Introduction
Hepatocellular carcinoma (HCC) is the sixth most
frequent diagnosed cancer and one of the most common cause of
cancer-related death worldwide (1,2).
Although the diagnostic and therapeutic techniques of HCC have
gradually improved, the morbidity and mortality of HCC continue to
show an upward trend (3–6). The potential molecular mechanisms are
critical for understanding the development and progression of HCC
(7). Therefore, we need to further
understand the pathogenesis of HCC and identify novel molecular
markers to improve the diagnosis and treatment of HCC.
Among the large scale of mammalian genome, only a
small fraction of transcripts represent protein-coding genes
(8), the involvement of non-protein
coding genes also act as critical regulators in pathogenesis and
growth of various human diseases including cancer processes
(9,10). MicroRNAs (miRNAs), a group of short
non-coding RNAs (20–25 nucleotides) that have been extensively
studied and identified to be crucial transcriptional regulators of
carcinogenesis (11,12). miRNAs regulated target gene
expression by induce messenger RNAs (mRNA) degradation and inhibit
the translation and stability of mRNAs through binding to the
3-untranslated region (3′-UTR) (13,14).
Numerous miRNAs were identified from thousands of cancer related
studies serving as promising biomarkers or therapeutic targets
(15,16). For instance, miR-21, miR-124,
miR-148a and miR-384 have been reported to be involved in the
progression of HCC (17–20). In recent years, emerging studies
showed that aberrant expression of long non-coding RNAs (lncRNAs),
a class of ncRNAs more than 200 nucleotides in length, was involved
in the tumorigenesis and development of various cancer, including
HCC (21–24). Present research demonstrated that
lncRNA function as competing endogenous RNAs (ceRNAs) or act as
molecular ‘sponges’ to regulate the concentration, expression and
biological functions of miRNAs (25). For example, Hu et al
(26) found that long non-coding
RNA GAS5 suppressed the migration and invasion of HCC cells through
negative regulation of miR-21 and its downstream targets.
Nevertheless, the underlying mechanism for the miRNA/lncRNA
trans-regulatory function in HCC remains unclear.
miR-26a has been reported to act as tumor suppressor
via targeting specific downstream genes in several human cancers,
such as pancreatic, breast, bladder and gastric cancer (27–30).
MEG3, one of the most significantly downregulated lncRNAs in HCC,
encodes a long non-coding RNA of ~1700 nucleotides and function as
a tumor-suppressor in HCC (31).
Notably, the study also demonstrated that miR-29a could regulate
the expression level of MEG3 through the modulation of DNMT3B
activity in HCC (31). In the
present study, we identified that both miR-26a and MEG3 were
significantly decreased in HCC compared to matched non-malignant
tissues and showed negative correlation between them. This leads to
the hypothesis that miR-26a could also silence the expression of
MEG3 in HCC via similar mechanism. Therefore, we conducted the
present study to investigate the role of miR-26a and examined
whether miR-26a suppresses HCC growth and metastasis by targeting
DNMT3B/MEG3 axis.
Materials and methods
Samples and cell line growth
conditions
Forty-six patients who received curative resection
for HCC at the First Affiliated Hospital of Xi'an Jiaotong
University between June 2015 and May 2016 were enrolled in the
study. None of the patients received any chemotherapy or
radiotherapy treatments before surgery. Tumor tissues and matched
adjacent non-malignant tissues (3–5 cm distal to the edge of tumor)
were immediately frozen in liquid nitrogen or stored at −80°C until
RNA extraction. All patients signed the informed consent and the
study was approved by the Institutional Ethics Committee. The human
HCC cell lines (HepG2, Hep3B, MHCC97-H and SMCC-7721) and normal
liver cell line (LO-2) were obtained from the Shanghai Institutes
of Biological Sciences (Shanghai, China). Cells were grown in
Dulbeccos modified Eagles medium (DMEM; Gibco-Invitrogen, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (FBS) and 1%
antibiotics, cultured at 37°C in 5% CO2 humidified
incubator.
RNA isolation and quantitative
real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from specimens and cultured
cells by using TRIzol reagent (Invitrogen) according to the
manufacturer's protocol. qRT-PCR were used to detect the expression
levels of miRNA, mRNA and lncRNA on Thermal Cycler Dice Real-Time
system. The required reagents, including Mir-X miRNA qRT-PCR SYBR
kit, PrimeScript RT Master Mix and SYBR Premix Ex Taq™ II, were
purchased from Takara Bio (Shiga, Japan). The primers for specific
genes were as follows: DNMT3B-F, 5′-GCTCTTACCTTACCATCG-3′;
DNMT3B-R, 5′-GATACTCTGAACTGTCTCC-3′. MEG3-F,
5′-GGCTGAAGAACTGCGGATG-3′; MEG3-R, 5′-CCAGGAAGGAGACGAGAGG-3′.
Bio-Rad CFX Manager 2.1 software was used for analysis of qRT-PCR.
All experiments were performed in triplicate and 2−ΔΔCt
was regarded as the analytic formula.
Cell transfection
hsa-miR-26a mimics and negative control (NC) were
purchased from Guangzhou RiboBio, Co., Ltd. (Guangzhou, China).
Small interfering RNA against DNMT3B and negative control (NC) were
designed by Shanghai GenePharma, Co., Ltd. (Shanghai China). The
sequences of the si-DNMT3B were as follows: sense
5′-GUACCAUGCUCUGGAGAAATT-3′ and antisense
5′-UUUCUCCAGAGCAUGGUACTT-3′. Both HepG2 and MHCC97-H cells were
seeded onto 6-well plates, ensured 80% confluence at the time of
transfection. Transfection was carried out using Lipofectamine 2000
(Invitrogen) method following the manufacturer's protocol.
Cell viability assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide) assay was used to measure cell
proliferation. In Brief, HepG2 and MHCC97-H cells were seeded in
96-well plates at a density of 6,000 cells/well, incubated in
appropriate medium. After transfection for 24, 48, 72 and 96 h,
cells were subsequently cultured in medium with 0.5 mg/ml MTT for 4
h. Following removal of the supernatant and 150 µl dimethyl
sulfoxide (DMSO) was added. Then the optical density (OD) at 490 nm
was determined by EnSpire Multimode plate reader (Perkin-Elmer,
Waltham, MA, USA).
Transwell assay
Transwell assay was performed to assess cell
migration and invasion after transfection for 48 h. For invasion,
the upper chambers were pre-coated with Matrigel (BD Biosciences,
San Jose, CA, USA). Cells (2×104) in serum-free medium
were seeded into the top chambers of an insert (8 µm pore size;
Merck Millipore, Billerica, MA, USA), which were soaked into the
bottom chambers filled with complete medium. For migration,
Matrigel was not needed to coat the upper membrane, then the same
invasion assay was performed. After 48 h of incubation, the
chambers were fixed by 4% paraformaldehyde and then stained with
0.1% crystal violet. The cells passing through the film were
observed under an optical microscope, 10 field-of-view counts were
selected and the mean was calculated.
Western blot analysis
Proteins were collected from cells using RIPA buffer
(Pierce, Rockford, IL, USA), then separated by 12% SDS-PAGE and
transferred to the PVDF membrane (Merck Millipore). The membrane
was blocked with 5% non-fat milk and incubated with primary
antibody DNMT3B (Abcam, Cambridge, MA, USA) and β-actin (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) with 1:1,000 dilution at 4°C
overnight. Then the membrane was washed extensively followed by
incubation with secondary antibodies (Zhuangzhi Biotech, Co., Ltd.,
Xi'an, China) at room temperature for 1 h. The results were
obtained with chemiluminescent (Pierce). ImageJ software-based
analysis was used to quantify the bands obtained through western
blot analysis.
Luciferase reporter assay
Cells were cultured in 96-well plates with 50–70%
confluence 24 h before transfection. A mixture of 120 ng plasmid
vector including the wild-type (WT) or mutant (Mut) 3UTR of DNMT3B
mRNA (Shanghai Genechem, Co., Ltd, Shanghai, China) together with
50 nM hsa-miR-26a mimics or negative control (Guangzhou RiboBio)
were co-transfected. Transfections were performed using 0.45 µl of
FuGENE (Promega, Madison, WI, USA). After transfection for 48 h,
the luciferase activity was measured by Dual-Glo Luciferase assay
system (Promega) with Renilla luciferase activity as
control.
Statistical analysis
Data are expressed as means ± SD. Statistical
analysis was performed using the SPSS statistics 20.0. Students
t-test was performed to compare the differences of the two groups.
ANOVA was performed for comparison for more than two groups.
Correlation between two groups was analyzed using Pearsons
correlation coefficient analysis. The level of significance was set
at P<0.05.
Results
miR-26a and MEG3 are downregulated in
HCC tissues
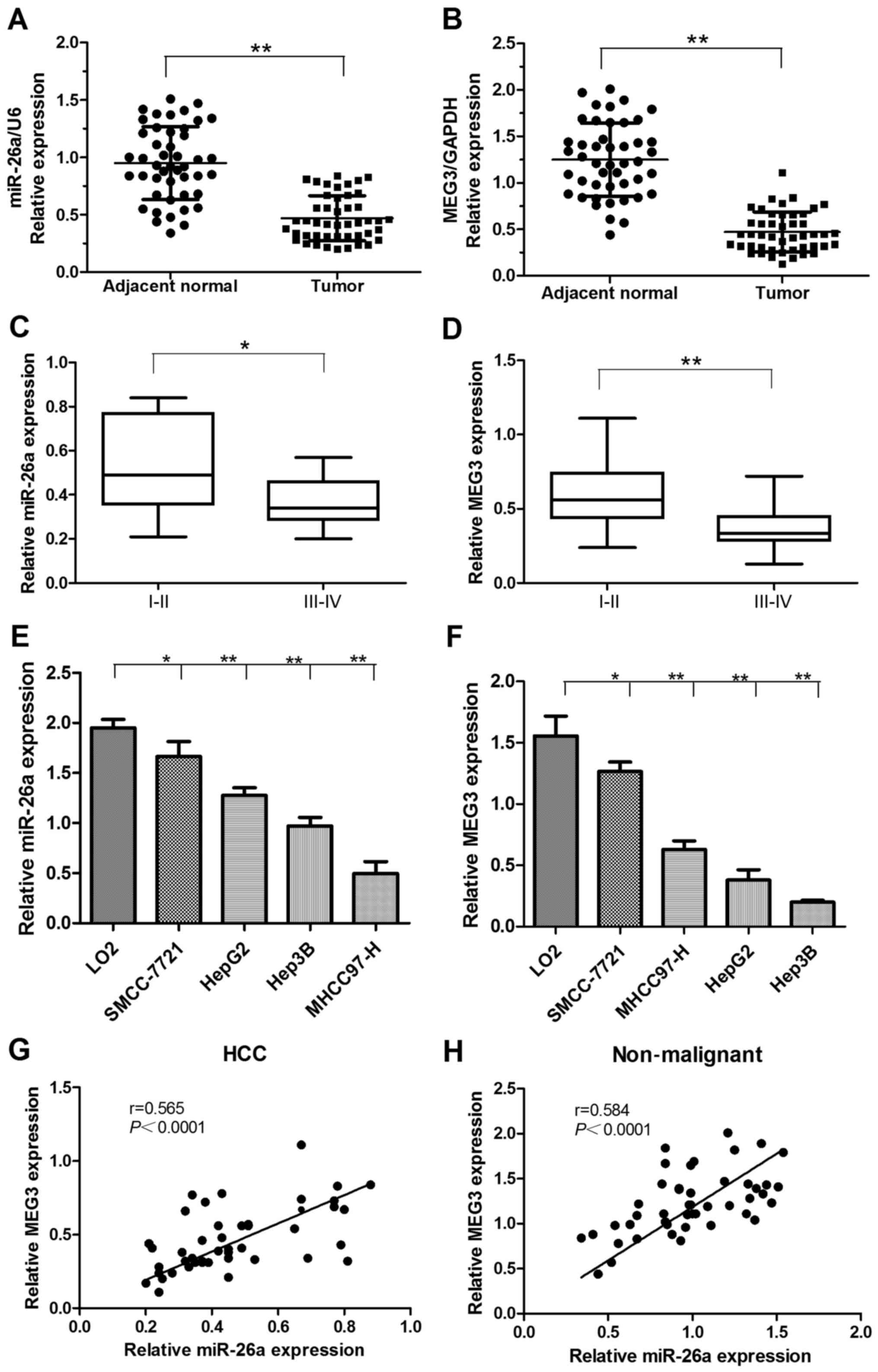
Expression of miR-26a and MEG3 was analyzed by
qRT-PCR in 46 HCC and matched adjacent non-malignant tissues. The
relative expression levels of miR-26a and MEG3 were significantly
downregulated in HCC tissues compared with matched non-malignant
specimens (Fig. 1A and B).
Moreover, the expression levels of miR-26a and MEG3 were negatively
correlated with the cancer clinical TNM stage, and the advanced TNM
stages were found with lower miR-26a and MEG3 expression (Fig. 1C and D). Furthermore, we
demonstrated that miR-26a and MEG3 expression levels in the HCC
cell lines were lower than normal liver LO2 cells (Fig. 1E and F). In addition, we analyzed
the relationship of their expression levels via Pearsons rank
correlation coefficient analysis. The result showed that the
expression level of miR-26a was positively correlated with MEG3
expression level in both HCC and matched non-malignant tissues
(Fig. 1G and H).
Relationship of miR-26a and MEG3
expression with clinicopathological features
The study revealed that the downregulation of
miR-26a and MEG3 in HCC were closely associated with the
clinicopathological parameters of HCC (Table I). The downregulation of miR-26a had
no significant association with patients age, sex, AFP or HBV
infection, but was significantly associated with tumor size,
histological differentiation and clinical TNM stage (Table I). As to MEG3 expression, the
decreased MEG3 levels were significantly associated with tumor size
and TNM stage. However, no statistically significant of its
expression was found with the age, sex, AFP, HBV infection or
histological differentiation (Table
I). These results suggested that downregulation of miR-26a and
MEG3 may contribute to the malignant progression of HCC.
 | Table I.The expression of miR-26a and lncMEG3
and its clinical characteristics in 46 HCC patients. |
Table I.
The expression of miR-26a and lncMEG3
and its clinical characteristics in 46 HCC patients.
|
|
| miR-26a | MEG3 |
|---|
|
|
|
|
|
|---|
| Clinical
factors | No. | Mean ± SD | P-value | Mean ± SD | P-value |
|---|
| All cases | 46 |
|
|
|
|
| Age (years) |
|
| 0.578 |
| 0.207 |
|
<59 | 20 | 0.489±0.177 |
| 0.432±0.190 |
|
|
≥59 | 26 | 0.457±0.213 |
| 0.504±0.230 |
|
| Sex |
|
| 0.855 |
| 0.737 |
|
Male | 35 | 0.474±0.191 |
| 0.478±0.216 |
|
|
Female | 11 | 0.462±0.217 |
| 0.453±0.215 |
|
| Tumor size
(cm) |
|
|
0.012a |
|
0.002a |
|
<5 | 27 | 0.615±0.177 |
| 0.566±0.207 |
|
| ≥5 | 19 | 0.350±0.112 |
| 0.378±0.118 |
|
| HBs Ag |
|
| 0.415 |
| 0.341 |
| + | 34 | 0.456±0.194 |
| 0.489±0.231 |
|
| − | 12 | 0.517±0.206 |
| 0.599±0.329 |
|
| AFP (µgl) |
|
| 0.058 |
| 0.081 |
|
<20 | 19 | 0.415±0.178 |
| 0.412±0.194 |
|
|
≥20 | 27 | 0.527±0.207 |
| 0.522±0.221 |
|
| Histological
differentiation |
|
|
0.009a |
| 0.304 |
| Well
and moderate | 22 | 0.427±0.190 |
| 0.505±0.244 |
|
|
Low | 24 | 0.595±0.165 |
| 0.439±0.179 |
|
| Clinical (AJCC)
stage |
|
|
0.013a |
|
0.004a |
|
I+II | 26 | 0.514±0.218 |
| 0.528±0.204 |
|
|
III+IV | 20 | 0.389±0.112 |
| 0.293±0.121 |
|
miR-26a inhibits HCC cell
proliferation, migration and invasion in vitro
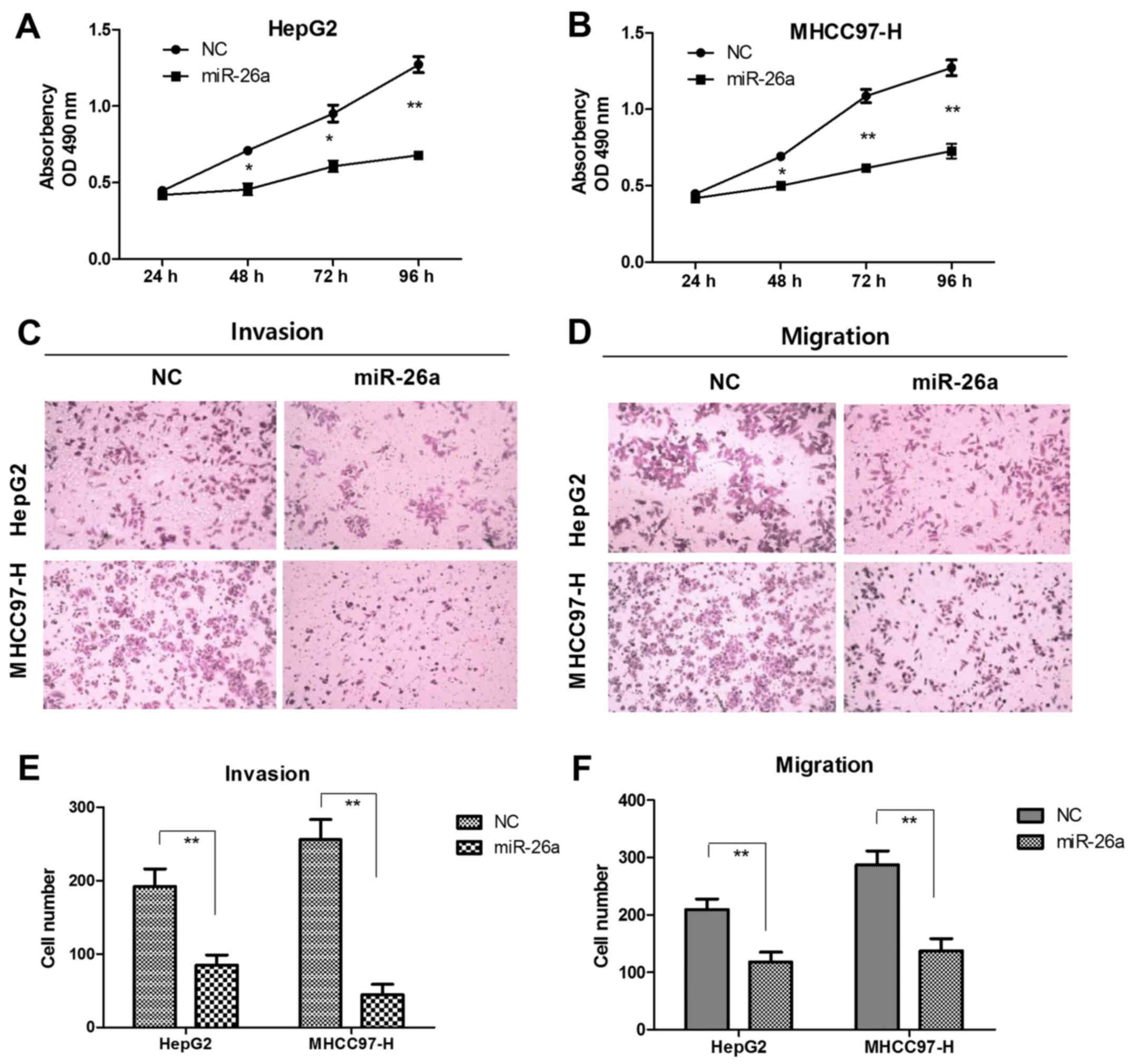
To further determine the potential biological
effects of miR-26a in HCC, we transfected the miR-26a mimics and
compared its performance on HepG2 and MHCC97-H cells. In contrast
to negative control group (NC), the ectopic expression of miR-26a
suppressed cell proliferation. Transfection of miR-26a mimics for
48, 72 or 96 h inhibited proliferation of HepG2 cells by 37.5%,
38.1% and 40.1% (Fig. 2A), MHCC97-H
cells by 30.2%, 43.2% and 44.1%, respectively (Fig. 2B). In Matrigel invasion assays
(Fig. 2C), upregulation of miR-26a
significantly decreased invasion of HepG2 cells (96 vs. 189 cells;
P<0.01) and MHCC97-H cells (45 vs. 258 cells; P<0.01)
(Fig. 2E). In migration assays
(Fig. 2D), mobility of HepG2 (109
vs. 226 cells; P<0.01) and MHCC97-H cells (119 vs. 288 cells;
P<0.01) (Fig. 2F) were
profoundly decreased after transfection of miR-26a mimics.
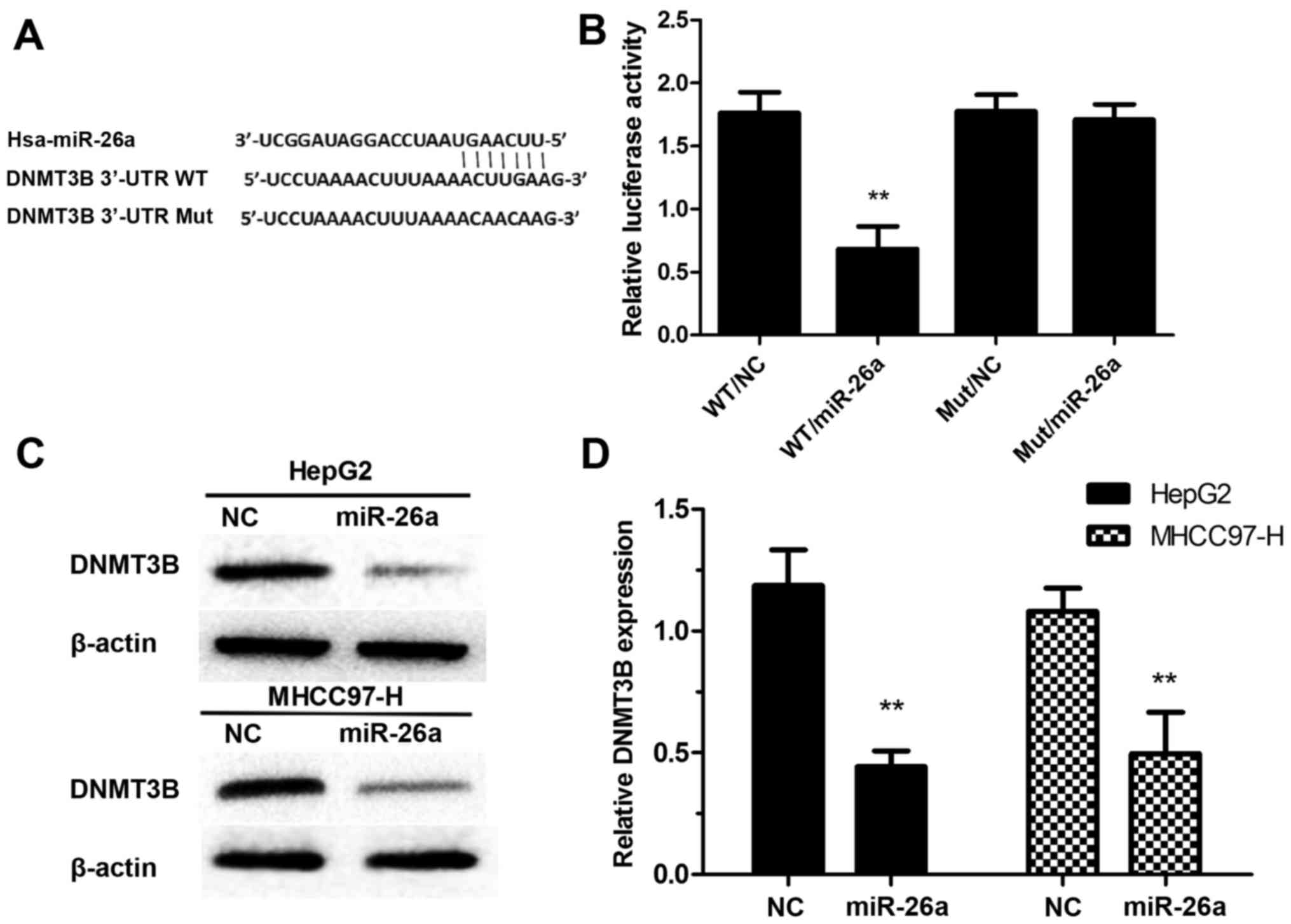
DNMT3B is a direct target of
miR-26a
We used the TargetScan (www.targetscan.org) and found a putative binding site
for miR-26a in the 3′-UTR of DNMTB (Fig. 3A). The luciferase reporter assay was
conducted to verify the prediction. The wild-type (WT) was
transfected with miR-26a mimics and negative control (NC), the
relative luciferase activity of WT/miR-26a was significantly
decreased compared with WT/NC. However, there was no significant
difference observed between Mut/miR-26a and Mut/NC (Fig. 3B). To verified the target
relationship between miR-26a and DNMT3B, we measured DNMT3B in HCC
cells after transfection of miR-26a mimics. We found that ectopic
expression of miR-26a suppressed the protein and mRNA expression
levels of DNMT3B in HepG2 and MHCC97-H cells (Fig. 3C and D).
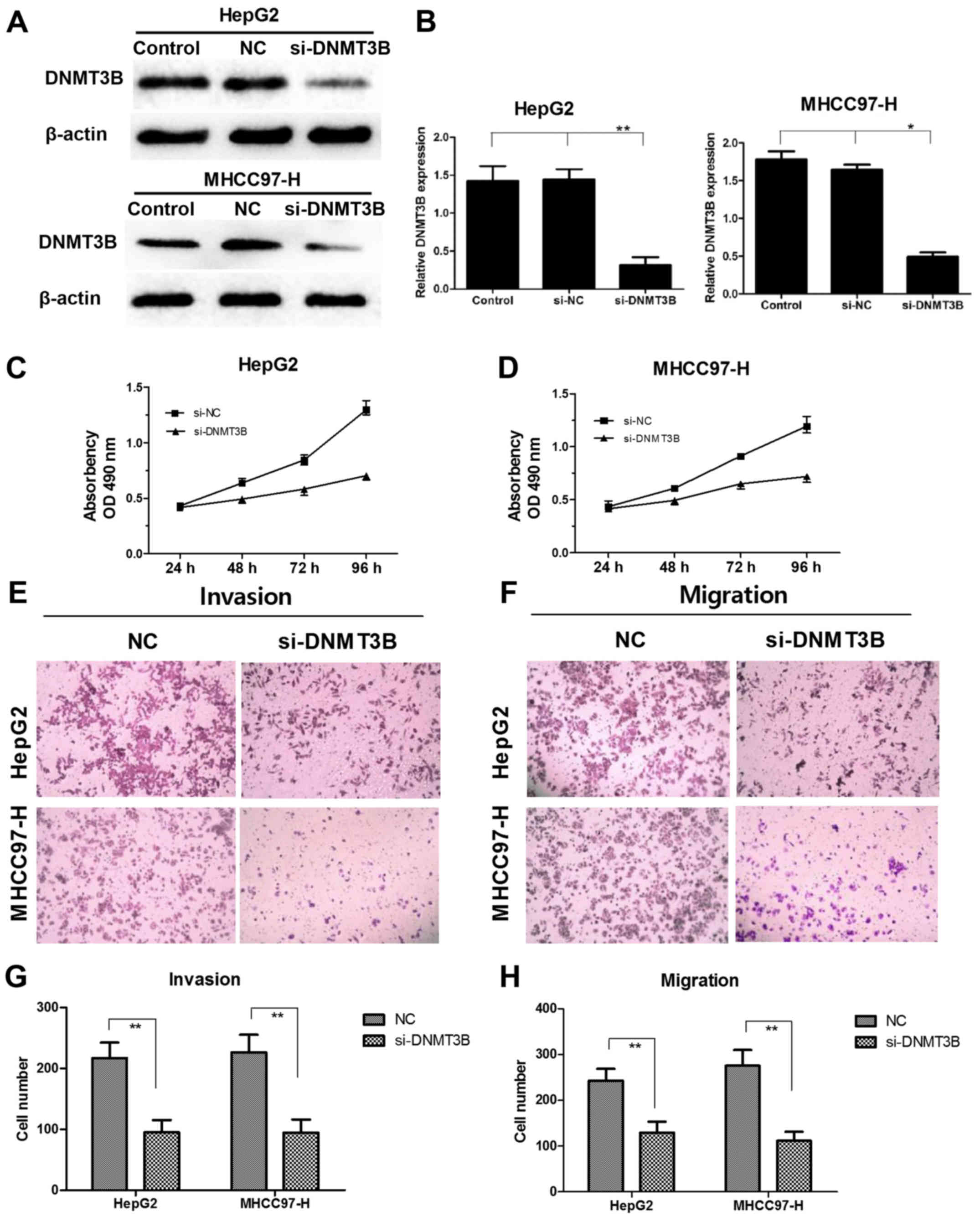
Knockdown of DNMT3B inhibits cell
proliferation, migration and invasion
To determine whether downregulation of DNMT3B had
similar effect with overexpression of miR-26a, a specific siRNA
against the DNMT3B gene transcript was introduced into HCC cells.
After transfection with siRNA-DNMT3B for 48 h, the mRNA and protein
expression level of DNMT3B was sharply decreased in HepG2 and
MHCC97-H cells (Fig. 4A). Moreover,
we found that siRNA-DNMT3B markedly suppressed cell proliferation
(Fig. 4B). In addition, the
migration and invasion of HCC cells were decreased compared to NC
group, respectively (Fig. 4C and
D). The effect was consistent with miR-26a overexpression, and
indicated that negative regulation of DNMT3B by miR-26a was
responsible for miR-26a-induced HCC cell progression at least in
part.
miR-26a upregulates MEG3 through
inhibition of the DNMT3B expression
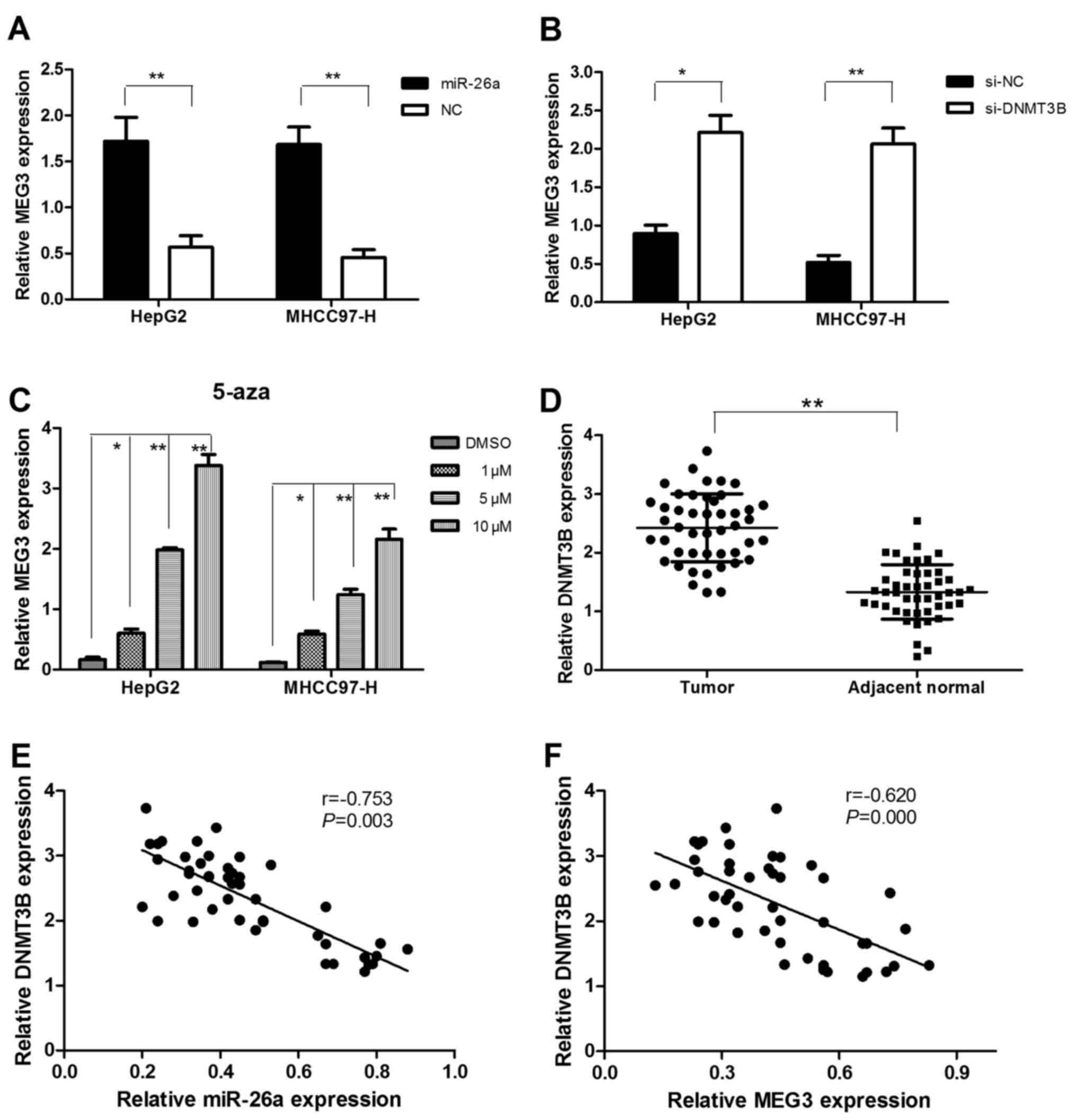
In vitro experiment, we found that
upregulated miR-26a could increase the expression of MEG3 in HepG2
and MHCC97-H cells (Fig. 5A).
Furthermore, knockdown of DNMT3B could also increase MEG3
expression in HepG2 and MHCC97-H cells (Fig. 5B). In combination with our previous
findings that miR-26a inhibited the expression of DNMT3B,
overexpression of miR-26a may increase the MEG3 expression through
reduction of the DNMT3B expression.
MEG3 expression is modulated by DNA
methylation
To ascertain the role of aberrant methylation in
deregulation of MEG3 in HCC cells, we assessed the effect of a DNA
demethylating agent (5-aza-CdR) on MEG3 expression by treating
HepG2 and MHCC97-H HCC cells with different densities (20, 40, 60
and 80 µM). The result showed that MEG3 expression was
significantly upregulated with increased density of 5-aza-CdR
compared with control (Fig. 5C). It
verified again that DNMT3B could imitate the effect of 5-aza-CdR to
downregulate the expression of MEG3 in HCC cells.
Upregulation of DNMT3B expression is
inversely correlated with miR-26a and MEG3 expression in HCC
To confirm molecular relationship observed in HCC
cells, we measured the DNMT3B expression in patient specimens and
found that DNMT3B was upregulated in 35 (35/46, 76.1%) HCC compared
to matched non-malignant tissues (Fig.
5D). Moreover, Pearsons correlation coefficient analysis
suggested that DNMT3B expression was inversely correlated with
miR-26a and MEG3 expression levels (Fig. 5E and F).
Discussion
Emerging evidence shows that deregulation of miRNAs
play a critical role in tumorigenesis and progression. However, the
effect and regulatory mechanisms are often controversial. miR-26a
acts as oncogene or tumor suppressor in several human cancers via
inhibiting its different target genes. Gao et al (28) found that miR-26a inhibited
proliferation and migration of breast cancer through regulating the
expression of MCL-1. The study of Deng et al (30) showed that miR-26a suppressed tumor
growth and metastasis by targeting FGF9 in gastric cancer. On the
contrary, some researchers also found that miR-26a was
significantly upregulated in glioblastoma and ovarian cancer
(32,33) and promoted lung cancer cell growth
through inhibition of PTEN/AKT1 pathway (34). Especially, the biological effect of
miR-26a involved in the carcinogenesis of prostate cancer was
reported to be in contrast (35,36).
Therefore, miR-26a possessed dual effects, depending on
organ-specific actions and peculiar target genes in different
cancer type. In the present study, miR-26a expression was
significantly downregulated in HCC compared with matched
non-malignant tissues, and low expression of miR-26a was
significantly associated with the tumor size, histological
differentiation and clinical stage. In vitro, overexpression
of miR-26a significantly suppressed the proliferation, migration
and invasion of HepG2 and MHCC97-H cells. These results confirmed
the inhibitory effect of miR-26a on growth and metastasis in
HCC.
miRNAs are suggested to regulate nearly 60% of
protein-encoding genes in mammals post-transcriptionally (37). Recent studies reported that loss of
combinations of some miRNAs was associated with hypermethylation
defect in breast cancer cell lines, and decreased expression of
regulatory miRNAs including miR-26a contributed to DNMT3B
overexpression (38). DNMT3B
belongs to methyltransferases family and is primarily responsible
for methylation of certain genomic regions (39). The overexpression of DNMT3B was
observed in some cancer types and contributed to tumor growth and
metastasis (40,41). Loss of DNMT3B regulation resulted in
complete or partial demethylation of the tumor-suppressive gene
MTSS1 promoters and contributed to HCC development (42). Therefore, investigating the
contribution of DNMT3B to the process of tumorigenesis is important
towards understanding the mechanisms through aberrant DNA
methylation in HCC. In the present study, we demonstrated that
DNMT3B expression was increased in HCC compared to the adjacent
non-malignant tissues, and verified that DNMT3B was a novel target
of miR-26. Moreover, knockdown of DNMT3B inhibited cell
proliferation, migration and invasion in HepG2 and MHCC97-H cells,
which limited the miR-26a-induced tumor suppressive effect. The
results suggested that miR-26a regulates proliferation, migration
and invasion of HCC cells via inhibition of DNMT3B.
A previous study revealed that MEG3 functions as a
tumor-suppressor in HCC, and miR-29a could modulate the expression
of MEG3 through the regulation of DNMT3B activity in HCC (31). Other researches reported that DNA
methylation could suppress the expression of MEG3 in meningioma and
pituitary tumors (43,44). Based on above studies, we assumed
that miR-26a could imitate the mechanism to silence the expression
of MEG3 in HCC through modulation of DNMT3B. Therefore, we
validated our hypothesis in vitro experiments, and the
results showed that overexpression of miR-26a increased the
expression of MEG3 through reducing expression of its potential
target gene DNMT3B in HCC cells. To better confirm the aberrant
methylation-dependent mechanism in deregulation of MEG3 in HCC, we
introduced the methylation-inhibitor 5-aza-CdR to measure the
expression of MEG3. The results showed that MEG3 expression was
robustly increased by incubation with 5-aza-CdR in HCC cells, and a
concentration-dependent relationship existed. In specimens, the
expression of MEG3 was downregulated in HCC compared with
non-malignant tissues and showed inverse correlation with either
miR-26a or DNMT3B expression in HCC tissues. Based on the above
observations, we demonstrated that miR-26a inhibits proliferation
and metastasis of HCC by regulating DNMT3B/MEG3 axis.
In summary, we confirmed the tumor suppressor role
of miR-26a in HCC, partly at least, via inhibition of DNMT3B/MEG3
axis. The present study complemented the molecular mechanisms of
miRNAs in the development of HCC, emphasizing epigenetic
modulation, in particular the association of DNA methylation
between expression of miRNAs and lncRNAs. A novel molecular
regulation axis-miR-26a/DNMT3B/MEG3 may become a promising
therapeutic target for HCC treatment. Further studies are needed to
investigate the upstream regulatory mechanism underlying miR-26a
expression and the downstream signaling pathways of DNMT3B in
HCC.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (nos. 30771895 and 81502095)
and the Key Program of International Cooperation Project of Shaanxi
Province, China (no. 2014KW23-04).
References
|
1
|
Bellissimo F, Pinzone MR, Cacopardo B and
Nunnari G: Diagnostic and therapeutic management of hepatocellular
carcinoma. World J Gastroenterol. 21:12003–12021. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomas MB, Jaffe D, Choti MM, Belghiti J,
Curley S, Fong Y, Gores G, Kerlan R, Merle P, ONeil B, et al:
Hepatocellular carcinoma: Consensus recommendations of the National
Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol.
28:3994–4005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Crissien AM and Frenette C: Current
management of hepatocellular carcinoma. Gastroenterol Hepatol (NY).
10:3994–4005. 2014.
|
|
4
|
Balogh J, Victor D III, Asham EH,
Burroughs SG, Boktour M, Saharia A, Li X, Ghobrial RM and Monsour
HP Jr: Hepatocellular carcinoma: A review. J Hepatocell Carcinoma.
3:41–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao ZC, Zheng SS, Wan YL, Jia CK and Xie
HY: The molecular mechanism underlying angiogenesis in
hepatocellular carcinoma: The imbalance activation of signaling
pathways. Hepatobiliary Pancreat Dis Int. 2:529–536.
2003.PubMed/NCBI
|
|
8
|
Kawaji H, Severin J, Lizio M, Forrest AR,
van Nimwegen E, Rehli M, Schroder K, Irvine K, Suzuki H, Carninci
P, et al: Update of the FANTOM web resource: From mammalian
transcriptional landscape to its dynamic regulation. Nucleic Acids
Res. 39:(Database). D856–D860. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amorim M, Salta S, Henrique R and Jerónimo
C: Decoding the usefulness of non-coding RNAs as breast cancer
markers. J Transl Med. 14:2652016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Beermann J, Piccoli MT, Viereck J and Thum
T: Non-coding RNAs in development and disease: Background,
mechanisms, and therapeutic approaches. Physiol Rev. 96:1297–1325.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arora A, Singh S, Bhatt AN, Pandey S,
Sandhir R and Dwarakanath BS: Interplay between metabolism and
oncogenic process: Role of microRNAs. Transl Oncogenomics. 7:11–27.
2015.PubMed/NCBI
|
|
13
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu X, Li Z, Yu J, Chan MT and Wu WK:
MicroRNAs predict and modulate responses to chemotherapy in
colorectal cancer. Cell Prolif. 48:503–510. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jansson MD and Lund AH: MicroRNA and
cancer. Mol Oncol. 6:590–610. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wen Y, Han J, Chen J, Dong J, Xia Y, Liu
J, Jiang Y, Dai J, Lu J, Jin G, et al: Plasma miRNAs as early
biomarkers for detecting hepatocellular carcinoma. Int J Cancer.
137:1679–1690. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hu S, Tao R, Wang S, Wang C, Zhao X, Zhao
H, Li L, Zhu S, He Y, Jiang X, et al: MicroRNA-21 promotes cell
proliferation in human hepatocellular carcinoma partly by targeting
HEPN1. Tumour Biol. 36:5467–5472. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zheng F, Liao YJ, Cai MY, Liu YH, Liu TH,
Chen SP, Bian XW, Guan XY, Lin MC, Zeng YX, et al: The putative
tumour suppressor microRNA-124 modulates hepatocellular carcinoma
cell aggressiveness by repressing ROCK2 and EZH2. Gut. 61:278–289.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang JP, Zeng C, Xu L, Gong J, Fang JH
and Zhuang SM: MicroRNA-148a suppresses the epithelial-mesenchymal
transition and metastasis of hepatoma cells by targeting Met/Snail
signaling. Oncogene. 33:4069–4076. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lai YY, Shen F, Cai WS, Chen JW, Feng JH,
Cao J, Xiao HQ, Zhu GH and Xu B: MiR-384 regulated IRS1 expression
and suppressed cell proliferation of human hepatocellular
carcinoma. Tumour Biol. 37:14165–14171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xue D, Zhou C, Lu H, Xu R, Xu X and He X:
LncRNA GAS5 inhibits proliferation and progression of prostate
cancer by targeting miR-103 through AKT/mTOR signaling pathway.
Tumour Biol. Oct 14–2016.(Epub Epub ahead of print). View Article : Google Scholar
|
|
22
|
Yim GW, Kim HJ, Kim LK, Kim SW, Kim S, Nam
EJ and Kim YT: Long non-coding RNA HOXA11 antisense promotes cell
proliferation and invasion and predicts patient prognosis in serous
ovarian cancer. Cancer Res Treat. Oct 11–2016.(Epub Epub ahead of
print). View Article : Google Scholar :
|
|
23
|
Wang J, Ye C, Xiong H, Shen Y, Lu Y, Zhou
J and Wang L: Dysregulation of long non-coding RNA in breast
cancer: an overview of mechanism and clinical implication.
Oncotarget. 8:5508–5522. 2016.
|
|
24
|
Liu YR, Tang RX, Huang WT, Ren FH, He RQ,
Yang LH, Luo DZ, Dang YW and Chen G: Long noncoding RNAs in
hepatocellular carcinoma: Novel insights into their mechanism.
World J Hepatol. 7:2781–2791. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia T, Liao Q, Jiang X, Shao Y, Xiao B, Xi
Y and Guo J: Long noncoding RNA associated-competing endogenous
RNAs in gastric cancer. Sci Rep. 4:60882014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu L, Ye H, Huang G, Luo F, Liu Y, Liu Y,
Yang X, Shen J, Liu Q and Zhang J: Long noncoding RNA GAS5
suppresses the migration and invasion of hepatocellular carcinoma
cells via miR-21. Tumour Biol. 37:2691–2702. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deng J, He M, Chen L, Chen C, Zheng J and
Cai Z: The loss of miR-26a-mediated post-transcriptional regulation
of cyclin E2 in pancreatic cancer cell proliferation and decreased
patient survival. PLoS One. 8:e764502013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao J, Li L, Wu M, Liu M and Xie X, Guo J,
Tang H and Xie X: MiR-26a inhibits proliferation and migration of
breast cancer through repression of MCL-1. PLoS One. 8:e651382013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin Y, Chen H, Hu Z, Mao Y, Xu X, Zhu Y,
Xu X, Wu J, Li S, Mao Q, et al: miR-26a inhibits proliferation and
motility in bladder cancer by targeting HMGA1. FEBS Lett.
587:2467–2473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deng M, Tang HL, Lu XH, Liu MY, Lu XM, Gu
YX, Liu JF and He ZM: miR-26a suppresses tumor growth and
metastasis by targeting FGF9 in gastric cancer. PLoS One.
8:e726622013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Braconi C, Kogure T, Valeri N, Huang N,
Nuovo G, Costinean S, Negrini M, Miotto E, Croce CM and Patel T:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo P, Lan J, Ge J, Nie Q, Guo L, Qiu Y
and Mao Q: MiR-26a enhances the radiosensitivity of glioblastoma
multiforme cells through targeting of ataxia-telangiectasia
mutated. Exp Cell Res. 320:200–208. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen W, Song M, Liu J, Qiu G, Li T, Hu Y
and Liu H: MiR-26a promotes ovarian cancer proliferation and
tumorigenesis. PLoS One. 9:e868712014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu B, Wu X, Liu B, Wang C, Liu Y, Zhou Q
and Xu K: MiR-26a enhances metastasis potential of lung cancer
cells via AKT pathway by targeting PTEN. Biochim Biophys Acta.
1822:1692–1704. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao S, Ye X, Xiao L, Lian X, Feng Y, Li F
and Li L: MiR-26a inhibits prostate cancer progression by
repression of Wnt5a. Tumour Biol. 35:9725–9733. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo K, Zheng S, Xu Y, Xu A, Chen B and Wen
Y: Loss of miR-26a-5p promotes proliferation, migration, and
invasion in prostate cancer through negatively regulating SERBP1.
Tumour Biol. 37:12843–12854. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Melo SA and Esteller M: Dysregulation of
microRNAs in cancer: Playing with fire. FEBS Lett. 585:2087–2099.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sandhu R, Rivenbark AG and Coleman WB:
Loss of post-transcriptional regulation of DNMT3b by microRNAs: A
possible molecular mechanism for the hypermethylation defect
observed in a subset of breast cancer cell lines. Int J Oncol.
41:721–732. 2012.PubMed/NCBI
|
|
39
|
Nishida N, Nishimura T, Nakai T, Chishina
H, Arizumi T, Takita M, Kitai S, Yada N, Hagiwara S, Inoue T, et
al: Genome-wide profiling of DNA methylation and tumor progression
in human hepatocellular carcinoma. Dig Dis. 32:658–663. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen WC, Chen MF and Lin PY: Significance
of DNMT3b in oral cancer. PLoS One. 9:e899562014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu Z, Wang L, Wang LE, Sturgis EM and Wei
Q: Polymorphisms of the DNMT3B gene and risk of squamous cell
carcinoma of the head and neck: A case-control study. Cancer Lett.
268:158–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fan H, Chen L, Zhang F, Quan Y, Su X, Qiu
X, Zhao Z, Kong KL, Dong S, Song Y, et al: MTSS1, a novel target of
DNA methyltransferase 3B, functions as a tumor suppressor in
hepatocellular carcinoma. Oncogene. 31:2298–2308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao J, Dahle D, Zhou Y, Zhang X and
Klibanski A: Hypermethylation of the promoter region is associated
with the loss of MEG3 gene expression in human pituitary tumors. J
Clin Endocrinol Metab. 90:2179–2186. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is associated
with meningioma pathogenesis and progression. Cancer Res.
70:2350–2358. 2010. View Article : Google Scholar : PubMed/NCBI
|