Introduction
In the United States, prostate cancer (PCa) has
become the most commonly diagnosed male cancer and the second
leading cause of cancer-related deaths, with an estimated 220,800
annual diagnoses and 27,540 annual deaths during 2015 (1). With increasing incidence and
mortality, PCa is a current global health issue. Although many
advanced therapies have significantly improved the survival of PCa
patients, the number of deaths is still substantial. Current
treatments are not effective once cancer metastasis occurs, as
there are no therapies that cure metastatic disease or prevent the
metastatic process. Epithelial-to-mesenchymal transition (EMT) is a
key link in the induction of pathological changes in cancer cells,
which plays a central role in cancer development and progression.
Carcinoma cells undergo EMT which is a reversible phenotypic change
in which epithelial cells lose cell-cell adhesion and epithelial
polarization and are thus converted to a mesenchymal phenotype,
gaining increased motility and invasiveness (3). In PCa, the EMT may contribute to
metastasis and intravasation from the organ of origin to distant
sites. Therefore, in order to more successfully treat PCa, a deeper
understanding of the biological mechanisms that are involved in
regulating PCa metastasis is necessary.
The RON and c-Met receptor are ~180-kDa
heterodimeric proteins composed of a 35-kDa α-chain and a 145-kDa
β-chain linked by disulfide bonds that share 60% similarity in
their functional domains (4,5). RON
and c-Met are the only members of the MET proto-oncogene family
expressed in humans, and they play important roles during embryonic
development, but are mostly dormant after birth. However, both
receptors are activated once tissue becomes cancerous. RON and
c-Met are activated by their corresponding ligands [macrophage
stimulating protein (MSP) for RON and hepatocyte growth factor
(HGF) for c-Met]. RON was firstly identified as a member of the MET
subfamily by Ronsin and colleagues in 1993 (6). Shortly afterwards, other researchers
found that MSP acts as a specific ligand for RON. c-Met was first
discovered in 1984 as an oncogene and its ligand HGF was identified
in 1991 (7). The MSP/RON and
HGF/c-Met signaling pathways have gained increasing attention in
the fields of tumor biology and therapy over the past 20 years.
They have been found to be necessary for the survival and growth of
cancer cells in many tumors (8).
Patients with RON+/c-Met+ tumors have
significantly poorer 10-year disease-free survival rates than those
with RON−/c-Met− tumors (11.8 vs. 79.3%,
respectively; P=0.009) (9). Thus,
RON/c-Met expression has potential as a prognostic biomarker for
cancer progression in some cancers, and it is crucial to understand
the mechanism of RON/c-Met signaling in PCa.
Foretinib is a high affinity multikinase of the
tyrosine kinase inhibitor (TKI) family that inhibits c-Met at a
half-maximal inhibitory concentration (IC50) of 0.4 nM
and RON at an IC50 of 3 nM. It has been demonstrated
that foretinib has antitumor activity in different tumor types
(10–13) and phase II clinical trials with
foretinib have been initiated to investigate its ability to treat
diverse cancers, including head and neck, renal, glioblastoma,
gastric and liver cancers (14–18).
To date, however, the ability of foretinib to treat PCa is still
unknown.
In the present study, we identified a markedly
positive co-expression of RON and c-Met in human PCa tissues by
immunohistochemical staining and quantitative real-time PCR. We
demonstrated that targeting RON and c-Met using foretinib
(GSK1363089) significantly suppressed metastasis and promoted the
reversal of EMT in PCa cells. Additionally, we further revealed
that RON and c-Met facilitate metastasis via ERK1/2 signaling.
Materials and methods
Cell culture
PCa cell lines (PC3, DU145, 22RV1 and LNCaP) were
purchased from the Chinese Academy of Sciences Cell Bank (Shanghai,
China). All the cell lines were maintained in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA,
USA) and 1% penicillin/streptomycin (Jinuo Biomedical Technology,
Co., Ltd., Hangzhou, China), in a 5% CO2 atmosphere at
37°C. Treatment with foretinib (Shanghai Selleck Chemicals, Co.,
Ltd., Shanghai, China), recombinant human HGF (Thermal Tech) and
recombinant human MSP (R&D Systems, Minneapolis, MN, USA) was
performed according to the manufacturers instructions.
Clinical samples and
immunohistochemical staining
Ten pairs of PCa and their corresponding peri-cancer
tissues were obtained from patients at The Second Affiliated
Hospital of Zhejiang University School of Medicine (Hangzhou,
China). Ethical approval was obtained from the Second Affiliated
Hospital of Zhejiang University School of Medicine Research Ethics
Committee, and written informed consent was obtained from all
patients. PCa tissue samples were used for quantitative reverse
transcription PCR (qRT-PCR) and sectioned at 5-µm sections for IHC.
The slides were incubated with human anti-RON (1:100), c-Met
(1:200) and secondary antibodies, and the sections were developed
in diaminobenzidine solution under a microscope and counterstained
with hematoxylin. Negative control slides omitting the primary
antibodies were included in all assays.
Western blot analysis
Western blot analysis was performed to detect the
levels of RON (1:5,000) (a gift from Professor Hangping Yao,
Zhejiang University, Hangzhou, China), c-Met (1:1,000; Cell
Signaling Technology, Danvers, MA, USA), E-cadherin (1:2,000; Cell
Signaling Technology), N-cadherin (1:1,000; Ruiying Biological,
Suzhou, China), AR (1:2,000; Cell Signaling Technology), p-ERK1/2
(1:1,000; Cell Signaling Technology), ERK1/2 (1:1,000; Cell
Signaling Technology) and GAPDH (1:5,000; Proteintech Group Inc.,
Chicago, IL, USA). Cell lysates were extracted with RIPA buffer
(Beyotime Institute of Biotechnology, Shanghai, China) with
proteinase inhibitors (Shanghai Selleck Chemicals) and a cocktail
of phosphatase inhibitors (Shanghai Selleck Chemicals). Protein
samples were separated by SDS-PAGE and transferred to an PVDF
membrane. After blocking the non-specific binding for 2 h with 5%
non-fat milk, the membranes were incubated overnight at 4°C with
the specific primary antibodies. On the following day, the
membranes were further incubated with HRP-conjugated secondary
antibodies for 2 h at room temperature and recorded with the
ChemiDoc XRS System (Bio-Rad Laboratories, Hercules, CA, USA).
Quantitative real-time PCR
Total RNA was isolated from tumor sample or cells
using TRIzol reagent (Takara Bio, Dalian, China) and reversely
transcribed using the PrimeScript RT-PCR kit (Takara Bio) according
to the protocol. qRT-PCR was performed on the ABI 7900 Prism HT
(Applied Biosystems, Foster City, CA, USA), followed by melting
curve analysis. The 2−∆∆Ct method was used to assess the
gene expression levels. Primers used are as follows: RON forward,
CTT TGACGTGAAGTACGTGGT and reverse, CGTATGGCTA CAAACACAGCAC; c-Met
forward, AGCGTCAACAGA GGGACCT and reverse, AGCGTCAACAGAGGGACCT;
GAPDH forward, GCACCGTCAAGGCTGAGAAC and reverse,
TGGTGAAGACGCCAGTGGA.
Immunofluorescence
Cells were cultured in a 24-well plate, and then
were fixed with 4% paraformaldehyde for 20 min, permeabilized with
0.1% Triton X-100 for 15 min and blocked with 5% bovine serum
albumin (BSA; Sharp, Solon, OH, USA). The samples were incubated
with primary antibody at 4°C overnight, and then were incubated
with DyLight 594 conjugated goat anti-rabbit IgG (1:200; Abbkine,
Inc., Redlands, CA, USA) at room temperature for 2 h. Nuclei were
stained with DAPI (Beyotime Institute of Biotechnology) for 15 min.
Following a final rinse of three times with phosphate-buffered
saline (PBS), the cells were imaged using inverted fluorescent
microscope (Leica Microsystems, Wetzlar, Germany).
Migration and invasion assays
A 24-well Transwell plate was used to assess the
migration and invasion of the PCa cells. Briefly, for the Transwell
migration assay, 5×104 cells in 200 µl serum-free
RPMI-1640 were seeded on the upper chamber, 800 µl of medium
containing 5% FBS was placed in the lower chamber of each well and
incubated at 37°C for 24 h. The cell invasion assay was carried out
similarly, except that Matrigel (BD Biosciences, San Jose, CA, USA)
was added to each well according to the manufacturers instructions
before cells were seeded on the upper chamber. After the time of
incubation, non-migrated or non-invaded cells were gently removed
from the upper chamber with a cotton swab. The cells in the lower
chamber were fixed using 4% paraformaldehyde and stained with 0.1%
crystal violet. The cells that were located on the underside of the
filter were counted in 5 randomly selected fields.
Wound healing assay
Wound healing assay was conducted to examine the
capacity of cell migration. Briefly, a wound was generated when the
cells reached 80–90% confluency in a 6-well plate by scratching the
surface with a 1,000-µl pipette tip. The cells were then incubated
in 1% FBS for 24 h, and then photographed using phase contrast
microscopy (Leica Microsystems). The distance between the wound
edges of the scratch area was analyzed using Adobe Photoshop 7.0.
All experiments were performed in triplicate.
Small interfering RNA
transfection
Small interfering RNA (siRNA) duplexes targeting RON
and c-Met genes (Shanghai GenePharma, Co., Ltd., Shanghai, China)
were transfected into PCa cells using Lipofectamine 2000 reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturers
instructions: siRNA duplexes targeting RON (forward,
5′-CCUGCUGGACACACUAAUUTT-3′ and reverse,
5′-AAUUAGUGUGUCCAGCAGGTT-3); c-Met (forward,
5′-GCAACAGCUGAAUCUGCAATT-3′ and reverse,
5′-GCAACAGCUGAAUCUGCAATT-3′); and scramble siRNA duplex (forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′). The silencing effect was measured by
western blotting and qRT-PCR 48 h post-transfection.
Cell scattering
Cells were plated in a 6-well plate at a density of
500 cells/well and allowed to form colonies for 7 days. Cell
colonies were incubated with MSP and HGF for 24 h and then
photographed. This protocol was according to a previous study
(19).
Statistical analysis
Statistical calculations and graphical presentation
were performed using GraphPad Prism 5.0. Data are expressed as mean
and SD, unless otherwise indicated. Continuous variables were
evaluated using an unpaired Students t-test for comparisons between
two groups. Statistical significance was confirmed at
P<0.05.
Results
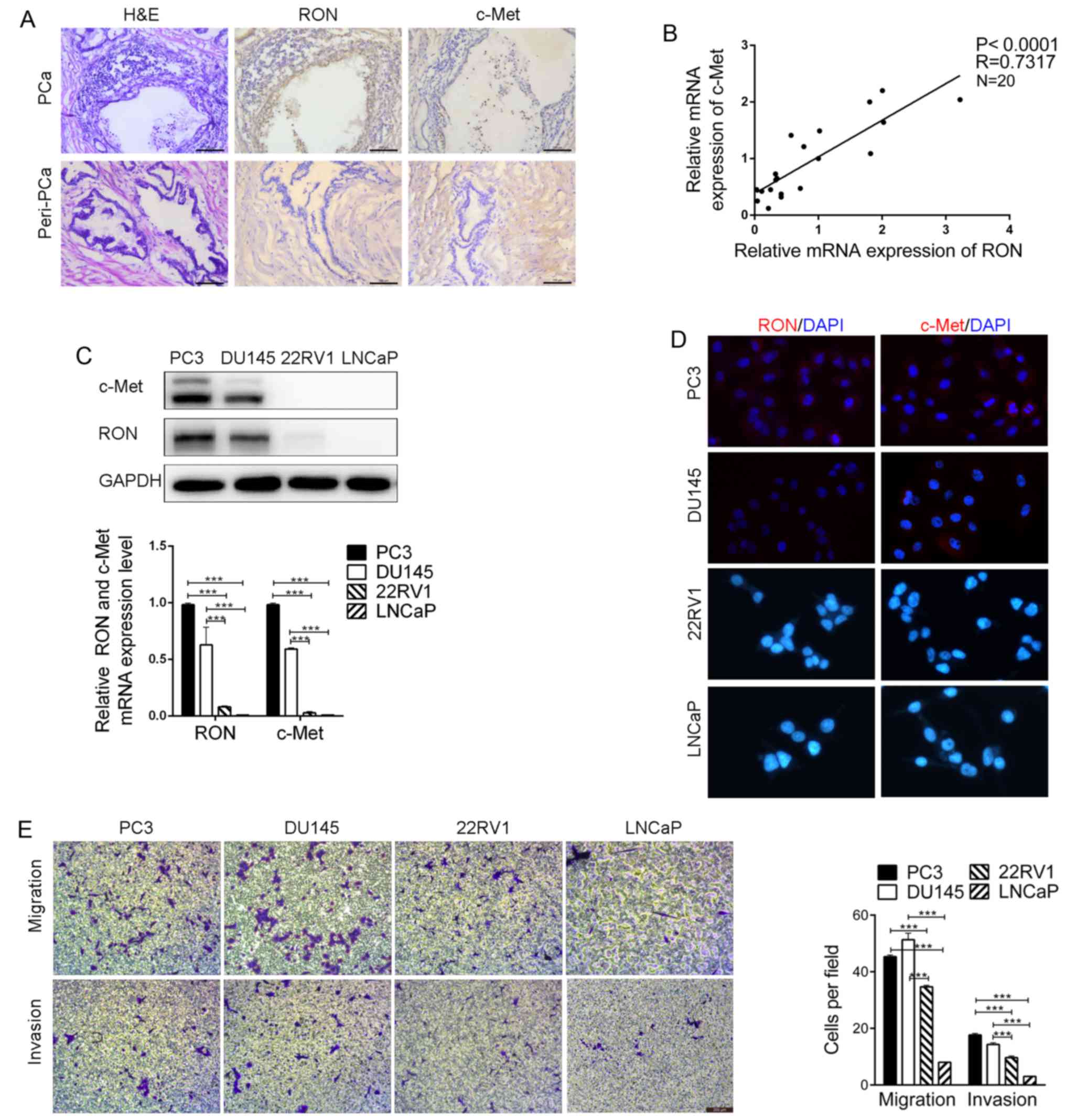
Robust RON and c-Met expression in
PCa
RON and c-Met expression has been assessed in a
variety of types of cancer, yet there has been no study concerning
the co-expression of RON and c-Met in PCa. To determine the
expression of RON and c-Met, we first analyzed RON and c-Met in PCa
tissues and non-cancerous prostate tissues by IHC. As shown in
Fig. 1A, high RON and c-Met
expression was observed in the PCa tissues and only minimal
expression was noted in the non-cancerous prostate tissues.
Moreover, a positive correlation was observed between the relative
expression levels of RON and c-Met mRNA in the prostate tissues
(Fig. 1B). To further explore the
role of RON and c-Met in PCa, we evaluated the expression of RON
and c-Met in PCa cell lines. As shown via western blotting, qRT-PCR
and immunofluorescent staining, RON and c-Met were highly expressed
in the PC3 and DU145 cells, but exhibited little to no expression
in the 22RV1 and LNCaP cells (Fig. 1C
and D). In contrast, 22RV1 and LNCaP cells displayed high
expression of AR, while PC3 and DU145 cells exhibited negligible
expression of this protein (data not shown). Further studies of the
migration and invasive potential of PCa cell lines revealed that
PC3 and DU145 cells, which have high RON and c-Met expression,
showed relatively higher migration and invasion abilities when
compared to the 22RV1 and LNCaP cells, two low RON and
c-Met-expressing prostate cancer cell lines (Fig. 1E). Therefore, the expression of RON
and c-Met was found to be positively correlated with the metastatic
potential of the PCa cell lines.
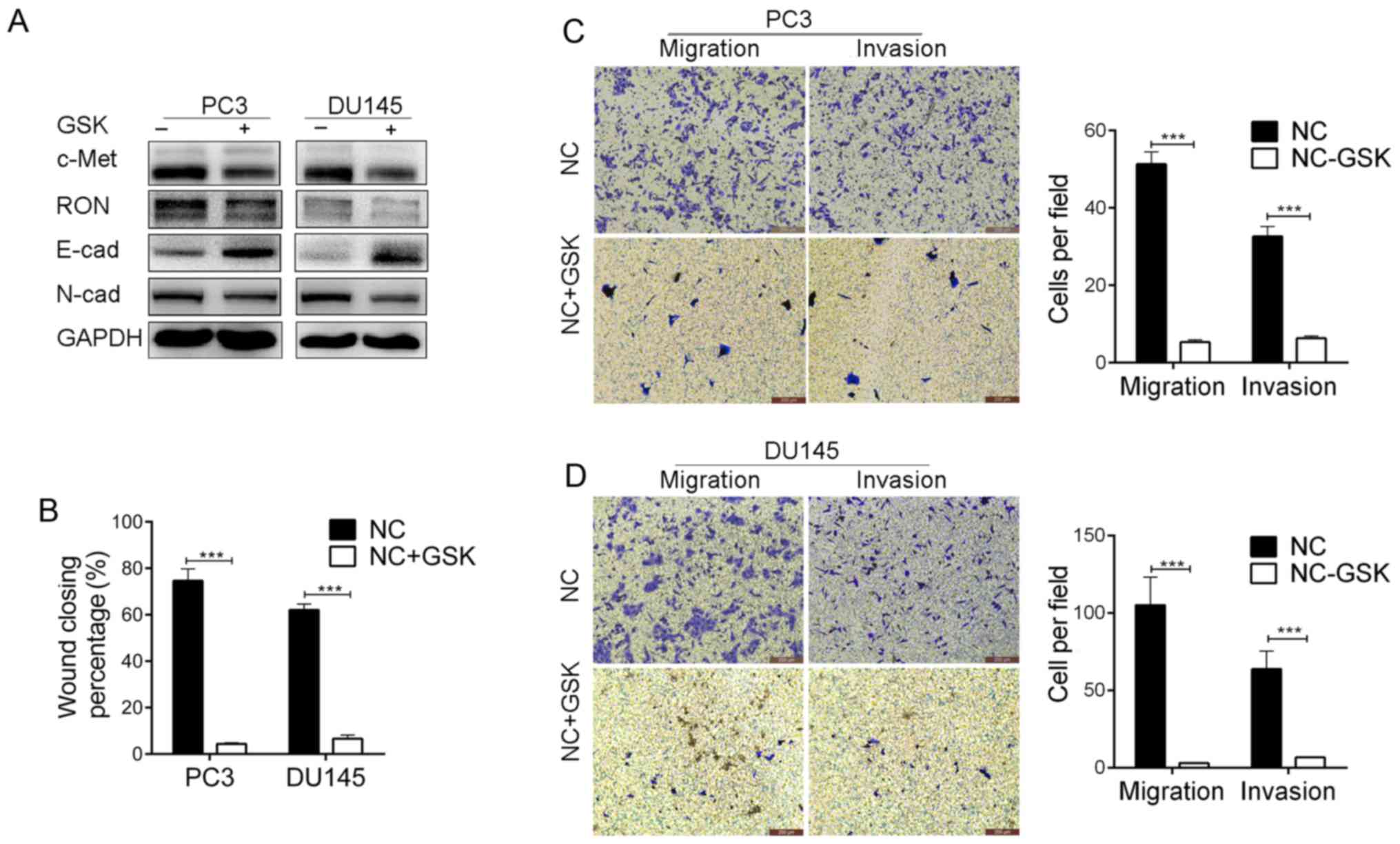
Inhibition of RON and c-Met using
foretinib suppresses PCa cell migration and invasion
To further investigate the role of RON and c-Met in
PCa, we next determined whether RON and c-Met could be therapeutic
targets for PCa. PC3 and DU145 cells were exposed to the RON and
c-Met inhibitor foretinib (GSK1363089) for 24 h. The effect of
foretinib on RON and c-Met expression was confirmed by western blot
analysis (Fig. 2A). Foretinib (20
nM) downregulated RON and c-Met expression in the cells, while also
leading to increased E-cadherin and decreased N-cadherin expression
(Fig. 2A). E-cadherin is a
canonical epithelial cell marker, while N-cadherin is canonically
associated with mesenchymal cells, suggesting that foretinib
treatment reversed the EMT in these cells. In contrast, these
responses to foretinib were not observed in the 22RV1 and LNCaP
cells (data not shown). The migratory and invasive potentials of
PC3 and DU145 cells were also extremely impaired after treatment
with foretinib (20 nM). As shown in Fig. 2B-D, foretinib (20 nM) significantly
impaired both the wound closure ability and invasive potential of
the PC3 and DU145 cells. These data suggest that RON and c-Met
mediate PCa metastasis, and foretinib may have potential as a
therapeutic agent for PCa metastasis.
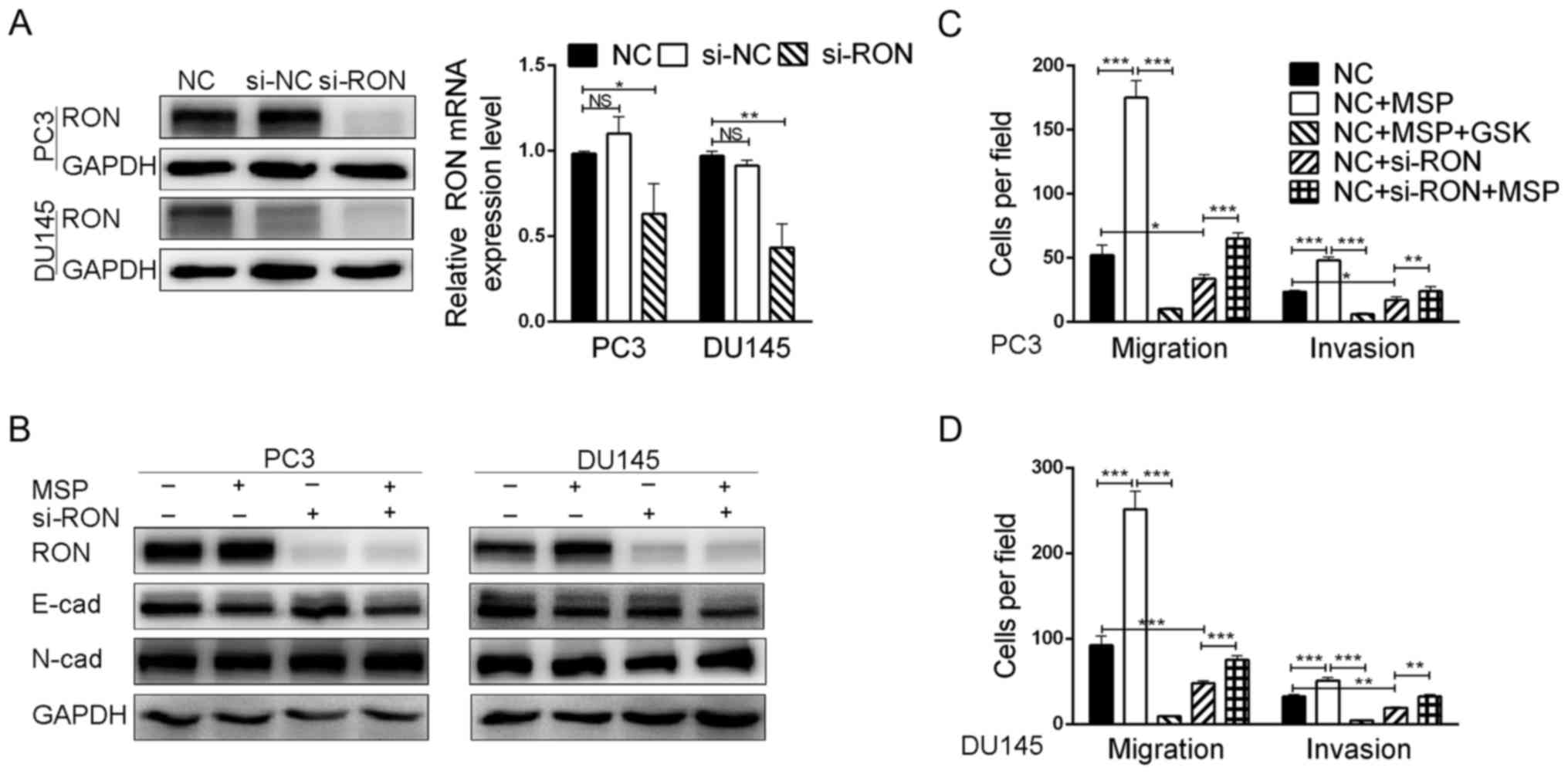
RON enhances cell migration and
invasion of PCa cells
We next investigated whether the knockdown of RON by
siRNA would phenocopy the functional effects of foretinib treatment
and promote the reversal of EMT by examining the expression of
characteristic epithelial and mesenchymal cell markers.
Transfection of the RON-targeting siRNA in the PC3 and DU145 cells
almost completely abrogated RON protein expression, and led to a
60–70% decrease in RON mRNA expression (Fig. 3A). Remarkably, the knockdown of RON
did not substantially alter E-cadherin and N-cadherin expression
compared with the control groups (Fig.
3B). Migration and invasion assays were carried out to evaluate
the effects of RON knockdown on cell migratory and invasive
capacity. Our data revealed that the knockdown of RON attenuated
the cell migratory and invasive abilities of the PC3 and DU145
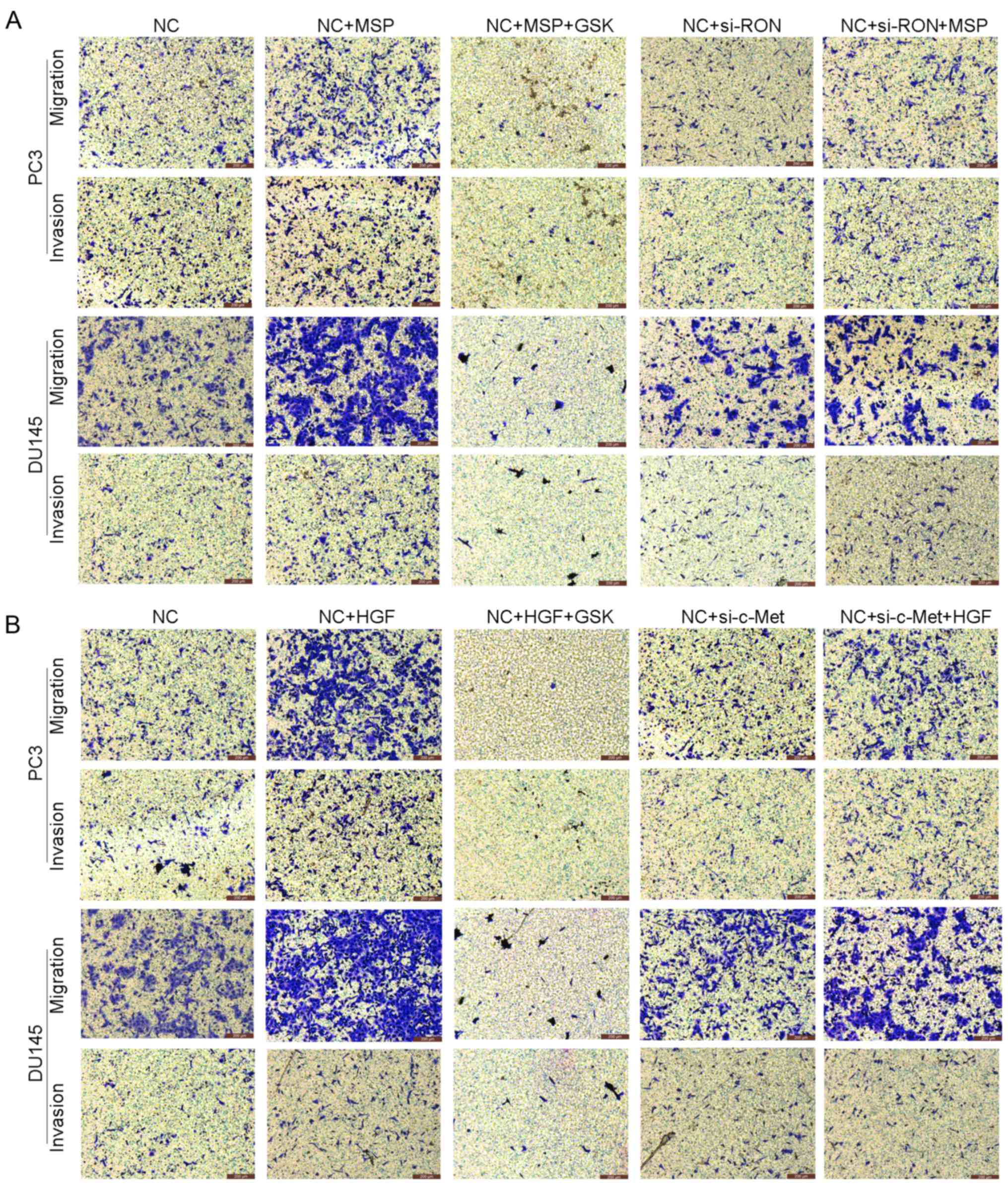
cells (Figs. 3C and D and 5A). Furthermore, in order to analyze the
function of RON, we exogenously activated RON with recombinant
human MSP (100 ng/ml) for 24 h. After treatment with MSP, the
invasive and migratory abilities of the PC3 and DU145 cells were
significantly increased (Figs. 3C and
D and 5A). Treatment with MSP
did not substantially alter E-cadherin and N-cadherin expression
(Fig. 3B), nor did it cause the
cells to adopt a spindle-shaped mesenchymal morphology (data not
shown).
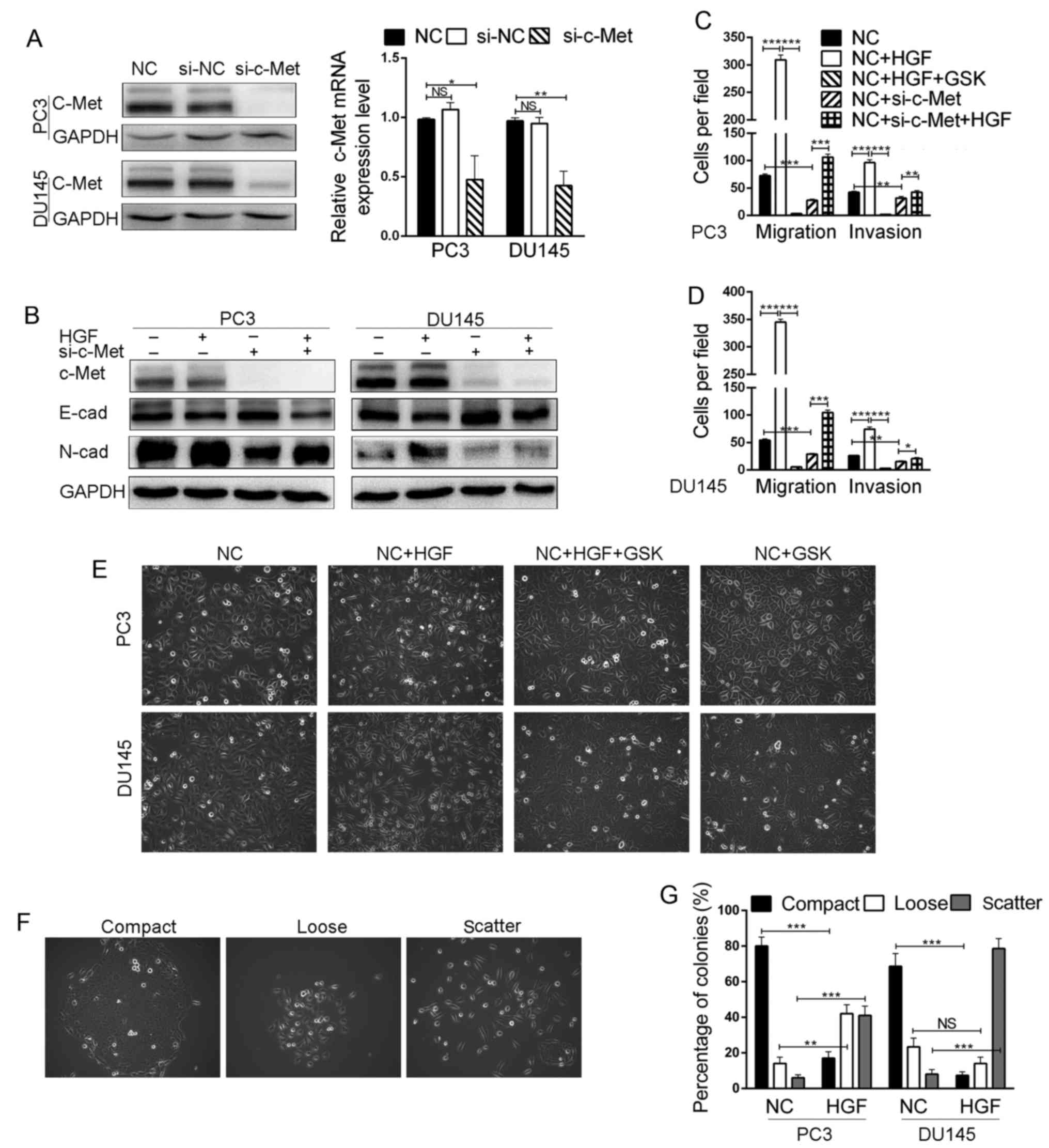
c-Met enhances cell migration and
invasion in PCa
To further understand the function of c-Met in PCa
progression, we decreased the expression of c-Met in the PC3 and
DU145 cells via siRNA-mediated knockdown. We confirmed the
efficiency of the siRNA by western blot analysis and qRT-PCR, and
observed almost complete loss of c-Met protein expression and a
60–70% decrease in c-Met mRNA expression in both the PC3 and DU145
cells (Fig. 4A). We next examined
the expression of characteristic epithelial and mesenchymal
markers. Remarkably, the knockdown of c-Met increased E-cadherin
and reduced N-cadherin expression in the PC3 and DU145 cells
compared to these levels in the control groups (Fig. 4B). Migration and invasion assays
were utilized to determine the effects of c-Met on the migratory
and invasive capacities of cells. The results showed that knockdown
of c-Met expression resulted in significant suppression of the
migration and invasion of both PC3 and DU145 cell lines (Figs. 4C and D and 5B). Following treatment with HGF, the
invasive and migratory abilities of the PC3 and DU145 cells were
significantly increased (Figs. 4C and
D and 5B). In addition,
treatment with HGF induced cell scattering and caused the cells to
adopt a spindle-shaped mesenchymal morphology (Fig. 4E), accompanied by increased
expression of N-cadherin and decreased expression of E-cadherin
(Fig. 4B). In contrast, treatment
with MSP had no effect (Fig. 3B).
Moreover, foretinib could inhibit these in vitro changes in
morphology, invasiveness and migratory potential caused by HGF in
PCa cells (Fig. 4C and E and
5B). These results indicate that
the HGF/c-Met signaling pathway may be a dominant factor in PCa
cell EMT and play an important role in regulating PCa cell
migration and invasion.
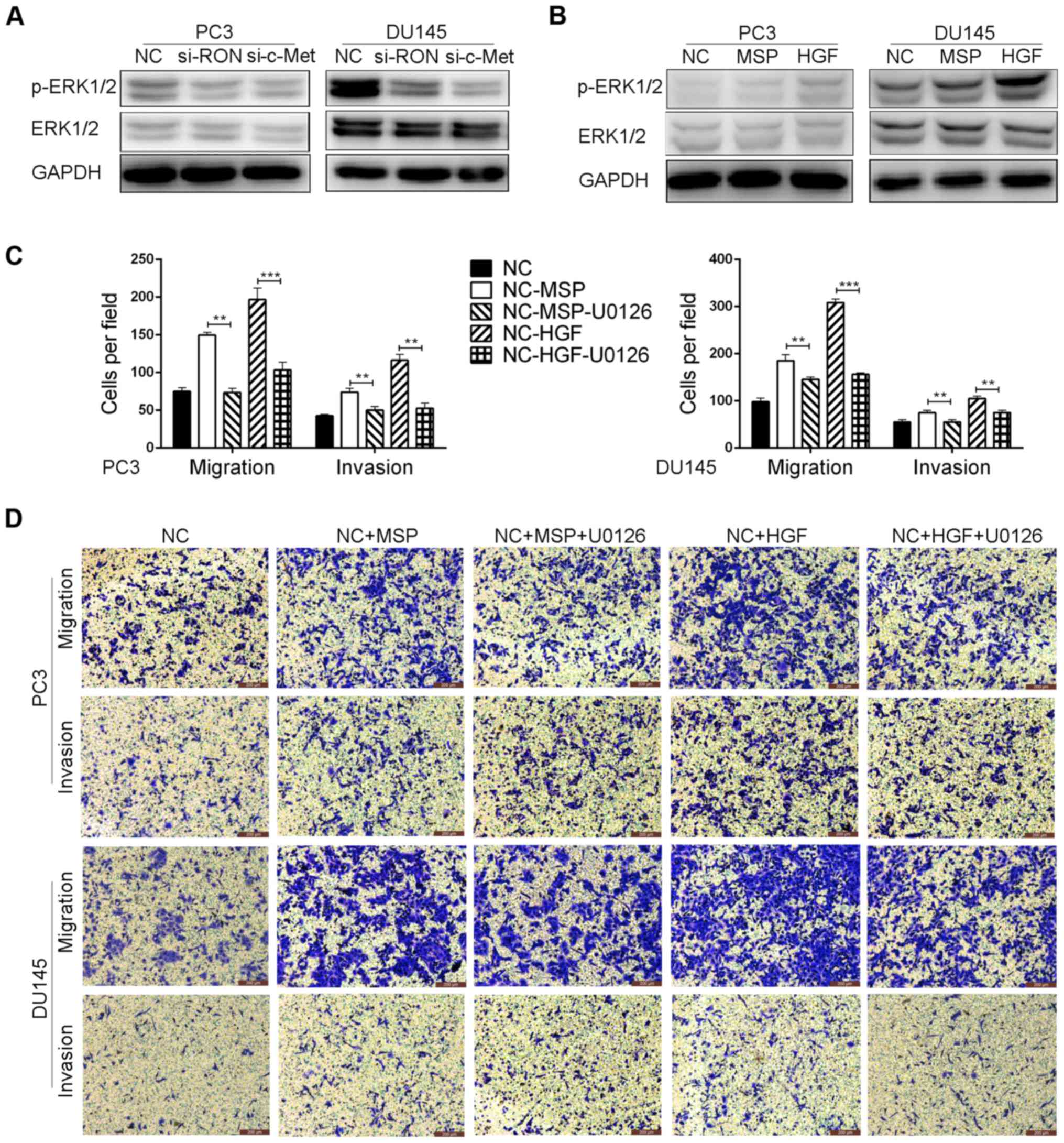
The ERK1/2 pathway mediates RON and
c-Met-induced PCa metastasis
A previous study revealed that RON and c-Met are
canonically mediated by ERK1/2 signaling (19). Therefore, we verified whether this
signaling pathway could be responsible for the induction of RON and
c-Met that mediates PCa metastasis. Our results demonstrated that
the levels of p-ERK1/2 were suppressed by the siRNA-mediated
knockdown of RON and c-Met and significantly increased following
activation by MSP or HGF (Fig. 6A and
B), compared with the control cells. In addition, PC3 and DU145
cells were pre-incubated with U0126, a highly selective inhibitor
of the ERK1/2 pathway and its upstream kinase. Blockade of the
ERK1/2 pathway by U0126 significantly suppressed the MSP- or
HGF-induced migration and invasion of the PC3 and DU145 cells
(Fig. 6C and D). These findings
suggest that the ERK1/2 pathway may play a critical role in RON-
and c-Met-induced metastasis.
Discussion
PCa is a male malignancy of global health concern
deserving intensive investigation. Metastasis is the leading cause
of the poor prognosis of PCa patients. Although substantial
progress has been made over the past decade towards uncovering the
molecular mechanism underlying PCa metastasis, it remains
incompletely understood. A better understanding of PCa metastasis
could contribute to more efficient treatments.
The high expression of RON or c-Met has been
reported in many tumors, including gastric, prostate, ovarian and
breast cancer, and has been identified as a marker for poor
prognosis (21). Moreover, a
previous study reported that co-expression of RON and c-Met in
node-negative breast cancer patients appears to result in an
aggressive phenotype compared with a disease-free group (9). In the present study, we showed
increased RON and c-Met expression in PCa and found that the
expression of RON mRNA was positively correlated with c-Met mRNA in
prostate tissues (Fig. 1A and B).
Although we did not find a relationship between RON and c-Met
expression with PCa grade severity in this limited sample size, our
results suggest that the co-expression of RON and c-Met in prostate
tissue may play a key role in PCa progression. Furthermore, we
found that RON and c-Met were robustly expressed in highly
metastatic PCa cell lines (PC3 and DU145), but barely expressed in
low-metastatic cell lines (22RV1 and LNCaP) (Fig. 1C-E). Such findings suggest that RON
and c-Met play a vital role in PCa metastasis and help to identify
suitable cell lines for the study of pathogenic mechanism of RON
and c-Met in PCa. Thus, RON and c-Met may be used as biomarkers to
predict cancer progression (20).
Although RON and c-Met share 60% structural
similarity and are co-expressed in various human cancers
contributing to tumor progression, the functions of RON and c-Met
have not been fully elucidated (22,23).
Due to the similar structure between RON and c-Met, TKIs that are
specific to either RON or c-Met have not yet been reported, with
most TKIs having dual-specific effects on both RON and c-Met
(20). Previous studies have
supported the concept that RON/c-Met inhibition might represent a
promising therapeutic option for suppressing the metastatic
progression of cancer. Therefore, we analyzed the effects of RON
and c-Met suppression on PCa invasive and migratory ability using
the RON and c-Met dual inhibitor foretinib (Fig. 2B-D). Our data showed that foretinib
significantly reduced metastasis and implies that RON and c-Met may
be potential therapeutic targets for PCa metastasis. However, the
cellular and molecular mechanism of RON and c-Met in cancer must be
fully investigated in further studies to achieve maximal efficacy
of TKIs.
As a proof of concept, we knocked down the
expression of RON and c-Met or activated RON and c-Met in PCa
cells. As expected, we found that silencing of RON and c-Met
attenuated the invasion and migration of the PCa cells (Figs. 3C and D, 4C and D and 5A
and B). However, knockdown of c-Met upregulated E-cadherin and
downregulated N-cadherin expression in both the PC3 and DU145 cell
lines (Fig. 4B), even though the
knockdown of RON did not produce the same effect (Fig. 3B). Activation of both RON with MSP
and c-Met with HGF led to enhanced invasion of PCa cells in
vitro (Figs. 3D and E, 4E and F and 5A
and B). Similarly, activation of c-Met with HGF led to the
upregulation of N-cadherin and downregulation of E-cadherin
expression in PCa, but activation of RON with MSP had no effect
(Figs. 3C and 4D). Cell scattering is a hallmark of cell
invasion and metastasis that consists of cell-cell dissociation,
cell spreading and motility. To determine the effect of stimulating
c-Met on scatter, we treated cells with HGF and found that this
resulted in a change from compact colonies to scattered colonies
(Fig. 4F and G). The above results
indicate that the downregulation of RON and c-Met further inhibited
the metastasis and reversal of the EMT of PCa cells to some extent
through the HGF/c-Met signaling pathway. Prior studies showed that
RON mediated the EMT in cancer cell lines via the RAS-ERK signaling
pathway in Madin-Darby canine kidney (MDCK) epithelial cells
(24,25). However, we found that MSP/RON
signaling is a significant contributor to invasiveness and
metastasis, but not EMT, in prostate cancer, similar to the effects
observed in breast, pancreatic and colon cancer. The molecular
mechanisms of PCa metastasis are complicated and require further
investigation.
We further screened for signaling pathways that were
involved in the regulation of RON- and c-Met-mediated PCa
metastasis. A previous study revealed that RON and c-Met are
canonically mediated by ERK1/2 signaling (20). Therefore, we assessed the expression
of active ERK1/2 upon RON and c-Met knockdown and found that
silencing of RON and c-Met decreased the levels of p-ERK1/2
(Fig. 6A), while activation of RON
and c-Met increased the levels of p-ERK1/2 (Fig. 6B). These findings were further
validated by treating cells with U0126, which significantly
suppressed the MSP- or HGF-induced migration and invasion of PC3
and DU145 cells (Fig. 6C and D).
Taken together, these results demonstrated that RON and c-Met can
promote PCa metastasis through the ERK1/2 pathway.
In conclusion, the present study indicated that RON
and c-Met expression are upregulated in PCa tissues and this
facilitates the metastasis of PCa cells via ERK1/2 signaling. To
the best of our knowledge, this is the first study to demonstrate
that foretinib has a marked antitumor metastatic effect on PCa
cells. Although the underlying mechanism remains to be fully
elucidated, this study suggests that targeting RON and c-Met may be
a rational therapeutic strategy for suppressing the metastasis of
PCa, especially for PCa patients who have increased expression of
RON and c-Met.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81271917)
and the Natural Science Foundation of Zhejiang Province (grant nos.
LY14H200002, LY15H200002 and LY16H160023). We thank the Clinical
Research Center from the Second Affiliated Hospital of Zhejiang
University School of Medicine for essential technical support. We
thank the American Journal Experts (AJE) for English language
editing.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA: A Cancer Journal for Clinicians. 65:5–29.
2015.PubMed/NCBI
|
|
2
|
Park JC and Eisenberger MA: Advances in
the Treatment of Metastatic Prostate Cancer. Mayo Clin Proc.
90:1719–1733. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 5:871–90. 2009. View Article : Google Scholar
|
|
4
|
Park M, Dean M, Kaul K, Braun MJ, Gonda MA
and Vande WG: Sequence of MET protooncogene cDNA has features
characteristic of the tyrosine kinase family of growth-factor
receptors. Proc Natl Acad Sci U S A. 84:pp. 6379–83. 1987;
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang K, Karnad A, Zhao S and Freeman JW:
Roles of c-Met and RON kinases in tumor progression and their
potential as therapeutic targets. Oncotarget. 6:3507–18. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ronsin C, Muscatelli F, Mattei MG and
Breathnach R: A novel putative receptor protein tyrosine kinase of
the met family. Oncogene. 8:1195–202. 1993.PubMed/NCBI
|
|
7
|
Birchmeier C, Birchmeier W, Gherardi E and
Woude GF Vande: Met, metastasis, motility and more. Nat Rev Mol
Cell Bio. 4:915–925. 2003. View
Article : Google Scholar
|
|
8
|
Wang J, Rajput A, Kan JLC, Rose R, Liu XQ,
Kuropatwinski K, Hauser J, Beko A, Dominquez I, Sharratt EA,
Brattain L, LeVea C, Sun FL, Keane DM, Gibson NW and Brattain MG:
Knockdown of Ron Kinase Inhibits Mutant Phosphatidylinositol
3-Kinase and Reduces Metastasis in Human Colon Carcinoma. J Biol
Chem. 284:10912–10922. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee WY, Chen HH, Chow NH, Su WC, Lin PW
and Guo HR: Prognostic significance of co-expression of RON and MET
receptors in node-negative breast cancer patients. Clin Cancer Res.
11:2222–8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Faria CC, Golbourn BJ, Dubuc AM, Remke M,
Diaz RJ, Agnihotri S, Luck A, Sabha N, Olsen S, Wu X, Garzia L,
Ramaswamy V, Mack SC, Wang X, Leadley M, Reynaud D, Ermini L, Post
M, Northcott PA, Pfister SM, Croul SE, Kool M, Korshunov A, Smith
CA, Taylor MD and Rutka JT: Foretinib Is Effective Therapy for
Metastatic Sonic Hedgehog Medulloblastoma. Cancer Res. 75:134–146.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kataoka Y, Mukohara T, Tomioka H,
Funakoshi Y, Kiyota N, Fujiwara Y, Yashiro M, Hirakawa K, Hirai M
and Minami H: Foretinib (GSK1363089), a multi-kinase inhibitor of
MET and VEGFRs, inhibits growth of gastric cancer cell lines by
blocking inter-receptor tyrosine kinase networks. Invest New Drugs.
30:1352–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huynh H, Ong R and Soo KC: Foretinib
demonstrates anti-tumor activity and improves overall survival in
preclinical models of hepatocellular carcinoma. Angiogenesis.
15:59–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
You WK, Sennino B, Williamson CW, Falcon
B, Hashizume H, Yao LC, Aftab DT and McDonald DM: VEGF and c-Met
blockade amplify angiogenesis inhibition in pancreatic islet
cancer. Cancer Res. 71:4758–68. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huynh H, Ong R and Soo KC: Foretinib
demonstrates anti-tumor activity and improves overall survival in
preclinical models of hepatocellular carcinoma. Angiogenesis.
15:59–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shah MA, Wainberg ZA, Catenacci DV,
Hochster HS, Ford J, Kunz P, Lee FC, Kallender H, Cecchi F, Rabe
DC, Keer H, Martin AM, Liu Y, Gagnon R, Bonate P, Liu L, Gilmer T
and Bottaro DP: Phase II study evaluating 2 dosing schedules of
oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients
with metastatic gastric cancer. Plos One. 8:e540142013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choueiri TK, Vaishampayan U, Rosenberg JE,
Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN,
Adams LM, Ottesen LH, Laubscher KH, Sherman L, McDermott DF, Haas
NB, Flaherty KT, Ross R, Eisenberg P, Meltzer PS, Merino MJ,
Bottaro DP, Linehan WM and Srinivasan R: Phase II and biomarker
study of the dual MET/VEGFR2 inhibitor foretinib in patients with
papillary renal cell carcinoma. J Clin Oncol. 31:181–6. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seiwert T, Sarantopoulos J, Kallender H,
McCallum S, Keer HN and Blumenschein G: Phase II trial of
single-agent foretinib (GSK1363089) in patients with recurrent or
metastatic squamous cell carcinoma of the head and neck. Invest New
Drug. 2:417–24. 2012.
|
|
18
|
Yau T, Yen CJ, Chen PJ, Chau Y, Lencioni
R, Kallender H, Ottesen LH and Poon RTP: A phase I/II study of
foretinib, an oral multikinase inhibitor targeting MET RON, AXL,
TIE-2, and VEGFR in advanced hepatocellular carcinoma (HCC). J
Hepatol. 54:S2682011. View Article : Google Scholar
|
|
19
|
Knubel KH, Pernu BM, Sufit A, Nelson S,
Pierce AM and Keating AK: MerTK inhibition is a novel therapeutic
approach for glioblastoma multiforme. Oncotarget. 5:1338–51. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shtutman M, Levina E, Ohouo P, Baig M and
Roninson IB: Cell Adhesion Molecule L1 Disrupts
E-Cadherin-Containing Adherens Junctions and Increases Scattering
and Motility of MCF7 Breast Carcinoma Cells. Cancer Res.
66:11370–11380. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao H, Zhou Y, Zhang R and Wang M: MSP-RON
signalling in cancer: pathogenesis and therapeutic potential. Nat
Rev Cancer. 13:466–481. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kretschmann KL, Eyob H, Buys SS and Welm
AL: The macrophage stimulating protein/Ron pathway as a potential
therapeutic target to impede multiple mechanisms involved in breast
cancer progression. Curr Drug Targets. 11:1157–68. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee WY, Chen HH, Chow NH, Su WC, Lin PW
and Guo HR: Prognostic significance of co-expression of RON and MET
receptors in node-negative breast cancer patients. Clin Cancer Res.
11:2222–8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Follenzi A, Bakovic S, Gual P, Stella MC,
Longati P and Comoglio PM: Cross-talk between the proto-oncogenes
Met and Ron. Oncogene. 19:3041–9. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Benvenuti S, Lazzari L, Arnesano A, Li CG,
Gentile A and Comoglio PM: Ron kinase transphosphorylation sustains
MET oncogene addiction. Cancer Res. 71:1945–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang D, Shen Q, Chen YQ and Wang MH:
Collaborative activities of macrophage-stimulating protein and
transforming growth factor-beta1 in induction of epithelial to
mesenchymal transition: roles of the RON receptor tyrosine kinase.
Oncogene. 23:1668–80. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma Q, Guin S, Padhye SS, Zhou YQ, Zhang RW
and Wang MH: Ribosomal protein S6 kinase (RSK)-2 as a central
effector molecule in RON receptor tyrosine kinase mediated
epithelial to mesenchymal transition induced by
macrophage-stimulating protein. Mol Cancer. 10:662011. View Article : Google Scholar : PubMed/NCBI
|