolation and
characterization of DNA adducts produced by anticancer platinum
complexes. Pharmacol Ther. 34:155–166. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hurwitz JL, McCoy F, Scullin P and Fennell
DA: New advances in the second-line treatment of small cell lung
cancer. Oncologist. 14:986–994. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martin SA, Lord CJ and Ashworth A: DNA
repair deficiency as a therapeutic target in cancer. Curr Opin
Genet Dev. 18:80–86. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Madhusudan S and Middleton MR: The
emerging role of DNA repair proteins as predictive, prognostic and
therapeutic targets in cancer. Cancer Treat Rev. 31:603–617. 2005.
View Article : Google Scholar : PubMed/NCBI
|