Introduction
Globally, breast cancer (BCa) is the most prevalent
form of cancer among women (1). In
particular, the basal form of cancer carrying the triple-negative
(TNBC) phenotype comprises 10–15% of all forms of BCa, and
manifests the worst prognosis and morbidity (2). TNBC demonstrates aggressive
proliferation and metastasis. Given its lack of the endocrine
receptors for estrogen, progesterone [estrogen receptor (ER), and
progesterone receptor (PR)], and the human epidermal growth factor
receptor-2 (HER-2), targeted therapy for this form of cancer
remains a challenge (3). These
cancer types express a high concentration of epidermal growth
factor receptor (EGFR). Disappointingly, while targeted therapies
against the EGFR have demonstrated a promising reduction in the
tumor mass, overall survival rate has remained unchanged.
Consequently, there is considerable impetus to investigate agents
such as phytochemicals and their potential to target TNBCs and
inhibit tumor progression and metastasis.
Signal transducers and activators of transcription
(STAT) constitute a family of seven transcription factors, STAT1,
STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. STATs are
overexpressed in a variety of cancers including breast cancer, head
and neck cancer, and hematologic malignancies (4,5). Among
the members of STAT family, STAT3 and STAT5b play significant roles
in the initiation, progression, and spread of malignant breast
tumors, and are termed oncogenes (6). STAT3 has been found to have strong
association with migration, invasion, and the metastatic capacity
of cells. Multiple genes involved in complex metastatic networks
are regulated by STAT3, making it a valuable target for the
inhibition of metastasis. The activation of STAT proteins is
mediated via the ligand associated tyrosine kinase, Janus kinase
(Jak), in particular, Jak-2 (7).
Upon phosphorylation of specific residues in STATs, homo- or
heterodimer formation ensues, followed by nuclear translocation.
Once in the nucleus, activated STAT molecules bind to specific
response elements in target gene promoters to transcriptionally
activate them (8). Not
surprisingly, the role of Jak2/STAT3 pathway in cell proliferation,
differentiation, apoptosis, and metastasis, has been the subject of
intense investigation (9,10).
Matrix metalloproteinases (MMPs) are a group of
proteins responsible for the degradation of the extracellular
matrix (ECM), and play a major role in cancer progression by their
impact on invasion, migration, and the metastasis of cancerous
cells (11). The control of MMP
activity is, potentially, a potent tool with which to prevent
cancer metastasis (12). MMPs are
mainly divided into six categories: the collagenase group,
gelatinase group, stromelysin group, matrilysin group,
membrane-type MMPs, and others (13). MMP2 is a member of gelatinases
group, which has been studied in various aspects of cancer
progression (14), including
migration, invasion, and metastasis. In endothelial cells,
MMP2 is found to increase angiogenesis by digesting the
extracellular membrane (15,16)
following chemokine stimulation. Xie et al have reported
that STAT3 activation-regulates brain tumor metastasis via MMP2
(17), and suggested that MMP2 is a
downstream target of STAT3 (17,18).
Furthermore, Lee et al suggest that the status of STAT3
controls the expression and secretion of MMP2 (19). These data led us to hypothesize that
the inhibition of STAT3 activation, and therefore MMP2, could be
exploited in TNBC cells in order to inhibit their metastatic
phenotype.
Silibinin is the primary active component of the
flavonoid silymarin, and is extracted from milk thistle seeds
(20,21). In the US and in Eastern European
countries, silibinin is used as a drug for toxic liver damage,
chronic hepatitis, and cirrhosis (22). However, recent studies show that
silibinin is a powerful antioxidant and scavenger, capable of
reducing lipid peroxidation (23,24),
with promise as a cancer cell growth inhibitor, cell cycle
regulator, and apoptosis inducer in prostate and colon carcinoma
cells (25,26). In particular, Bosch-Barrera and
Menendez have reported the importance of STAT3 in regulating tumor
growth, highlighting the potential of silibinin as a novel clinical
approach with which to inhibit Jak2/STAT3 pathway and related
effectors in breast, colon, prostate, and lung cancers (27). Furthermore, silibinin pre-treatment
confers a favorable effect on the phosphorylation of STAT3, as well
as the expression of MMP2 (28).
In the present study, we focused on the modulation
of Jak2/STAT3 pathway using silibinin, and its inhibition of in
vitro metastasis by diminishing MMP2 expression and activity in
TNBC cells. We found that silibinin has an anti-proliferative
effect on TNBC cells, and inhibits their invasive as well as
migratory abilities by downregulating MMP2 gene expression,
following inhibition of the JAK2/STAT3 signaling pathway in TNBC
cells.
Materials and methods
Antibodies and cell culture
reagents
Dulbecco's modified Eagle's medium (DMEM) was
purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Penicillin-streptomycin solution and fetal bovine serum (FBS) were
purchased from Hyclone (South Logan, UT, USA). Trypsin-EDTA (0.05%)
was obtained from Gibco-BRL (Grand Island, NY, USA). Antibodies
specific for phosphorylated STAT3 (Tyr705), STAT3, MMP9, and
β-actin, together with secondary antibody reagents (goat anti-mouse
and rabbit IgG-horseradish peroxidase), were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against MMP2
were purchased from Enogene Biotech Co. (NY, USA), with an
anti-Jak2 antibody obtained from Millipore (Billerica, MA, USA). An
anti phosphor Jak2 antibody (Tyrosine residue 1007/1008) was
purchased from Cell Signaling Technology (Beverly, MA, USA).
Silibinin was purchased from Sigma Co. (diluted in DMSO).
Cell culture and drug treatments
MDA-MB-231 human breast cancer cells were cultured
in DMEM containing 10% FBS, 2 mM glutamine, and 100 U/ml penicillin
and streptomycin. Cell culture was at 37°C, in a 5% CO2
gassed incubator. For each experiment, cells were resuspended in
medium at a density of 2.5×105 cells/ml. Unless
otherwise specified, cells were treated with 200 µM silibinin for
24 h.
Inhibition of cell proliferation
Cell viability was assayed by MTT assay. Briefly,
cells were resuspended in DMEM one day before drug treatment, at a
density of 3×103 cells per well in 96-well culture
plates. Culture medium was replaced with fresh medium containing
dimethyl sulfoxide (DMSO) as a vehicle control. Cells were
incubated with increasing concentrations of silibinin (from 50 to
350 µM). Following drug treatment, MTT (5 mg/ml) was added, with
culture dishes incubated for 4 h at 37°C. The resulting formazan
product was dissolved in DMSO and its absorbance read at 550 nm on
an Ultra Multifunctional Microplate Reader (Tecan, Durham, NC,
USA). All measurements were performed in triplicate, with
experiments repeated at least three times.
Western blotting
Whole cell lysates from MDA-MB-231 cells treated (or
not) with silibinin were prepared on ice using
radioimmunoprecipitation (RIPA) lysis buffer containing phosphatase
and protease inhibitors. Cells were disrupted by aspiration through
a 23-gauge needle and centrifuged at 15,000 rpm for 10 min at 4°C
to remove cellular debris. Protein concentrations were measured
using the Bradford method (Thermo Fisher Scientific, MA, USA).
Equal amounts of protein were resolved by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then
transferred onto nitrocellulose membranes. The blots were blocked
for 1 h with 5% skimmed milk in TBS-T buffer. Membranes were then
probed overnight at 4°C with the relevant primary antibody,
followed by washing with TBS-T and incubated for 1 h with the
horseradish peroxidase-conjugated secondary antibodies. Detection
was performed using the ECL Plus detection kit [Amersham Pharmacia
Biotech (Piscataway, NJ, USA)] and a LAS-4000 imaging device
(Fujifilm, Japan). Blot stripping was with Restore western blot
stripping buffer.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted using the RNeasy Mini kit
and QIAprep Spin Miniprep kit (Qiagen) according to the
manufacturer's protocol, with RNA quantified spectrophotometrically
at 260 nm. Then, RT-PCR analyses for MMP2, MMP9, and
18S RNA were performed. cDNA was synthesized from total RNA
using a first strand cDNA synthesis kit (Bioneer, Korea) and oligo
d(T) primers. The RT-PCR Premix kit with primers for MMP2, MMP9,
and 18S amplification, were synthesized by Bioneer (Daejeon,
Korea). PCR amplification to generate a 472-bp MMP2 fragment
was with the following primers: MMP2 sense,
5′-GGCCCTGTCACTCCTGAGAT-3′; antisense, 5′-GGCATCCAGGTTATCGGGGA-3′.
The PCR conditions were 94°C for 5 min, and then 32 cycles of 94°C
for 30 sec, 58°C for 30 sec, 72°C for 45 sec, followed by 72°C for
7 min. PCR amplification to generate a 455-bp MMP9 fragment
involved the following primers and PCR conditions: MMP9
sense, 5′-CCTGCCAGTTTCCATTCATC-3′; antisense,
5′-GCCATTCACGTCGTCCTTAT-3′, 94°C for 5 min, and then 30 cycles of
94°C for 40 sec, 60°C for 40 sec, 68°C for 50 sec, followed by 72°C
for 7 min. Finally, a 489-bp amplified 18S mRNA fragment was
generated using the following primers and PCR conditions:
18S sense, 5′-CGGCTACCACATCCAAGGAA-3′ and antisense,
5′-CCGGCGTCCCTCTTAATC-3′, 94°C for 5 min, and then 30 cycles of
94°C for 40 sec, 60°C for 40 sec, and 68°C for 50 sec, followed by
72°C for 7 min. PCR products were resolved by electrophoresis on a
1% agarose gel with PCR products visualized by ethidium bromide
staining.
Electrophoretic mobility shift assay
(EMSA)
STAT3 DNA binding activity was detected using EMSA
(Promega Corp., Madison, WI, USA). The electrophoretic mobility
shift assay (EMSA) kit, oligonucleotide probes (STAT3), and
reporter lysis buffer were from Promega. MDA-MB-231 cells were
grown to ~90% confluence with nuclear protein extracts prepared
using the Nuclear Extract kit (Affymetrix, CA, USA). EMSA was
performed using the EMSA gel shift kit (Panomics, Fremont, CA,
USA), according to the manufacturer's protocol. Briefly, nuclear
protein was subject to hybridization to a double stranded,
biotin-labeled oligonucleotide probe, containing the
consensus-binding site for STAT3 (sense strand,
5′-GATCCTTCTGGGAATTCCTAGATC-3′). The protein-DNA complexes were
resolved on a 6% non-denaturing PAGE gel and then transferred to a
Pall Biodyne B nylon membrane (Pall Life Sciences, Pensacola, FL,
USA). Signal detection was with streptavidin-HRP and a
chemiluminescent substrate.
Small interfering RNA (siRNA)
analyses
MDA-MB-231 cells (1×105) were cultured in
6-well plates, and grown to 70% confluence. Cells were then
transfected with the ON-TARGETplus SMARTpool siRNA, targeting
STAT3, or ON-TARGETplus non-target siRNA (Dharmacon, Chicago, IL,
USA), as a control. Transfection was with the DharmaFECT
transfection reagent (Dharmacon, Lafayette, CO, USA), according to
the manufacturer's instructions. Following transfection for 48 h,
cells were cultured in serum-free medium for a further 24 h, and
then exposed to 200 µM silibinin for 24 h. Levels of STAT3,
p-STAT3, MMP2, and β-actin were quantified by western blotting.
Wound healing assay
MDA-MB-231 cells were cultured in 6-well plates at a
concentration of 1×105 cells/well in DMEM media (i.e.,
serum containing), and then incubated for 24 h in a humidified
chamber. After forming a confluent monolayer, the cell layer was
scratched with a pipette tip and washed with PBS to remove cell
debris. Cells were untreated (controls) or exposed to 100 or 200 µM
silibinin. Images of the scratch sites (wounds) were captured at 0
and 24 h, and the average area of the wound calculated using ImageJ
software.
Matrigel invasion assay
The Transwell invasion assay was performed using
Matrigel pre-coated, ready to use invasion chambers (BD Biocoat,
MA, USA). Cells suspended at 5×104 were added to the
inserts. Drug-containing media was then added to the receiver
plate, and the inserts placed onto it. After a 24-h incubation in a
humidified chamber at 37°C, the cells that had invaded the apical
surface of the inserts were identified using crystal violet. The
plates were then incubated in ambient conditions for 24 h followed
by a wash and then fixation using formaldehyde. The cells on the
upper surface were removed using a cotton swab and the invaded
cells quantified using a microscope. Cells were counted in four
fields of view.
Statistical analyses
All experiments were performed at least three times
with results expressed as means ± SEM. Statistical analyses were
with ANOVA and Student's t-test (SAS 9.3 program). One-way analysis
of variance (ANOVA) was performed with Duncan's multiple range
test. A p-value of <0.05 was considered to be statistically
significant.
Results
Silibinin inhibits MDA-MB-231 cell
proliferation
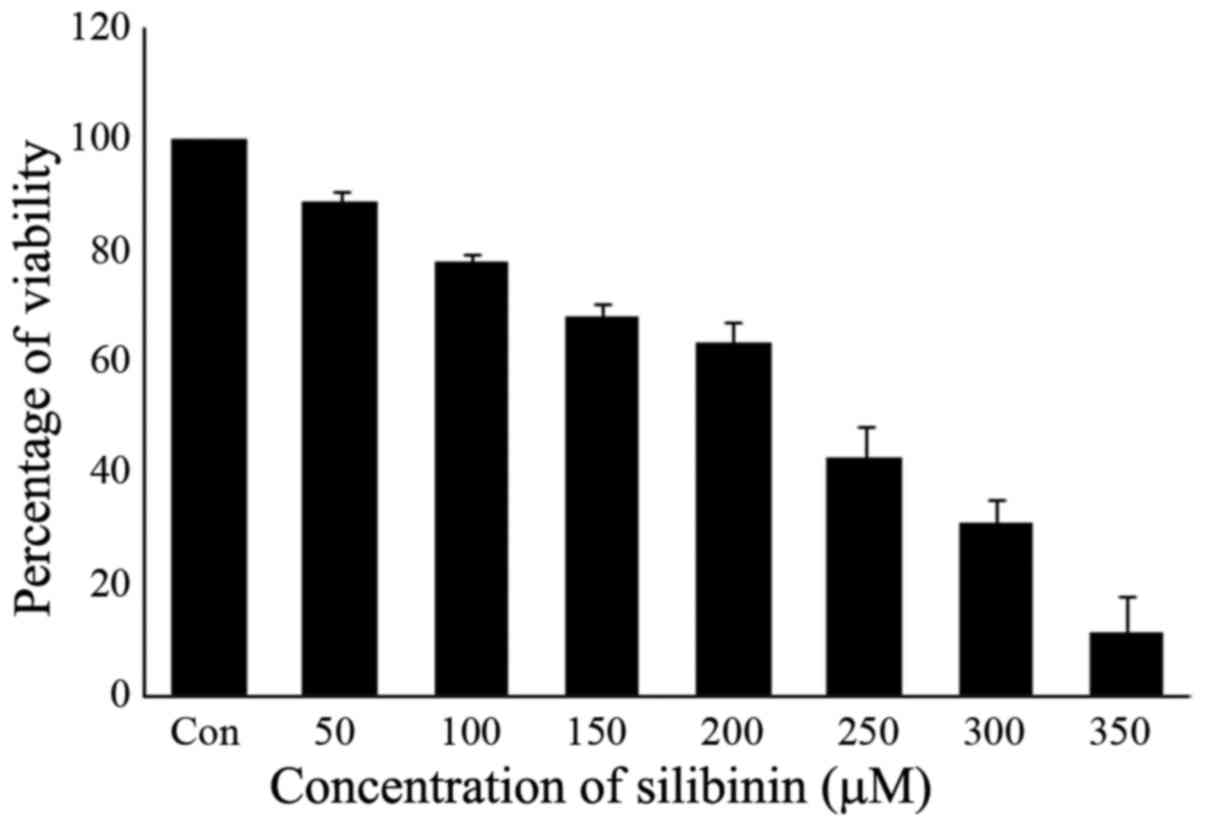
The effect of silibinin on cell proliferation was
examined by the MTT assay. The human triple-negative breast cancer
cell line MDA-MB-231 was exposed to increasing concentrations of
silibinin (50, 100, 150, 200, 250, 300 and 350 µM) for a period of
24 h. The number of silibinin-treated cells during the logarithmic
phase of growth was compared to that of control cells. Exposure to
silibinin substantially decreased viability in a dose-dependent
manner, with IC50 values ranging from 200 to 250 µM
following a 24-h exposure (Fig. 1).
Therefore, a 200 µM concentration of silibinin was taken as the
IC50, with this concentration used in subsequent
experiments.
Silibinin suppresses the expression
and phosphorylation of Jak2 and STAT3 proteins in a dose and
time-dependent manner
We previously reported that STAT3 is a potent
signaling molecule associated with tumor development and
progression (29). In this study,
our aim was to analyze the role of silibinin in the expression of
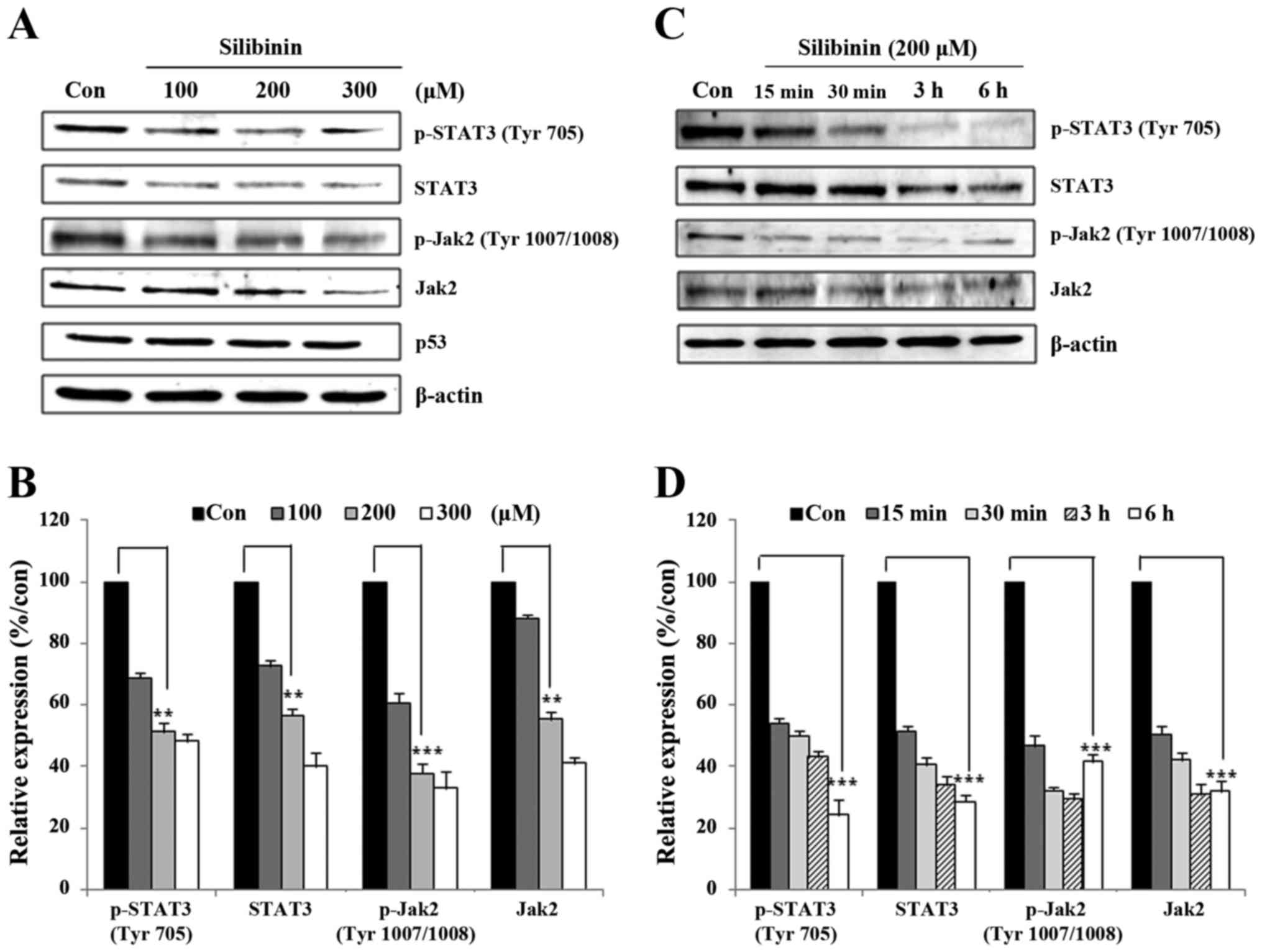
JAK2/STAT3-related proteins. Initially, MDA-MB-231 cells were
exposed to silibinin for 24 h at different doses, and then for
varying time periods. Whole cell lysates were prepared using 1X
RIPA lysis buffer and immunoblotted using antibodies with Jak2,
STAT3, and their respective phospho forms. As seen in Fig. 2A, silibinin suppressed the
expression of Jak2, STAT3, and phospho proteins in a dose-dependent
manner. Compared with the control group, 200 µM silibinin provoked
an almost 50% reduction in the expression of these proteins
(Fig. 2B). A time course experiment
(15 min to 6 h) showed that the expression of STAT3 and phospho
proteins continuously decreased in a time-dependent manner.
Concurrent with the dose-dependent inhibition experiments, the
time-dependent analysis also showed a 50% reduction in the
expression and phosphorylation of Jak2/STAT3 signaling proteins
(Fig. 2D). These data suggest that
silibinin has the capacity to regulate the activity of the
Jak2/STAT3 signaling pathway.
Silibinin inhibits the binding of
STAT3 to the MMP2 gene promoter
Recent studies have shown that STAT3 activation
regulates MMP2 expression (18).
Upon activation, STAT translocates to the nucleus and binds
specific response elements in target gene promoters in order to
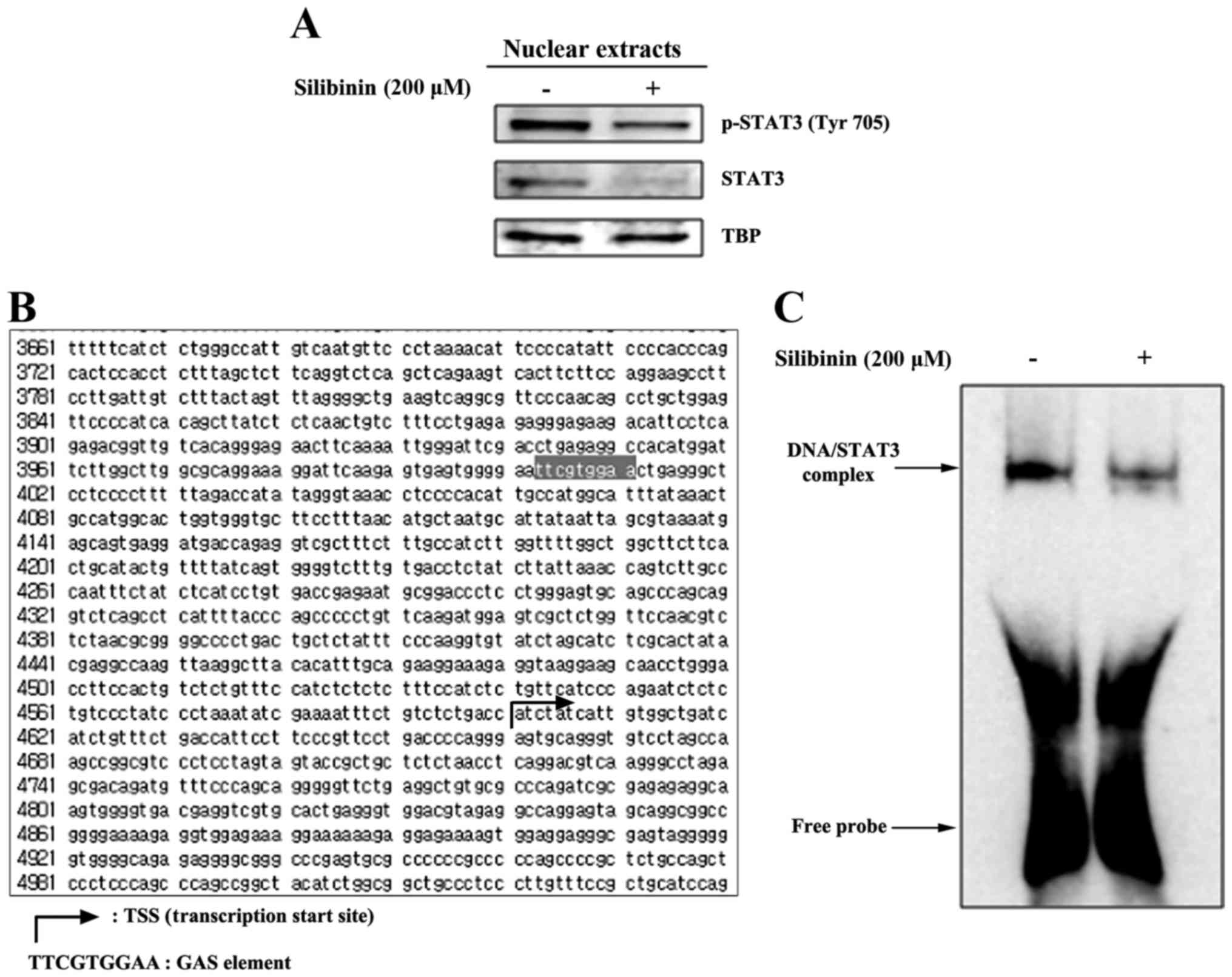
exert transcriptional control. Therefore we assayed levels of STAT3
and pSTAT3 in nuclear extracts prepared from silibinin-treated and
control, non-treated TNBC cells. As shown in Fig. 3A, there was a reduction in the level
of STAT3 and pSTAT3 in nuclear extracts derived from the 200 µM
silibinin-treated groups. We then analyzed the MMP2 gene
promoter for the presence of a GAS element, the DNA-binding
sequence for the STAT family of transcription factors. Having
located this element [in a domain upstream of the transcription
start site (TSS) (Fig. 3B)]
DNA-binding was then studied using EMSA. These data revealed that
exposure to silibinin provokes an inhibition of STAT3-DNA binding
(Fig. 3C), indicating that
silibinin, via STAT3, transcriptionally regulates MMP2 gene
expression.
Silibinin mediates a dose-dependent
downregulation of MMP2 and MMP9 expression at both the mRNA and
protein levels
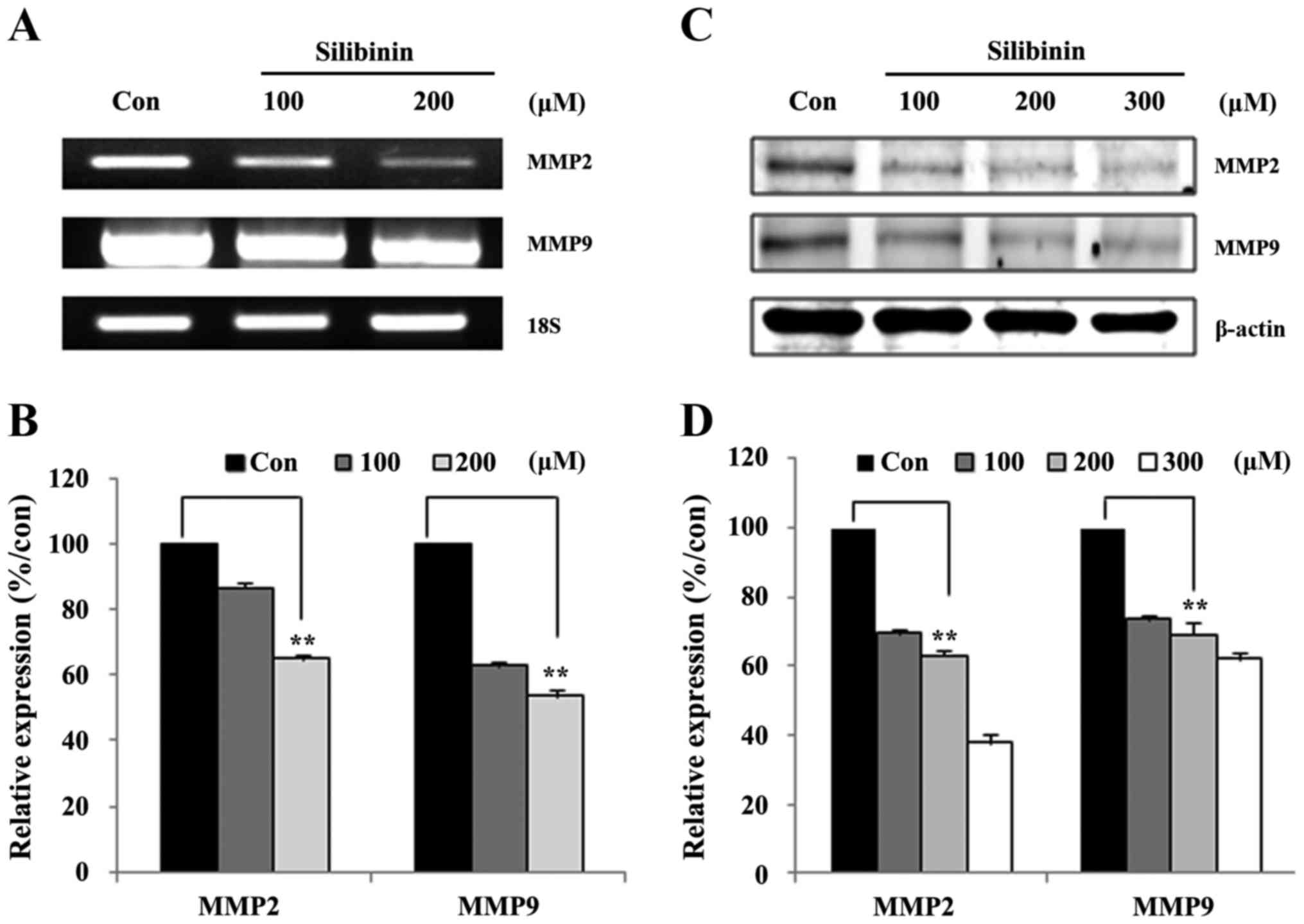
In the above section we showed that silibinin
suppressed Jak2/STAT3 signaling in a dose- and time-dependent
manner, and inhibited STAT3-DNA binding. We hypothesized that the
inhibition of STAT3-DNA binding should provoke a downregulation of
MMP2 gene expression. In order to confirm this, the expression of
STAT3 downstream targets were assayed at both the mRNA (Fig. 4A), and protein levels (Fig. 4C). Transcriptional level analyses
revealed that treatment with silibinin led to a dose-dependent
decrease in the transcription of both MMP2 and MMP9 (Fig. 4A). The silibinin-treated groups
manifested an ~40–60% inhibition of MMP2 mRNA expression when
compared to non-treated groups (Fig.
4B). Similar to the transcriptional data, at the protein level,
dose-dependent reductions in the expression of both MMP2 and MMP9
were evident (Fig. 4C) Membranes
were then probed overnight at 4°C with the relevant primary
antibody, followed by washing with TBS-T and incubated for 1 h with
the horseradish peroxidase-conjugated secondary antibodies with
silibinin-treated groups incurring a 40–60% inhibition of MMP2
protein expression (Fig. 4D).
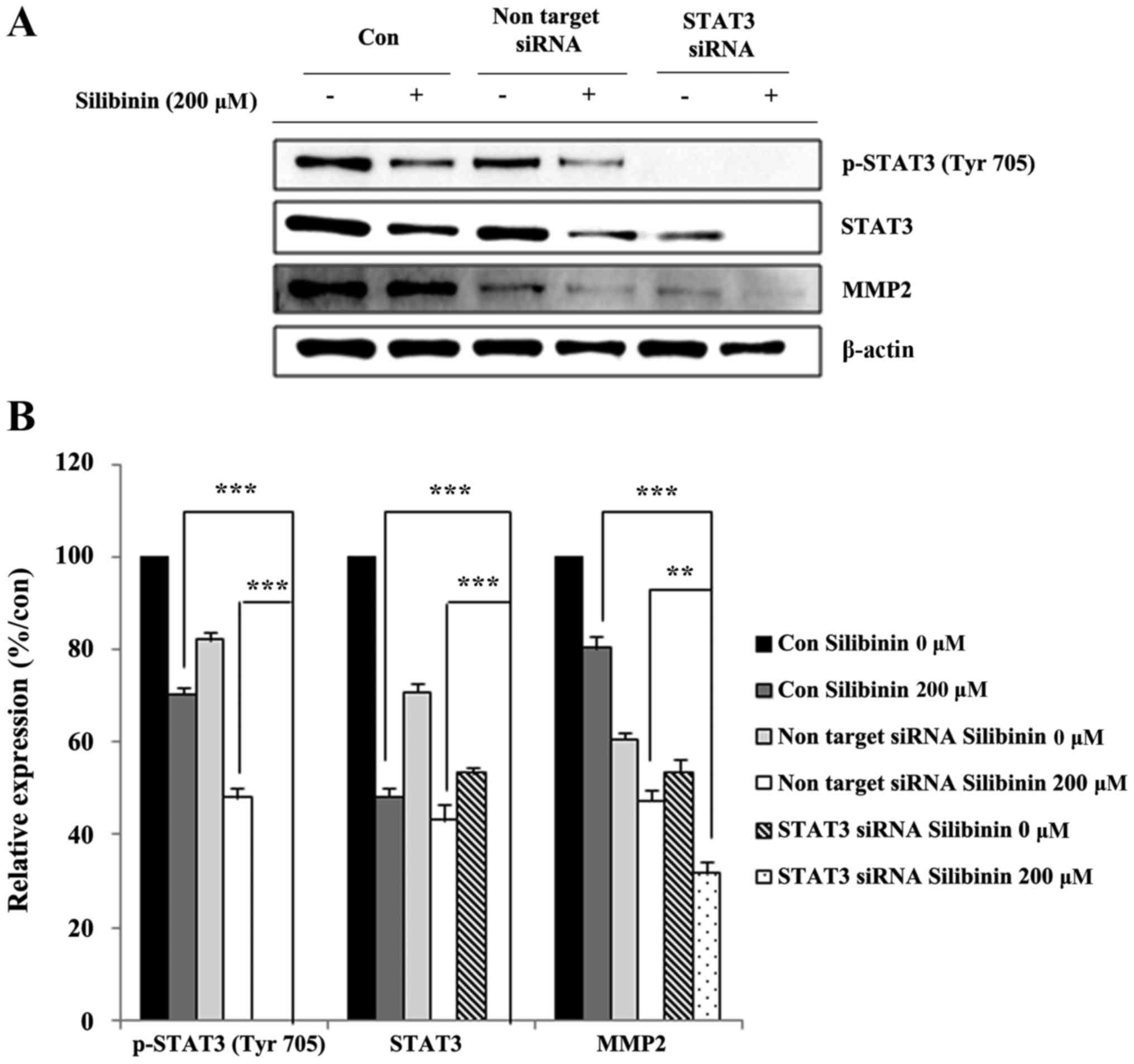
Silibinin inhibits MMP2 expression via
inhibition of STAT3
In order to confirm crosstalk between STAT3 and MMP2
expression, STAT3 was knocked-down using an siSTAT3 construct, and
our analyses repeated. STAT3 knock-down cells were exposed (or not,
for controls) to silibinin for a predetermined time, then whole
cell lysates prepared and analyzed for their expression of MMP2.
STAT3 knock-down effectively eliminated STAT3 expression, with
levels of MMP2 also markedly reduced (Fig. 5A). The relative expression of
protein, with respect to vehicle controls, was performed to confirm
the effect of STAT3 on the inhibition of MMP2 expression following
exposure to silibinin (Fig. 5B).
These data showed that silibinin could inhibit cell migration and
invasion via inhibition of MMP2 in a STAT3-dependent manner.
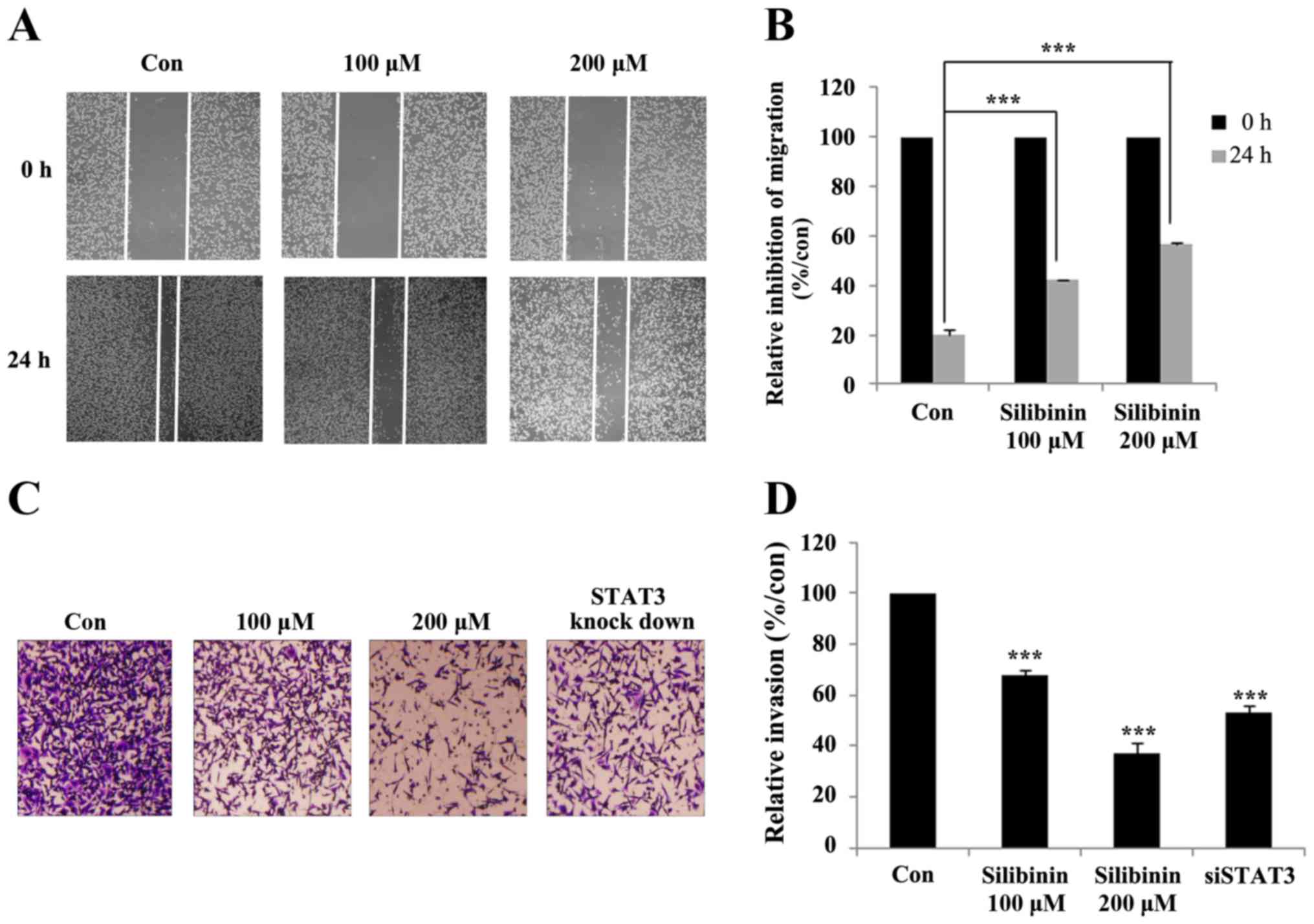
Silibinin suppresses cell migration
and invasion via STAT3 and MMP2
In the previous section we showed that knock-down of
STAT3 lead to a decline in the expression of MMP2. MMP2 is a major
determinant of cancer invasion, including cell migration and
invasion (30). The ability of
silibinin to inhibit cancer cell migration was subsequently studied
with the help of a wound-healing assay (Fig. 6A). Here the relative inhibition of
wound closure was used as a proxy measure for migration. The
non-treated control group showed a relatively high degree of cell
migration and wound closure, which was diminished in the
silibinin-treated group (Fig. 6B).
The crossing of the extracellular membrane is an essential step for
the dissemination of cancer cells from the primary tumor to distant
locations. The ability of silibinin to inhibit this invasion
process was studied using a Matrigel invasion assay.
Silibinin-treated cells showed a promising inhibition of invasion
compared to untreated controls. The role of STAT3 in silibinin
inhibition of invasion was then studied using STAT3 knock-down
cells. The results confirmed that silibinin and STAT3 play an
important role in inhibiting the invasive capacity of TNBC cells
(Fig. 6C). Compared with the
control group, both 200 µM silibinin-treated, and the STAT3
knock-down group, showed a 50% reduction in migration rate
(Fig. 6D). These results confirmed
that silibinin inhibits cell migration and invasion by inhibiting
STAT3 phosphorylation and MMP2 expression.
Discussion
Triple-negative breast cancer (TNBC) is an
aggressive subtype of breast cancer that lacks endocrine receptor
expression, and carries a mutated p53 gene. TNBCs are typified by
their poor differentiation (attributed to rapid growth), are highly
metastatic, and result in poor survival (31). While TNBCs express elevated levels
of EGFR at their surface, targeted therapies to the EGFR does not
improving overall survival of the patients, despite achieving a
rapid shrinkage in tumor size (32). These findings have promoted our
examination of the metastatic phenotype of TNBCs. In the present
study, we used silibinin as an inhibitor of the metastatic
potential of TNBC cells. It is already reported that silibinin has
an inhibitory role in the proliferation, invasion, and migration of
various cancer cell lines (33,34).
We found that silibinin also inhibited proliferation of the
triple-negative breast cancer cell line MDA-MB-231, with an
IC50 of ~200 µM for a treatment period of 24 h (Fig. 1). Even though this concentration is
high in terms of physiologic level, dietary feeding of silibinin
0.5% w/w for a period of 7 weeks was shown to inhibit the growth of
pancreatic cancer xenografts with no apparent changes in body
weight or feeding habits (35).
Similarly, the same 0.5% w/w concentration of silibinin inhibited
the growth of ectopically implanted and established PC-3 tumor
xenografts, without any gross sign of toxicity as monitored by
feeding habits (i.e., appetite) and weight gain (36).
An understanding of the molecular mechanisms that
underpin the anti-metastatic role of silibinin are essential if we
are to develop new therapeutic strategies as well as drug
combinations. For that, we checked the role of silibinin in the
modulation of Jak2/STAT3 pathway in MDA-MB-231 cells. Jak2 is the
receptor associated ligand-dependent tyrosine kinase responsible
for phosphorylation of STAT3 (6,7,9). Upon
phosphorylation, STAT3 is activated and then regulates a broad
range of downstream target genes, most related to malignant
transformation and metastasis (6,37,38).
Silibinin at a concentration of 200 µM inhibited Jak2 expression
and phosphorylation, resulting in the inhibition of phosphorylation
of its downstream substrate, STAT3 (Fig. 2A and C).
STAT3 phosphorylation at Y705 provokes its homo- or
hetero-dimerization, and then nuclear translocation in order to
bind gene-specific response elements in target gene promoters
(i.e., MMP2) (39). The MMPs play
an important role in various aspects of cancer such as gene
expression, interaction with the ECM, and cell motility (40), all of which can affect the growth
and differentiation of cells, their migration, and invasion
(11,12,40).
In recent years, the targeting of MMPs has received considerable
attention as a new treatment strategy with which to combat
metastasis (14,41). MMP2 is responsible for the migration
and invasive phenotype of cancer cells by digesting the
extracellular membrane and providing space for cells to migrate or
invade. Analysis of the MMP2 gene promoter sequence revealed
a GAS element downstream of the transcription start site (Fig. 3B), with gel shift analyses
indicating the DNA binding activity of STAT3 to the MMP2
gene promoter, which could be inhibited by silibinin (Fig. 3C). Moreover, analyses at the mRNA
and protein levels showed inhibition of both following exposure to
silibinin (Fig. 4A and C). In order
to confirm the role of STAT3 in the inhibition of MMP2 expression,
we knocked-down STAT3 and then re-MMP2 expression levels. Knocking
out of STAT3 effectively eliminated the expression of MMP2
(Fig. 5B).
Cell migration and invasion are the two major steps
in the dissemination of cancers from their primary to secondary
locations (39,42). Accordingly, our aim was to inhibit
the migration and invasive capacity of MDA-MB-231 cells using
silibinin. Exposure to silibinin appears to inhibit cell migration
and invasion in a dose- (Fig. 6A and
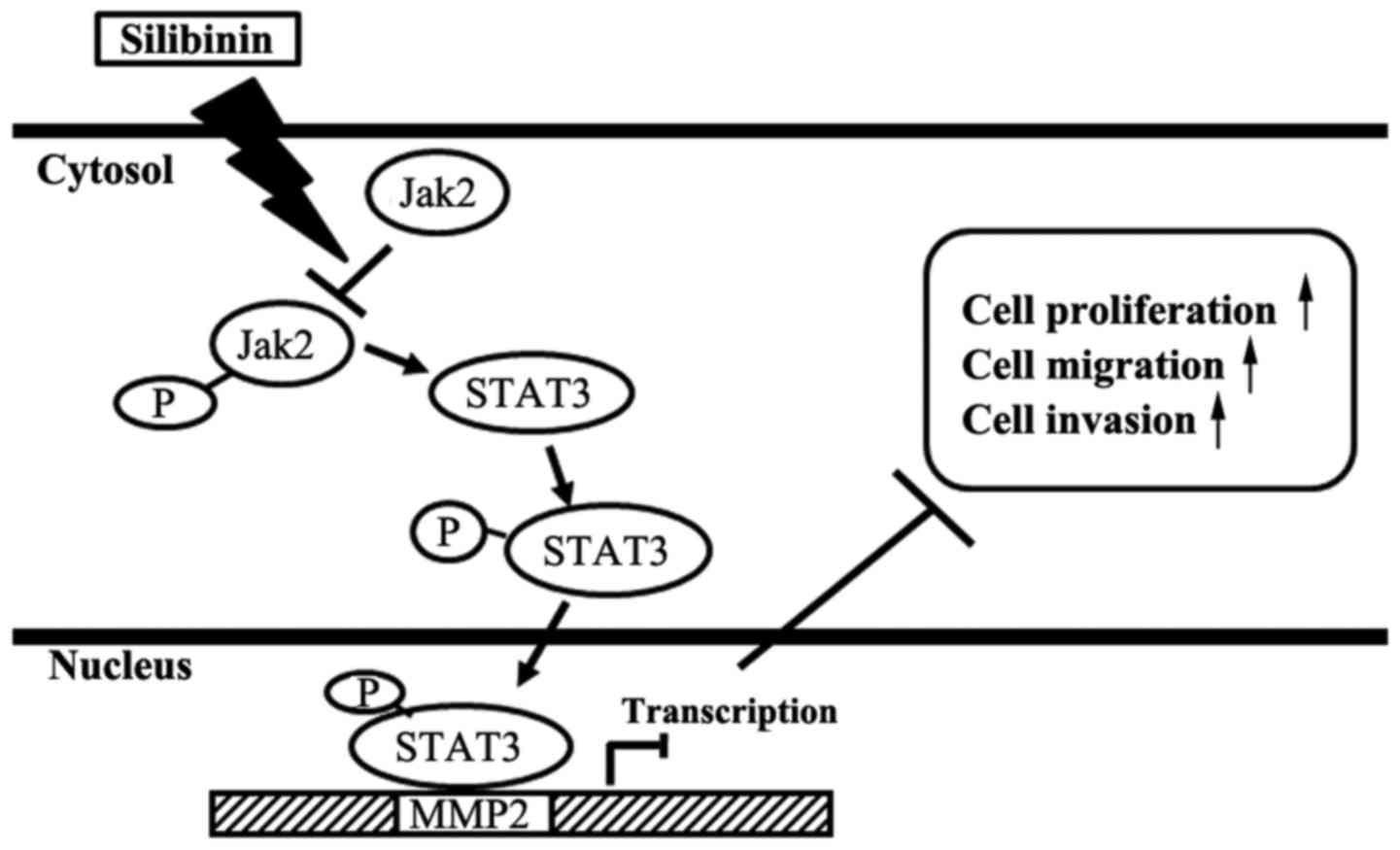
C), and STAT3-dependent manner (Fig. 6C). Overall, the treatment with
silibinin inhibited the gene specific transcriptional activation of
MMP2 through the inhibition of Jak2 and STAT3 phosphorylation. This
phosphorylation inhibition lead to the inhibition of Jak2/STAT3
pathway by blocking the STAT3 nuclear translocation, DNA-binding
and resulted in the inhibition of proliferation, migration and
invasion of TNBC cells (Fig.
7).
In conclusion, silibinin inhibits the cell
proliferation of TNBC cells, together with their migration, and
invasive potential. These results are consistent with our
hypothesis that silibinin downregulates MMP2 gene expression
via Jak2/STAT3 pathway. These data lead us to suggest that
silibinin be used as an experimental drug for the management of
metastatic cancer, with promise shown in terms of increasing
patient survival.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF); Ministry of Education (2013R1A1A2057942).
References
|
1
|
Cleator S, Heller W and Coombes RC:
Triple-negative breast cancer: Therapeutic options. Lancet Oncol.
8:235–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuo WH, Chang YY, Lai LC, Tsai MH, Hsiao
CK, Chang KJ and Chuang EY: Molecular characteristics and
metastasis predictor genes of triple-negative breast cancer: A
clinical study of triple-negative breast carcinomas. PLoS One.
7:e458312012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen JQ and Russo J: ERalpha-negative and
triple negative breast cancer: Molecular features and potential
therapeutic approaches. Biochim Biophys Acta. 1796:162–175.
2009.PubMed/NCBI
|
|
4
|
Garcia R, Bowman TL, Niu G, Yu H, Minton
S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parsons S, et
al: Constitutive activation of Stat3 by the Src and JAK tyrosine
kinases participates in growth regulation of human breast carcinoma
cells. Oncogene. 20:2499–2513. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garcia R, Yu CL, Hudnall A, Catlett R,
Nelson KL, Smithgall T, Fujita DJ, Ethier SP and Jove R:
Constitutive activation of Stat3 in fibroblasts transformed by
diverse oncoproteins and in breast carcinoma cells. Cell Growth
Differ. 8:1267–1276. 1997.PubMed/NCBI
|
|
6
|
Siveen KS, Sikka S, Surana R, Dai X, Zhang
J, Kumar AP, Tan BK, Sethi G and Bishayee A: Targeting the STAT3
signaling pathway in cancer: Role of synthetic and natural
inhibitors. Biochim Biophys Acta. 1845:136–154. 2014.PubMed/NCBI
|
|
7
|
Imada K and Leonard WJ: The Jak-STAT
pathway. Mol Immunol. 37:1–11. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darnell JE Jr: STATs and gene regulation.
Science. 277:1630–1635. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thomas SJ, Snowden JA, Zeidler MP and
Danson SJ: The role of JAK/STAT signalling in the pathogenesis,
prognosis and treatment of solid tumours. Br J Cancer. 113:365–371.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bowman T, Garcia R, Turkson J and Jove R:
STATs in oncogenesis. Oncogene. 19:2474–2488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rundhaug JE: Matrix metalloproteinases,
angiogenesis, and cancer: commentary re: A. C. Lockhart et al.,
Reduction of wound angiogenesis in patients treated with
BMS-275291, a broad spectrum matrix metalloproteinase inhibitor.
Clin. Cancer Res. 9:00-00, 2003. Clin Cancer Res 9. 551–554.
2003.
|
|
12
|
Verma RP and Hansch C: Matrix
metalloproteinases (MMPs): Chemical-biological functions and
(Q)SARs. Bioorg Med Chem. 15:2223–2268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Visse R and Nagase H: Matrix
metalloproteinases and tissue inhibitors of metalloproteinases:
Structure, function, and biochemistry. Circ Res. 92:827–839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brooks PC, Strömblad S, Sanders LC, von
Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP and
Cheresh DA: Localization of matrix metalloproteinase MMP-2 to the
surface of invasive cells by interaction with integrin αvβ3. Cell.
85:683–693. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rojiani MV, Alidina J, Esposito N and
Rojiani AM: Expression of MMP-2 correlates with increased
angiogenesis in CNS metastasis of lung carcinoma. Int J Clin Exp
Pathol. 3:775–781. 2010.PubMed/NCBI
|
|
16
|
Zheng H, Takahashi H, Murai Y, Cui Z,
Nomoto K, Niwa H, Tsuneyama K and Takano Y: Expressions of MMP-2,
MMP-9 and VEGF are closely linked to growth, invasion, metastasis
and angiogenesis of gastric carcinoma. Anticancer Res.
26A:3579–3583. 2006.
|
|
17
|
Xie TX, Huang FJ, Aldape KD, Kang SH, Liu
M, Gershenwald JE, Xie K, Sawaya R and Huang S: Activation of stat3
in human melanoma promotes brain metastasis. Cancer Res.
66:3188–3196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F,
Sawaya R and Huang S: Stat3 activation regulates the expression of
matrix metalloproteinase-2 and tumor invasion and metastasis.
Oncogene. 23:3550–3560. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee JW, Kwak HJ, Lee JJ, Kim YN, Lee JW,
Park MJ, Jung SE, Hong SI, Lee JH and Lee JS: HSP27 regulates cell
adhesion and invasion via modulation of focal adhesion kinase and
MMP-2 expression. Eur J Cell Biol. 87:377–387. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Valenzuela A and Garrido A: Biochemical
bases of the pharmacological action of the flavonoid silymarin and
of its structural isomer silibinin. Biol Res. 27:105–112.
1994.PubMed/NCBI
|
|
21
|
Singh RP and Agarwal R: Cosmeceuticals and
silibinin. Clin Dermatol. 27:479–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Loguercio C and Festi D: Silybin and the
liver: From basic research to clinical practice. World J
Gastroenterol. 17:2288–2301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kosina P, Kren V, Gebhardt R, Grambal F,
Ulrichová J and Walterová D: Antioxidant properties of silybin
glycosides. Phytother Res. 16 Suppl 1:S33–S39. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dehmlow C, Murawski N and de Groot H:
Scavenging of reactive oxygen species and inhibition of arachidonic
acid metabolism by silibinin in human cells. Life Sci.
58:1591–1600. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tyagi AK, Singh RP, Agarwal C, Chan DC and
Agarwal R: Silibinin strongly synergizes human prostate carcinoma
DU145 cells to doxorubicin-induced growth Inhibition,
G2-M arrest, and apoptosis. Clin Cancer Res.
8:3512–3519. 2002.PubMed/NCBI
|
|
26
|
Agarwal C, Singh RP, Dhanalakshmi S, Tyagi
AK, Tecklenburg M, Sclafani RA and Agarwal R: Silibinin upregulates
the expression of cyclin-dependent kinase inhibitors and causes
cell cycle arrest and apoptosis in human colon carcinoma HT-29
cells. Oncogene. 22:8271–8282. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bosch-Barrera J and Menendez JA: Silibinin
and STAT3: A natural way of targeting transcription factors for
cancer therapy. Cancer Treat Rev. 41:540–546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tyagi A, Agarwal C, Dwyer-Nield LD, Singh
RP, Malkinson AM and Agarwal R: Silibinin modulates TNF-α and IFN-γ
mediated signaling to regulate COX2 and iNOS expression in
tumorigenic mouse lung epithelial LM2 cells. Mol Carcinog.
51:832–842. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lim EJ, Hong DY, Park JH, Joung YH, Darvin
P, Kim SY, Na YM, Hwang TS, Ye SK, Moon ES, et al:
Methylsulfonylmethane suppresses breast cancer growth by
down-regulating STAT3 and STAT5b pathways. PLoS One. 7:e333612012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Björklund M and Koivunen E:
Gelatinase-mediated migration and invasion of cancer cells. Biochim
Biophys Acta. 1755:37–69. 2005.PubMed/NCBI
|
|
31
|
Bauer KR, Brown M, Cress RD, Parise CA and
Caggiano V: Descriptive analysis of estrogen receptor
(ER)-negative, progesterone receptor (PR)-negative, and
HER2-negative invasive breast cancer, the so-called triple-negative
phenotype: A population-based study from the California cancer
Registry. Cancer. 109:1721–1728. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dent R, Hanna WM, Trudeau M, Rawlinson E,
Sun P and Narod SA: Pattern of metastatic spread in triple-negative
breast cancer. Breast Cancer Res Treat. 115:423–428. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gluz O, Liedtke C, Gottschalk N, Pusztai
L, Nitz U and Harbeck N: Triple-negative breast cancer - current
status and future directions. Ann Oncol. 20:1913–1927. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chu SC, Chiou HL, Chen PN, Yang SF and
Hsieh YS: Silibinin inhibits the invasion of human lung cancer
cells via decreased productions of urokinase-plasminogen activator
and matrix metalloproteinase-2. Mol Carcinog. 40:143–149. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nambiar D, Prajapati V, Agarwal R and
Singh RP: In vitro and in vivo anticancer efficacy of silibinin
against human pancreatic cancer BxPC-3 and PANC-1 cells. Cancer
Lett. 334:109–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Singh RP, Deep G, Blouin MJ, Pollak MN and
Agarwal R: Silibinin suppresses in vivo growth of human prostate
carcinoma PC-3 tumor xenograft. Carcinogenesis. 28:2567–2574. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Darvin P, Joung YH, Sp N, Kang DY, Byun
HJ, Hwang DY, Cho KH, Park KD, Lee HK and Yang YM: Sorghum
polyphenol suppresses the growth as well as metastasis of colon
cancer xenografts through co-targeting jak2/STAT3 and PI3K/Akt/mTOR
pathways. J Funct Foods. 15:193–206. 2015. View Article : Google Scholar
|
|
39
|
Darvin P, Baeg SJ, Joung YH, Sp N, Kang
DY, Byun HJ, Park JU and Yang YM: Tannic acid inhibits the
Jak2/STAT3 pathway and induces G1/S arrest and mitochondrial
apoptosis in YD-38 gingival cancer cells. Int J Oncol.
47:1111–1120. 2015.PubMed/NCBI
|
|
40
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bauvois B: New facets of matrix
metalloproteinases MMP-2 and MMP-9 as cell surface transducers:
Outside-in signaling and relationship to tumor progression. Biochim
Biophys Acta. 1825:29–36. 2012.PubMed/NCBI
|
|
42
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|