Introduction
Prohibitin 1 (PHB) is multifunctional protein and
highly conserved among yeast, plants, worms, flies, and mammals
(1–5). The eukaryotic mitochondrial PHB
complex, which associates to form a ring-like structure, has a role
as a holdase type of chaperone specifically required in situations
of mitochondrial stress (6,7). Despite much evidence that PHB is
located at the mitochondrial inner membrane, various cellular
functions have been proposed for this protein outside the
mitochondria. PHB was found at the plasma membrane of human
intestinal epithelial cells where it functions as a binding site
for the Vi capsular polysaccharide of Salmonella typhi
(8). It has also been found to be
the target for a proapoptotic peptide in adipose vasculature
(9). Furthermore, it has been
implicated in mediating cellular Ras-Raf signaling at the membrane
(10). PHB also binds to a wide
range of proteins including retinoblastoma protein, E2F
transcription factor, Brg1/Brm and p53 (11,12).
In breast cancer cell lines, PHB co-localizes in the nucleus with
E2F1, retinoblastoma protein and p53, and the interaction of PHB
with E2F1 and retinoblastoma protein can be complex (13–15).
The coiled-coil domain of PHB is important in the interaction of
PHB with E2F1 and HDAC1 (16).
Recent reports suggest that recruitment of Brg1/Brm to E2F
responsive promoters is required for repression of E2F mediated
transcription by PHB and involves the JNK pathway (17). Brg1/Brm are involved in chromatin
remodeling and has been implicated in transcriptional activation by
the estrogen receptor (18). PHB
was also shown to repress the activity of estrogen receptor α
(19). These findings indicate that
PHB has several roles in cell cycle progression, the regulation of
transcription, and in cell surface signaling. PHBs are required for
embryonic development and in tissues that undergo cellular
proliferation (20).
Wide investigation of PHB expression revealed
that it is constitutively expressed in normal mammalian cells such
as hepatocytes, smooth muscle cells, chondrocytes, spermatocytes
and oocytes (21). Higher levels of
PHB expression were found in regenerating liver cells,
chemically-induced carcinoma, hyperplastic hepatic nodules and
hepatocellular carcinomas, and in cancer cells and primary tumor
samples (7,22–24).
The expression of PHB increases in yeast cells during
diauxic shift when cells switch from non-oxidative to oxidative
metabolism (7). There are
MYC-binding elements in the promoter region of PHB, and the
c-MYC oncogene is overexpressed in many cancer cells
(25). In a B-cell lymphoma line,
overexpression of PHB protects the cells from apoptosis, via
regulation of E2F activity (26).
Also retinoic acid or hexamethylene bisacetamide can stimulate the
translocation of PHB to the nucleus in tumor cells (27).
Among cancers, acute myeloid leukemia (AML) is a
heterogeneous human disease and involves a multi-step process
characterized by alteration in different pathways affecting cell
proliferation and myeloid differentiation (28,29).
Although in chronic myeloid leukemia samples with different
β-catenin levels lead to divergent effects in a Bcr-Abl model of
leukemogenesis (30), no β-catenin
mutations have been found in AML. Translocation products, such as
AML1-ETO (eight twenty-one) and promyelocytic leukemia zinc
finger-retinoic acid receptor α (PLZF-RARα) activate the Wnt
pathway through plakoglobin activation (31). Siapati et al demonstrated the
relevance of the Wnt pathway in AML cell lines (32).
In this study, we investigated Wnt-mediated
PHB expression in leukemic samples containing ALL and AML,
and found that Wnt signaling can activate transcription of
PHB via the TCF-4/LEF-1 binding motif in the promoter of
PHB in a MYC-independent manner. These data suggest that the
elevated level of PHB in leukemic cells is the result of Wnt
signals which are upregulated in leukemic cells.
Materials and methods
Cell culture and chemicals
HeLa cells were grown in DMEM (WelGene, Daegu,
Korea) supplemented with 10% (v/v) fetal bovine serum (JR
Scientific, Inc., Woodland, CA, USA), 1% (v/v)
penicillin/streptomycin (P4333, Sigma-Aldrich Korea, Seoul, Korea).
Leukemic cells provided by patients and Raji cell from ATCC were
grown in RPMI-1640 (WelGene) with same supplements as with DMEM.
For inhibition of c-MYC binding to DNA, 10058-F4
([Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, F3680,
Sigma-Aldrich Korea) was dissolved in 0.5% (v/v) DMSO (D5879,
Sigma-Aldrich Korea). Chemicals were administered to the cells in
1% serum condition and cells were incubated for 5–8 h.
Plasmid construction for promoter
assay and site-directed mutagenesis
To quantify the transcription amount of PHB,
we cloned the promoter region of PHB from genomic library of HeLa
cells using primers, PHBF3 (5′-CTCAAGCTTTTCCAAATAAAA-3′) and PHBR4
(5′-ACTGGATCCACATGAATTCCC-3′). The amplified 650-bp fragment was
treated with Klenow and BamHI and cloned into the
pcDNA-luciferase plasmid. HeLa cells were transfected by the
resulting PHB-Luc promoter reporter and the promoter activity was
analyzed using a luciferase assay. To mutate the transcription
binding sites in PHB promoter, the PCR product of the promoter
region was subcloned into pGEM®-T Easy Vector system
(A1360, Promega Korea, Seoul, Korea) and site-directed mutations
were introduced using the QuickChange® Site-Directed
Mutagenesis method (Agilent Technologies, Inc., Wilmington, DE,
USA). To remove candidate sites for TCF-4 binding to the promoter,
the following primers were used; PHBm1F
(5′-GTTGTAGTTCAGATGCTAAGAGCC-3′) and PHBm1R
(5′-GGCTCTTAGCATCTGAACTACAAC-3′); PHBm2F
(5′-GGAAGCGTGGAGATTGGAAAGCGG-3′) and PHBm2R
(5′-CCGCTTTCCAATCTCCACGCTTCC-3′). To mutate the binding site of
c-MYC, the following primers were used; PHBm4F
(5′-TACAGGATAGGCATATGCATTTAGCCCC-3′) and PHBm4R
(5′-GGGGCTAAATGCATATGCCTATCCTGTA-3′). After PCR reaction with PHB
promoter in T-vector and DpnI-digestion, the transformants
were identified by sequencing. After identification of mutated
sequences, the promoter regions were subcloned into the PHB-Luc
plasmid by exchange of the promoter region digested with
XhoI and BamHI restriction enzymes.
Determination of RNA amounts
Total RNAs from cells were extracted with TRIzol™
(Invitrogen) and first-strand cDNAs were synthesized using AMV
reverse transcriptase (F-570S, Finzymes, Espoo, Finland) at 42°C
for 1 h. For real-time PCR, the first-strand cDNA mixtures
corresponding to 5 µg of total RNA served as templates for PCR
analysis with the SYBR® premix Ex Taq™ (Takara, Kyoto,
Japan). Reactions were conducted on a Corbett RG-6000 apparatus
(Corbett Life Science, New South Wales, Australia). To optimize the
PCR conditions in terms of primers, annealing temperatures, PCR
efficiency, and standard curves were used. The PCR conditions were
as follows: cycles of 95°C for 10 sec, 58°C for 10 sec and 72°C for
15 sec. To calculate the relative expression levels of genes of
interest, the Ct value of GAPDH (glyceraldehyde-3-phosphate
dehydrogenase) expression was used as a normalizer. Amplification
of cDNAs was performed using primers: GDH1
(5′-TGAGAACGGGAAGCTTGTCA-3′) and GDH2 (5′-GGAAGGCCATGCCAGTGA-3′);
PHBF1 (5′-TCATTTTCTCATCCCGTGGGTA-3′) and PHBR1
(5′-TGCCTGGAGACCAGCTCTCT-3′); MYCF1 (5′-CGTCTCAGAGAAGCTGGCCT-3′)
and MYCR1 (5′-TGGCCTCCAGCAGAAGGTGA-3′); CCND1F
(5′-GGCGGAGGAGAACAAACAGA-3′) and CCND1R
(5′-TGGCACAGAGGGCAACGA-3′).
Immunoblot assay
Cultivated cells were harvested and lysed using
lysis buffer [1% (v/v) Triton X-100, 200 mM HEPES (pH 7.9), 300 mM
NaCl, 100 mM KCl, 10 mM EDTA, 10 µg/ml aprotinin, 100 µg/ml
leupeptin, and 10 µM PMSF]. Twenty micrograms of total protein
lysate was electrophoresed in an acrylamide gel and transferred to
a TransBlot® membrane (162–0145, Bio-Rad, Hercules, CA,
USA). Membranes were blocked in 5% (w/v) milk, incubated with
primary antibodies for 1 h at room temperature, washed and
incubated for 1 h with the appropriate HRP-conjugated secondary
antibodies (sc-2004 and sc2005, Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA). The blots were washed in PBS containing 0.05%
(v/v) Tween-20® (P1379, Sigma-Aldrich Korea) and an ECL
kit (34080, Pierce Biotechnology, Rockford, IL, USA) was used prior
to detection of protein bands. The antibodies used were: rabbit
anti-prohibitin (sc-28259, Santa Cruz Biotechnology, Inc.), rabbit
anti-β-catenin (sc-7199, Santa Cruz Biotechnology, Inc.), rabbit
anti-GSK-3β (sc-9166, Santa Cruz Biotechnology, Inc.), rabbit
anti-p-GSK-3β (sc-11757-R, Santa Cruz Biotechnology, Inc.), mouse
anti-c-Myc (sc-40, Santa Cruz Biotechnology, Inc.), and mouse
anti-β-actin monoclonal (sc-8432, Santa Cruz Biotechnology, Inc.)
to determine equal loading.
Promoter analysis
For the luciferase reporter assay, 0.4 µg of PHB-Luc
and 0.2 µg of CMV-LacZ were cotransfected into HeLa cells in 6-well
plates (Corning Inc., Corning, NY, USA) using the HilyMax
Transfection reagent (Dojindo, Kumamoto, Japan). Luciferase assay
was performed 48 h after transfection using a Luciferase assay
system (E1501, Promega Korea) and a Lumat LB9501 instrument
(Berthold, Bad Wildbad, Germany). For normalization, the
β-galactosidase activity in transfected cells was determined
spectrophotometrically at 420 nm using 0.2% (w/v) 2-nitrophenyl
β-D-galactopyranoside (N1127, Sigma-Aldrich Korea). Transfections
were performed in duplicate and repeated a minimum of three
times.
The binding of transcription factors to promoter
region of the PHB was monitored using a ChIP assay kit
(Upstate Biotechnology, Lake Placid, NY, USA). After cultivation of
Raji cells in a 100-mm dish (Corning, Inc.) with chemicals, 37%
formaldehyde was added to the culture media for 10 min to
cross-link DNA and proteins. After washing the cells with PBS, cell
pellets were resuspended in lysis buffer from the kit with protease
inhibitors. The cells were sonicated and the supernatants were used
in the ChIP assay according to the manufacturer's protocol. The
fragmented chromatin was pulled-down using a rabbit anti-β-catenin
antibody (sc-7199, Santa Cruz Biotechnology, Inc.; 1:2,000) and
163- and 250-bp products were amplified with primers: PHBpF
(5′-CCGCCGCTGCATAGCCTTT-3′) and PHBpR (5′-TGCGCATGAGCTCCTCTGC-3′)
for PHB; CCND1PF (5′-AGGCGCGGCGCCTCAGGC-3′) and CCND1PR
(5′-TGGAGGCTCCAGGACTTTGCA-3′) for the cyclin D1 gene,
respectively.
Results
High-levels of PHB expression in
leukemic cells
It was reported that PHB is one of the
targets of c-MYC, and an oncogenic transcription factor (19). Since PHB expressions was
increased in several acute and chronic myeloid leukemic cells in
our preliminary study, we checked the relationship between
c-MYC and PHB expression patterns in clinical
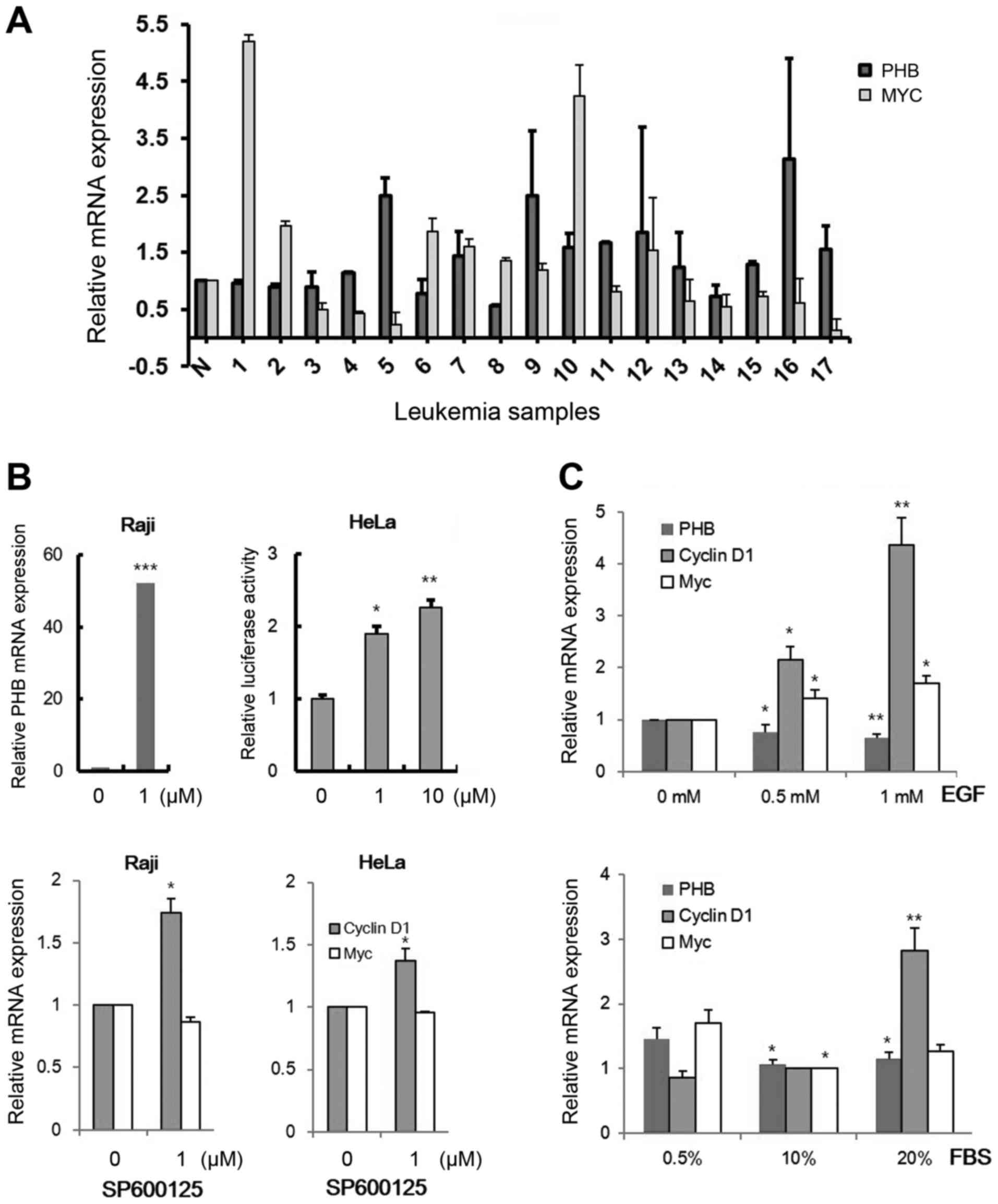
leukemic cells (Fig. 1A). Increased
PHB transcripts were detected in 9 samples (5, 7, 9, 10, 11,
12, 13, 16 and 17) among 17 clinical samples (52.9%) and the
enhanced levels of both c-MYC and PHB expression were
observed in only 3 samples, which are sample numbers 7, 9 and 10
(17.6%). We also confirmed PHB expression by cell
growth-promoting signals activating c-MYC activity (33). To screen a cell line expressing low
level of PHB, we tested 15 leukemic cells, selected the Raji cell
line (data not shown) and checked the transcriptional change in PHB
by several oncogenic signals (Fig. 1B
and C). PHB has been known to be associated with mitochondria
function and JNK-1 reduces mitochondria content via promoting the
nuclear localization of DAF-16/FOXO complex (34). Moreover, JNK regulates the protein
stability of c-Myc which is a transcription factor to induce PHB
transcription (35,36). Therefore we tested whether JNK
inhibitor regulates PHB expression. The PHB mRNA level was
increased by treatment with 1 µM of SP600125, a JNK inhibitor, in
Raji cells (Fig. 1B, left panel).
This regulation was also detected in HeLa cells using a
promoter-reporter system (PHB-Luc WT) after transfection (Fig. 1B, right panel). EGF ligand and serum
slightly decreased PHB transcription in HeLa cells (Fig. 1C). These results showed that other
factors but not routine signals for cell growth could exist to
activate PHB expression in leukemic cells.
Wnt signaling enhances PHB expression
in leukemic cells
Because accumulation of β-catenin in the nucleus has
been identified in chronic and acute leukemia (37,38),
we investigated the effect of β-catenin accumulation on PHB
expression. We monitored the expression levels of PHB with
lithium chloride (LiCl) treatment, which blocks GSK-3β activity and
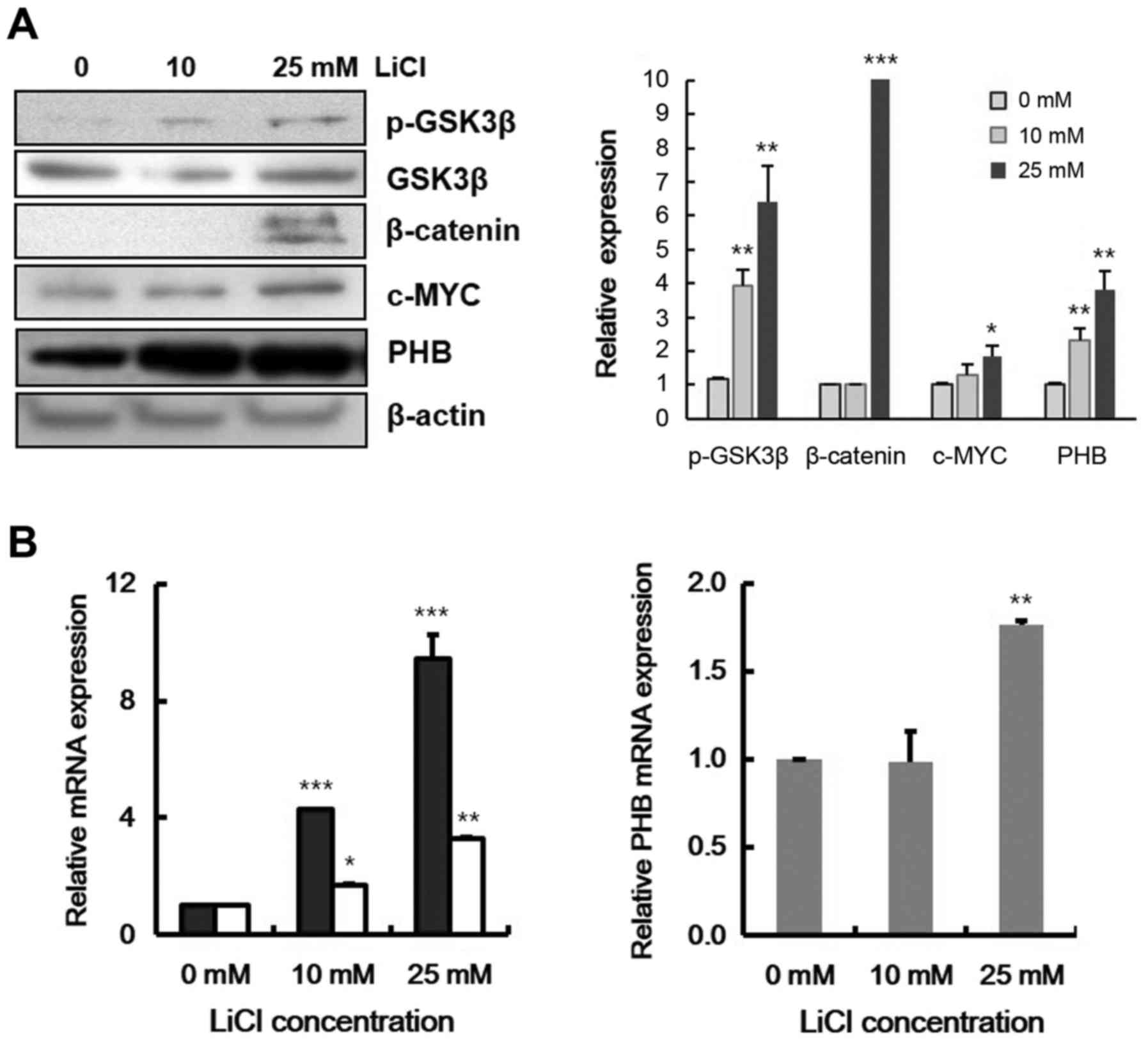
ultimately affects β-catenin accumulation (39). After treatment Raji cells with LiCl,
components of the Wnt signaling pathway were checked by immunoblot
assay. With LiCl, GSK-3β was phosphorylated while β-catenin, c-MYC
and PHB protein levels were increased (Fig. 2A). The transcriptional levels of
β-catenin target genes, c-MYC and CCND1, were
increased (Fig. 2B, left panel),
and also at 25 mM LiCl, transcription of PHB was
significantly increased by 2-fold (Fig.
2B, right panel). These data indicate that PHB expression was
induced by Wnt signaling in leukemic cells.
TCF-4/LEF-1 binds to the PHB
promoter
Because PHB expression was responsive to Wnt
signaling via β-catenin, we searched for TCF-4/LEF-1 binding
sequences in the promoter region of PHB. From information in
the Transcriptional Regulatory Element Database (TRED:http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=home),
two candidate sequences were found (Fig. 3A). In contrast to the identified
TCF-4/LEF-1 binding motives, GCTTTGATCTT or GCCATCTG, these sites
have a half-conserved sequence. TCFbs1, between −127 and −122, is
CATCTG and TCFbs2, between −39 and −34, is GATCTT. To confirm
TCF-4/LEF-1 binding on the PHB promoter, we cloned the promoter
region in a luciferase reporter vector (Fig. 3B). With LiCl treatment in HeLa cell,
wild-type of PHB promoter activity was increased (Fig. 3C). To identify the active site of
β-catenin in promoter region, the nucleotide sequences of the PHB
promoter were changed by site-directed mutagenesis (Fig. 3B). In PHB pm1, CATCTG was changed to
CAGATG, and GATCTT to GAGATT in PHB pm2. In the absence of LiCl,
the activity of PHB-Luc pm1 reporter was decreased by 2-fold
compared with that of PHB-Luc wild-type (WT) promoter. Also, in the
presence of LiCl, activities of PHB-Luc pm1 and pm1/2 reporter were
not increased whereas wild-type and pm2 PBH-Luc activities were
increased (Fig. 3C). These results
indicated that TCFbs1 is the β-catenin-working site and PHB
is a direct target of Wnt signaling. To confirm β-catenin binding
in this region, using primers covering these sites (Fig. 3B, arrows), the binding of β-catenin
to this region was checked by a chromatin immunoprecipitation assay
in Raji cells. Fig. 3D indicated
that with LiCl, the accumulated β-catenin bound to this region
through the TCF-4/LEF-1 binding sequence (TCFbs1) and PHB
was one of the direct Wnt target genes. Although the region pulled
down by anti-β-catenin antibody was amplified in the absence of
LiCl, due to a basal level of β-catenin in the Raji cells, the
amount of PCR product of this region was increased by LiCl
treatment. These data suggest that TCFbs1 is a ubiquitous working
site of β-catenin and PHB is a direct target gene of Wnt
signaling.
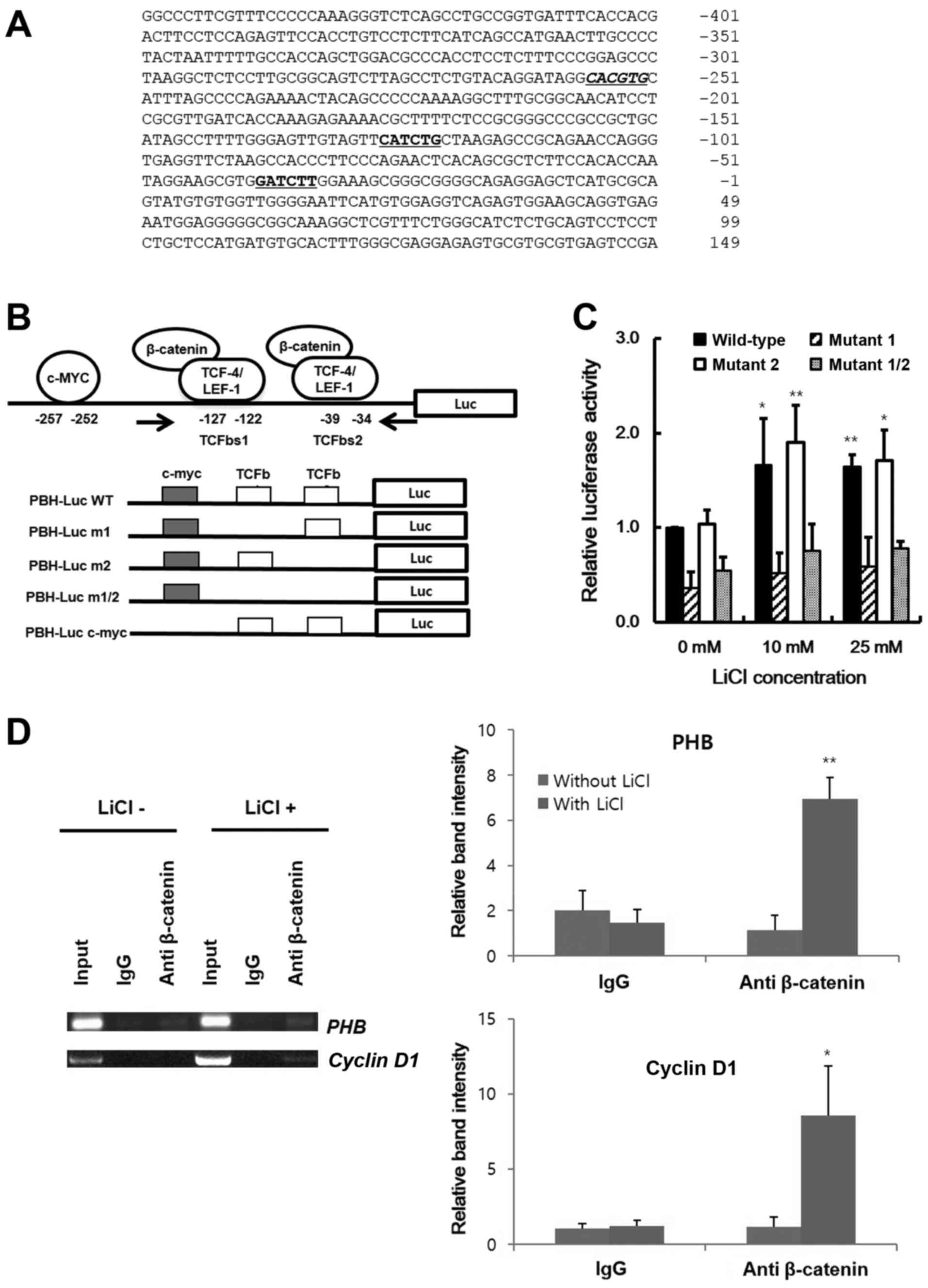 | Figure 3.Enhancement of PHB transcription by
activated β-catenin. (A) The nucleotide sequence of PHB promoter of
human. The candidate c-MYC and TCF-4/LEF-1 binding sites are
indicated by italics and underlined sequences, respectively. The
numbering starts from the transcription start site as +1. (B)
Schematic representation of the cloned promoter region of PHB to
construct the reporter vector (PHB-Luc) and its mutants. The
candidate sites of TCF-4/LEF-1 are represented by TCFbs1 and
TCFbs2, and the primers for identification of the pulled-down
genomic fragments in the chromatin immunoprecipitation are
indicated by arrows. The mutant m1 and m2 contain the modified
TCFbss at −127 to −122 and −39 to −34 in promoter region,
respectively. The double mutant m1/2 contains the modified
sequences at both sites. (C) Identification of TCF-4/LEF-1 binding
sequence in PHB promoter. After treatment of LiCl for 24 h in HeLa
cells transfected with PHB-Luc or its mutants, the emitted
luminescence was monitored with a luminometer. Black, diagonal,
white and gray bars represent the relative activity of PHB-Luc
promoter reporter of wild-type, m1, m2 and double mutant m1/2,
respectively. (D) Binding of β-catenin on PHB promoter. After
chromatin immunoprecipitation using genomic DNA from Raji cells by
anti-β-catenin antibody, the candidate regions were amplified with
the PCR primer set in (B). The cyclin D1 promoter region was used
as a positive control. Data are presented as mean ± SD (n=3).
*p<0.05, **p<0.01 and ***p<0.001. |
Effects of c-MYC and β-catenin on PHB
expression
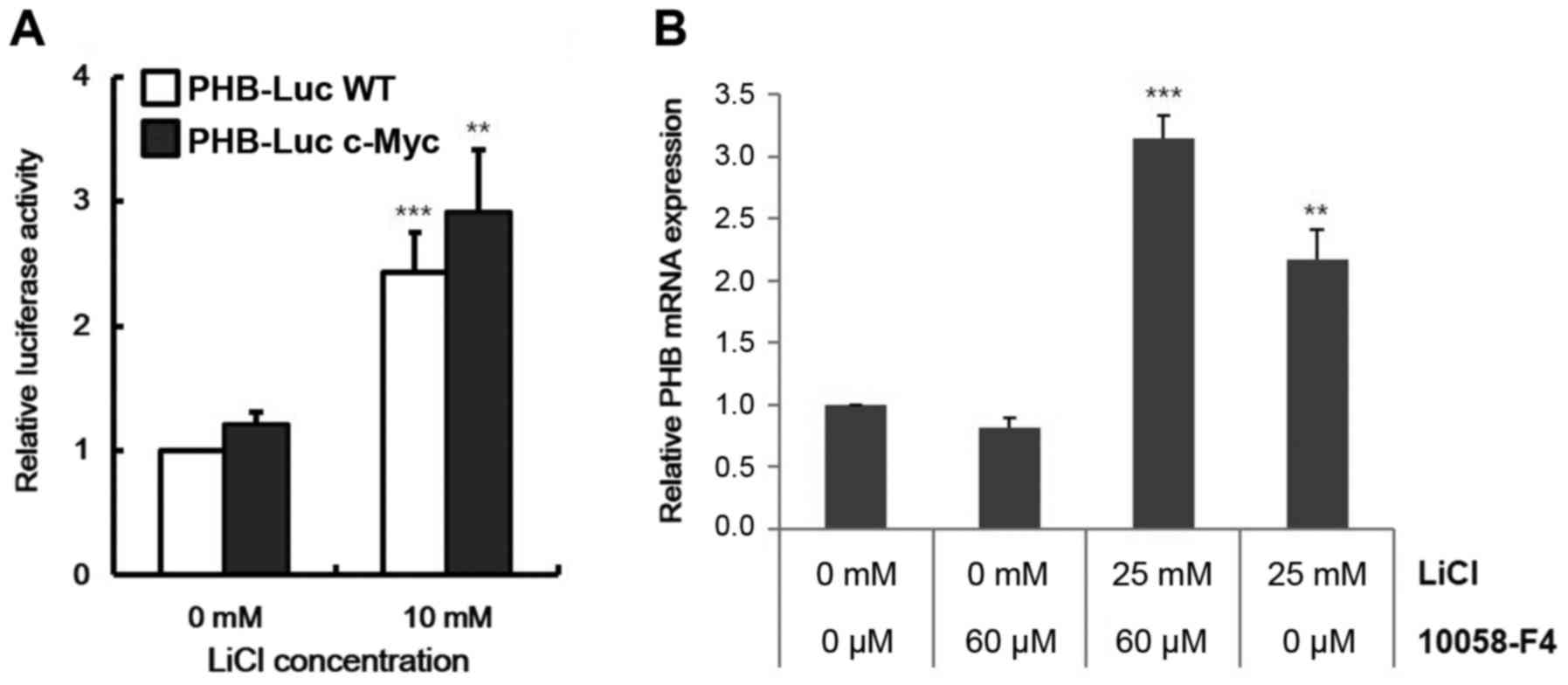
Because c-MYC is a known target gene of Wnt
signaling and it can activate PHB expression (25), we checked the dependency of c-MYC on
PHB transcription from Wnt signaling. Despite removal of the
c-MYC binding site (−257 to −252 in Fig. 3A and B), CACGTG in the promoter
region, the PHB transcript was increased by LiCl in HeLa
cells (Fig. 4A). These data
indicated c-MYC-independent activation of PHB expression by
Wnt signaling. To quantify the contributions of c-MYC and β-catenin
on regulation of PHB transcription, we analyzed transcript
level of PHB in Raji cells with treatment by 10058-F4, a
c-myc inhibitor, that disrupts formation of the c-MYC/MAX
heterodimer. Although PHB expression was slightly repressed
by 10058-F4, Wnt signaling overcame this repression (Fig. 4B). These data indicate that Wnt
signaling operates directly on the promoter of PHB.
Discussion
Although PHB has the potential to act as a tumor
suppressor, antiproliferative protein and regulator of cell-cycle
progression, recent studies have reported that overexpression of
PHB was found in various tumor cells including various leukemic
cells. Based on our proteomic analysis from AML patients, PHB level
was greatly elevated (data not shown). Despite its role as a tumor
suppressor, PHB level can be increased in phorbol ester-treated
chronic leukemic B-lymphocytes (40). We suggested that development of AML
or ALL can be associated with altered expression of specific
mitochondrial proteins such as PHB, as shown in chronic leukemic
B-lymphocytes. Many research projects reported the Wnt signal is
required for the development of leukemic cells (41–43).
The elevated PHB level in leukemic cells can be led by leukemic
driven forces and we searched PHB-inducing signals using promoter
investigation (Fig. 3A).
We focused on the regulatory signals involved in
PHB expression that promote cell proliferation such as EGF
and Wnt-mediated signals. In our data, PHB could be
expressed by Wnt signaling which is relevant in AML/ALL cells
(32). AML translocation products,
such a RUNX1-RUNX1T1, or epigenetic inactivation of pathway like
Wnt inhibitory factor (WIF1) activate the Wnt pathway increasing
TCF-4/LEF-1-dependent transcription (31,44).
In the activated Wnt pathway of leukemic cells, PHB could be
overexpressed via the TCF-4/LEF-1 binding motif in its promoter
region (Fig. 3). Although it was
reported that PHB promoter had a c-MYC binding region and
that the PHB co-localized with c-FOS and c-MYC (25,45),
our data showed that the role of c-MYC activity in expression of
PHB was less than β-catenin activity in leukemic cells
(Fig. 4). Additionally although we
assessed the role of a sequence similar to the E-box sequence
located at −384 in the PHB promoter, CACCTG, the response to
LiCl of the mutant promoter in this region was similar to that of
the region, −257 to −252 (data not shown). These data indicate that
the −257 to −252 region is a major functional site of the c-MYC
response to Wnt signaling.
The elevated level of PHB provides several arguments
as to its role in tumorigenesis. Clearly, the expression was
downregulated by growth signals (Fig.
1C) and this reinforces its role as a tumor suppressor. We
analyzed the mRNA sequences in several leukemic cells. Although
several mutations were found in some leukemic cells, most of their
sequences were normal (data not shown). These data suggest that the
increased transcription of PHB in leukemic cells by Wnt
signaling remains as yet unknown. PHB may be a common
differentially expressed nuclear matrix protein in some tumor
cells. PHB was distributed mainly in the regions of the nuclear
membrane and cytoplasm in several cell types (45). Also PHB exists as a component of the
nuclear matrix, and expression of PHB was downregulated
during differentiation (27,46).
In SK-N-SH cells, the expression of PHB is relatively strong
compared with retinoic acid-treated cells. By c-Jun-N-terminal
kinase-1 (JNK-1) activity, phosphorylated PHB could suppress
differentiation by inhibiting MEF2- and MRF-mediated gene
transcription (47,48).
In this study, we found that PHB has TCF-4 binding
site in the promoter region and its expression is elevated by LiCl.
However, mutant 1 reporter without TCF-4 binding site was not
affected by LiCl treatment and Fig.
3D shows that the accumulated β-catenin bound to PHB promoter
region through the TCF-4/LEF-1 binding sequence (TCFbs1) in the
treatment of LiCl. Thus indicating that PHB is one of the
direct Wnt target genes. Although we found that PHB is a
direct target gene of Wnt signal pathway and it is overexpressed in
acute leukemic cells, the effect of excess PHB remains unknown.
Further studies are needed to determine the effects of PHB
overexpression in leukemia.
Acknowledgements
This study was supported by the National Research
Foundation of Korea (NRF) grant funded by the Korean Government
(2013-R1A1A1007596 and 2015-M3A9C7030181). Also, this study was
financially supported by Chonnam National University (grant no.
2012-0828).
References
|
1
|
McClung JK, Jupe ER, Liu XT and Dell'Orco
RT: Prohibitin: Potential role in senescence, development, and
tumor suppression. Exp Gerontol. 30:99–124. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Snedden WA and Fromm H: Characterization
of the plant homologue of prohibitin, a gene associated with
antiproliferative activity in mammalian cells. Plant Mol Biol.
33:753–756. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Loukas A and Maizels RM: Cloning and
characterisation of a prohibitin gene from infective larvae of the
parasitic nematode Toxocara canis. DNA Seq. 9:323–328. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eveleth DD Jr and Marsh JL: Sequence and
expression of the Cc gene, a member of the dopa decarboxylase gene
cluster of Drosophila: Possible translational regulation. Nucleic
Acids Res. 14:6169–6183. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sato T, Saito H, Swensen J, Olifant A,
Wood C, Danner D, Sakamoto T, Takita K, Kasumi F, Miki Y, et al:
The human prohibitin gene located on chromosome 17q21 is mutated in
sporadic breast cancer. Cancer Res. 52:1643–1646. 1992.PubMed/NCBI
|
|
6
|
Back JW, Sanz MA, De Jong L, De Koning LJ,
Nijtmans LG, De Koster CG, Grivell LA, Van Der Spek H and Muijsers
AO: A structure for the yeast prohibitin complex: Structure
prediction and evidence from chemical crosslinking and mass
spectrometry. Protein Sci. 11:2471–2478. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nijtmans LG, Artal SM, Grivell LA and
Coates PJ: The mitochondrial PHB complex: Roles in mitochondrial
respiratory complex assembly, ageing and degenerative disease. Cell
Mol Life Sci. 59:143–155. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sharma A and Qadri A: Vi polysaccharide of
Salmonella typhi targets the prohibitin family of molecules in
intestinal epithelial cells and suppresses early inflammatory
responses. Proc Natl Acad Sci USA. 101:pp. 17492–17497. 2004;
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kolonin MG, Saha PK, Chan L, Pasqualini R
and Arap W: Reversal of obesity by targeted ablation of adipose
tissue. Nat Med. 10:625–632. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rajalingam K and Rudel T: Ras-Raf
signaling needs prohibitin. Cell Cycle. 4:1503–1505. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mishra S, Murphy LC, Nyomba BL and Murphy
LJ: Prohibitin: A potential target for new therapeutics. Trends Mol
Med. 11:192–197. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mishra S, Murphy LC and Murphy LJ: The
prohibitins: Emerging roles in diverse functions. J Cell Mol Med.
10:353–363. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang S, Fusaro G, Padmanabhan J and
Chellappan SP: Prohibitin co-localizes with Rb in the nucleus and
recruits N-CoR and HDAC1 for transcriptional repression. Oncogene.
21:8388–8396. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fusaro G, Dasgupta P, Rastogi S, Joshi B
and Chellappan S: Prohibitin induces the transcriptional activity
of p53 and is exported from the nucleus upon apoptotic signaling. J
Biol Chem. 278:47853–47861. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang S, Nath N, Adlam M and Chellappan S:
Prohibitin, a potential tumor suppressor, interacts with RB and
regulates E2F function. Oncogene. 18:3501–3510. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Joshi B, Ko D, Ordonez-Ercan D and
Chellappan SP: A putative coiled-coil domain of prohibitin is
sufficient to repress E2F1-mediated transcription and induce
apoptosis. Biochem Biophys Res Commun. 312:459–466. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang S, Zhang B and Faller DV: BRG1/BRM
and prohibitin are required for growth suppression by estrogen
antagonists. EMBO J. 23:2293–2303. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
DiRenzo J, Shang Y, Phelan M, Sif S, Myers
M, Kingston R and Brown M: BRG-1 is recruited to
estrogen-responsive promoters and cooperates with factors involved
in histone acetylation. Mol Cell Biol. 20:7541–7549. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He B, Feng Q, Mukherjee A, Lonard DM,
DeMayo FJ, Katzenellenbogen BS, Lydon JP and O'Malley BW: A
repressive role for prohibitin in estrogen signaling. Mol
Endocrinol. 22:344–360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Artal-Sanz M, Tsang WY, Willems EM,
Grivell LA, Lemire BD, van der Spek H and Nijtmans LG: The
mitochondrial prohibitin complex is essential for embryonic
viability and germline function in Caenorhabditis elegans. J Biol
Chem. 278:32091–32099. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arraztoa JA, Zhou J, Marcu D, Cheng C,
Bonner R, Chen M, Xiang C, Brownstein M, Maisey K, Imarai M, et al:
Identification of genes expressed in primate primordial oocytes.
Hum Reprod. 20:476–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Coates PJ, Nenutil R, McGregor A, Picksley
SM, Crouch DH, Hall PA and Wright EG: Mammalian prohibitin proteins
respond to mitochondrial stress and decrease during cellular
senescence. Exp Cell Res. 265:262–273. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ryu JW, Kim HJ, Lee YS, Myong NH, Hwang
CH, Lee GS and Yom HC: The proteomics approach to find biomarkers
in gastric cancer. J Korean Med Sci. 18:505–509. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu Z, Wu J and Zha X: Up-regulation of
prohibitin 1 is involved in the proliferation and migration of
liver cancer cells. Sci China Life Sci. 54:121–127. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Menssen A and Hermeking H:
Characterization of the c-MYC-regulated transcriptome by SAGE:
Identification and analysis of c-MYC target genes. Proc Natl Acad
Sci USA. 99:pp. 6274–6279. 2002; View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fusaro G, Wang S and Chellappan S:
Differential regulation of Rb family proteins and prohibitin during
camptothecin-induced apoptosis. Oncogene. 21:4539–4548. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li QF, Liang Y, Shi SL, Liu QR, Xu DH,
Jing GJ, Wang SY and Kong HY: Localization of prohibitin in the
nuclear matrix and alteration of its expression during
differentiation of human neuroblastoma SK-N-SH cells induced by
retinoic acid. Cell Mol Neurobiol. 31:203–211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gilliland DG, Jordan CT and Felix CA: The
molecular basis of leukemia, Hematology Am Soc Hematol Educ Progam.
2004.80–97
|
|
29
|
Hu Y, Chen Y, Douglas L and Li S:
Beta-catenin is essential for survival of leukemic stem cells
insensitive to kinase inhibition in mice with BCR-ABL-induced
chronic myeloid leukemia. Leukemia. 23:109–116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao C, Blum J, Chen A, Kwon HY, Jung SH,
Cook JM, Lagoo A and Reya T: Loss of beta-catenin impairs the
renewal of normal and CML stem cells in vivo. Cancer Cell.
12:528–541. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Müller-Tidow C, Steffen B, Cauvet T,
Tickenbrock L, Ji P, Diederichs S, Sargin B, Köhler G, Stelljes M,
Puccetti E, et al: Translocation products in acute myeloid leukemia
activate the Wnt signaling pathway in hematopoietic cells. Mol Cell
Biol. 24:2890–2904. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Siapati EK, Papadaki M, Kozaou Z, Rouka E,
Michali E, Savvidou I, Gogos D, Kyriakou D, Anagnostopoulos NI and
Vassilopoulos G: Proliferation and bone marrow engraftment of AML
blasts is dependent on β-catenin signalling. Br J Haematol.
152:164–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gururajan M, Chui R, Karuppannan AK, Ke J,
Jennings CD and Bondada S: c-Jun N-terminal kinase (JNK) is
required for survival and proliferation of B-lymphoma cells. Blood.
106:1382–1391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Artal-Sanz M and Tavernarakis N: Opposing
function of mitochondrial prohibitin in aging. Aging (Albany, NY).
2:1004–1011. 2010. View Article : Google Scholar
|
|
35
|
Yu K, Ravera CP, Chen YN and McMahon G:
Regulation of Myc-dependent apoptosis by p53, c-Jun N-terminal
kinases/stress-activated protein kinases, and Mdm-2. Cell Growth
Differ. 8:731–742. 1997.PubMed/NCBI
|
|
36
|
Noguchi K, Yamana H, Kitanaka C, Mochizuki
T, Kokubu A and Kuchino Y: Differential role of the JNK and p38
MAPK pathway in c-Myc- and s-Myc-mediated apoptosis. Biochem
Biophys Res Commun. 267:221–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jauregui MP, Sanchez SR, Ewton AA, Rice L,
Perkins SL, Dunphy CH and Chang CC: The role of beta-catenin in
chronic myeloproliferative disorders. Hum Pathol. 39:1454–1458.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jost E, Gezer D, Wilop S, Suzuki H, Herman
JG, Osieka R and Galm O: Epigenetic dysregulation of secreted
Frizzled-related proteins in multiple myeloma. Cancer Lett.
281:24–31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stambolic V, Ruel L and Woodgett JR:
Lithium inhibits glycogen synthase kinase-3 activity and mimics
wingless signalling in intact cells. Curr Biol. 6:1664–1668. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Woodlock TJ, Bethlendy G and Segel GB:
Prohibitin expression is increased in phorbol ester-treated chronic
leukemic B-lymphocytes. Blood Cells Mol Dis. 27:27–34. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Y, Krivtsov AV, Sinha AU, North TE,
Goessling W, Feng Z, Zon LI and Armstrong SA: The Wnt/beta-catenin
pathway is required for the development of leukemia stem cells in
AML. Science. 327:1650–1653. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim Y, Thanendrarajan S and Schmidt-Wolf
IGH: Wnt/β-catenin: A new therapeutic approach to acute myeloid
leukemia, Leuk Res Treatment. 2011.428960
|
|
43
|
Ashihara E, Takada T and Maekawa T:
Targeting the canonical Wnt/β-catenin pathway in hematological
malignancies. Cancer Sci. 106:665–671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chim CS, Chan WW, Pang A and Kwong YL:
Preferential methylation of Wnt inhibitory factor-1 in acute
promyelocytic leukemia: An independent poor prognostic factor.
Leukemia. 20:907–909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xu DH, Tang J, Li QF, Shi SL, Chen XF and
Liang Y: Positional and expressive alteration of prohibitin during
the induced differentiation of human hepatocarcinoma SMMC-7721
cells. World J Gastroenterol. 14:5008–5014. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhao CH and Li QF: Altered profiles of
nuclear matrix proteins during the differentiation of human gastric
mucous adenocarcinoma MGc80-3 cells. World J Gastroenterol.
11:4628–4633. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Feng W, Webb P, Nguyen P, Liu X, Li J,
Karin M and Kushner PJ: Potentiation of estrogen receptor
activation function 1 (AF-1) by Src/JNK through a serine
118-independent pathway. Mol Endocrinol. 15:32–45. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun L, Liu L, Yang XJ and Wu Z: Akt binds
prohibitin 2 and relieves its repression of MyoD and muscle
differentiation. J Cell Sci. 117:3021–3029. 2004. View Article : Google Scholar : PubMed/NCBI
|