Introduction
Spindle cell tumors are clinically heterogeneous but
morphologically similar neoplasms that can occur anywhere. The term
is descriptive and based on the tumor cells' long and slender
microscopic appearance (https://librepathology.org/wiki/Spindle_cell_lesions).
The diagnosis of spindle cell tumors relies on histological and
morphological features supported by ancillary investigations which
include immunohistochemistry, cytogenetics, fluorescence in
situ hybridization (FISH), and/or molecular genetics. The
diagnosis is prognostically imprecise and even sometimes fails to
distinguish benign from low-grade malignant tumors (1,2).
Cytogenetic and molecular genetic analyses of
spindle cell tumors have led to the recognition of several distinct
karyotypic entities, presumably corresponding to equally distinct
pathogenetic subgroups, characterized by chromosomal translocations
and also fusion genes that identify specific tumor types (3). For example, congenital fibrosarcomas
carry the translocation t(12;15)(p13;q25) which results in the
generation of an ETV6-NTRK3 fusion gene (4). Dermatofibrosarcoma protuberans,
another subtype of spindle cell sarcoma, is characterized
cytogenetically by supernumerary ring chromosomes or the
translocation t(17;22)(q22;q13) (5). Either change results in formation of a
COL1A1-PDGFB fusion gene in which PDGFB exon 1 is
deleted and replaced by a variable segment of the COL1A1
gene (5). A subset of inflammatory
myofibroblastic tumor, a neoplasm composed of myofibroblastic
spindle cells and infiltrating inflammatory cells, harbor clonal
chromosomal rearrangements of chromosome band 2p23 (6). These rearrangements target the
ALK gene which may serve as the 3′-partner in fusions with
various translocation partners bringing about ALK tyrosine kinase
activation (6).
Solitary fibrous tumor, another rare spindle cell
tumor, is now defined genetically as carrying a submicroscopic
inversion of the long arm of chromosome 12 (12q13) resulting in
fusion of the two neighboring genes NAB2 and STAT6
(7–10). This creates a chimeric transcription
factor in which the NAB2 repressor domain is substituted by a
carboxyl-terminal STAT6 transactivation domain or near-full-length
STAT6 (7–10).
In spite of all these genetic-pathologic
correlations, other spindle cell tumors exist that have not yet
been found to have characteristic, let alone pathognomonic, genetic
or pathogenetic features. By way of example, Fruth et al
(11) reported a laryngeal spindle
cell sarcoma which did not fit into any of the existing spindle
cell sarcoma sub-entities: The initially benign-appearing
mesenchymal tumor first changed its clinical phenotype without
corresponding histological signs of malignancy but later assumed
more aggressive histological features. Alaggio et al
(12) described two spindle cell
tumors with EWSR1-WT1 fusion and favorable prognosis.
According to the authors, the tumors could represent ‘an
unrecognized subgroup of tumors with spindle cell morphology,
bearing the same translocation as desmoplastic small round cell
tumor, but characterized by a more favorable clinical course’. In a
previous study of ours, we described a spindle cell sarcoma that
could not be further sub-classified, but which carried a ring
chromosome composed of chromosome 12 material, several fusion genes
mapping to 12q, and amplification of MDM2 (13). Nord et al (14) reported a spindle cell sarcoma of the
heart with a ring chromosome, amplification of the MDM2
gene, and homozygous deletion of CDKN2A. Finally, Lestou
et al (15) reported a case
of spindle cell sarcoma in the lower abdominal wall with a complex
karyotype, ring chromosomes, amplification of chromosome 18, and
co-amplification of 12p11 and 12q13-q22 in the ring chromosomes.
The examples above show that continuous examination of tumors with
spindle cell morphology is likely to reveal yet other genetic
subgroups that may, in the future, be seen to correspond to
meaningful clinical and even, when suitable therapeutics are
construed against the pathogenetic mechanisms involved, be also
therapeutically decisive.
In the present study, we analyzed genetically a
pediatric spindle cell tumor. The cytogenetic analysis showed that
the tumor cells carried a t(3;17)(p21;q12) chromosomal
translocation and RNA sequencing identified a SETD2-NF1
fusion gene caused by the translocation.
Materials and methods
Ethics statement
The study was approved by the Regional Committee for
Medical and Health Research Ethics, South-East Norway (REK sør-øst;
http://helseforskning.etikkom.no) and
written informed consent was obtained from the patient for
publication of the case details. The ethics committee's approval
included a review of the consent procedure. All patient information
has been de-identified.
Case history
A 16-year-old male presented with a mass in the left
deltoid region. After analysis of a needle biopsy, surgical
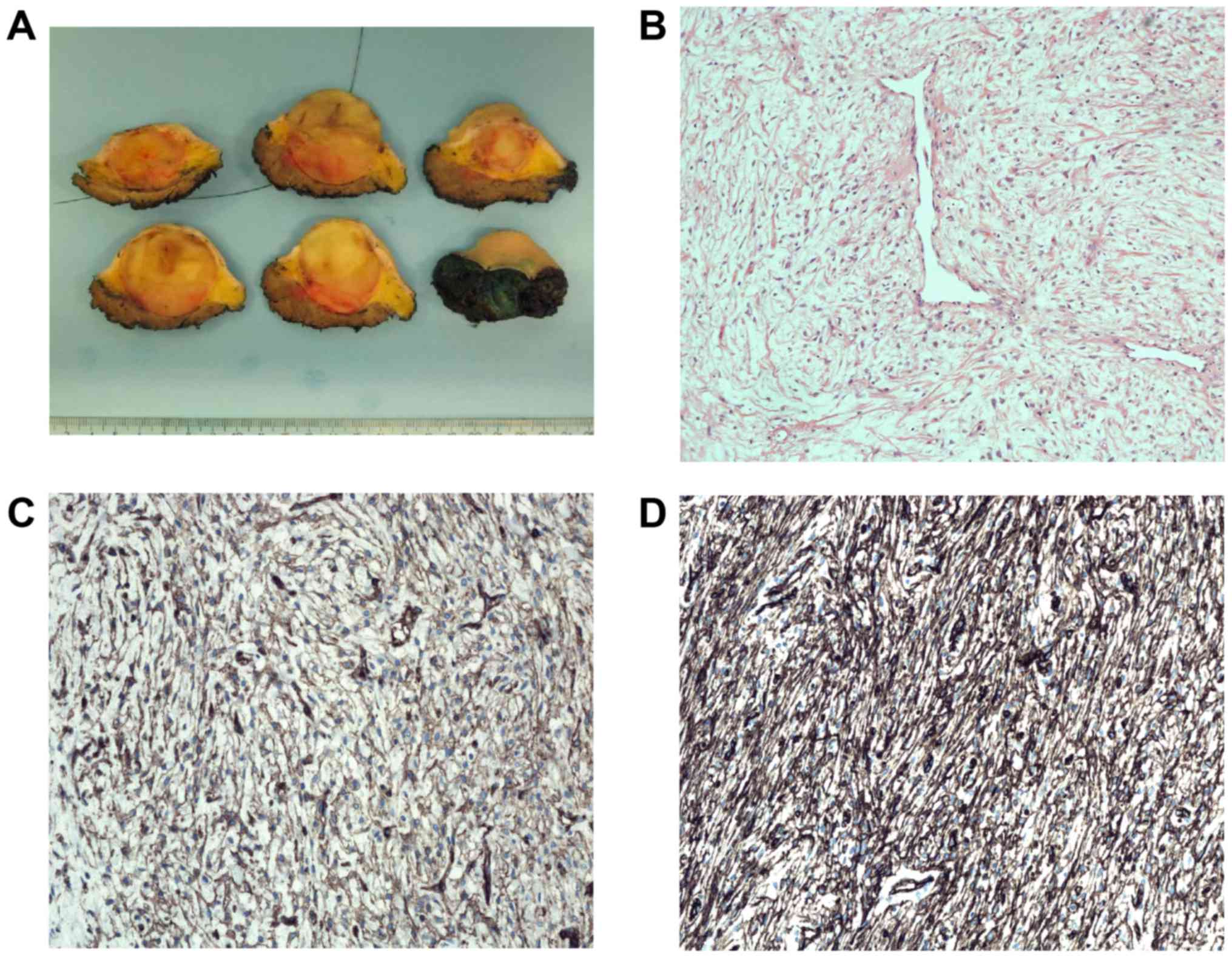
resection was performed. Macroscopic examination disclosed a 4.2 cm
large, well demarcated tumor (Fig.
1A). Microscopic examination revealed a moderately cellular
tumor with spindle cells without clear atypia intermingled with
loose intercellular matrix, partly with myxoid tissue and collagen
(Fig. 1B). Dilated vessels were
seen. There were some necrotic areas but very few mitotic figures
(0–1/10 high power fields). Immunohistochemistry demonstrated
positive focal staining for CD34 and CD99 (Fig. 1C and D), but negativity for
cytokeratin cocktail (AE1/AE3, EMA, S-100, SMA, and desmin; data
not shown). There was no nuclear STAT6 staining and the molecular
analysis did not show presence of the NAB2-STAT6 fusion
transcript which is pathognomonic for solitary fibrous tumor. FISH
analysis was negative for rearrangement of the FUS gene. The
histological diagnosis could therefore not be more precise than
spindle cell tumor of uncertain malignancy. Three years after
treatment, no local recurrence has developed and the patient is in
remission.
G-banding and karyotyping
Both a core needle preoperative biopsy and fresh
tissue from a representative area of the tumor in the surgical
specimen were received and analyzed cytogenetically as part of our
diagnostic routine. The samples were disaggregated mechanically and
enzymatically with collagenase II (Worthington, Freehold, NJ, USA).
The resulting cells were cultured and harvested using standard
techniques (16). Chromosome
preparations were G-banded with Wright stain and examined. The
karyotype was written according to The International System for
Human Cytogenetic Nomenclature (ISCN) 2013 guidelines (17).
High-throughput paired-end
RNA-sequencing analysis
Tumor tissue adjacent to that used for cytogenetic
analysis and histologic examination was frozen and stored at −80°C.
Total RNA was extracted using miRNeasy Mini kit according to the
manufacturer's instructions (Qiagen Nordic, Oslo, Norway). Tumor
tissue was disrupted and homogenized in Qiazol Lysis Reagent
(Qiagen Nordic) using a 5 mm stainless steel bead and TissueLyser
II (Qiagen Nordic). Subsequently, total RNA was purified using
QIAcube (Qiagen Nordic). The RNA quality was evaluated using the
Experion Automated Electrophoresis System (Bio-Rad Laboratories,
Oslo, Norway). The RNA quality indicator (RQI) was 9.9. Total RNA
(3 µg) was sent for high-throughput paired-end RNA-sequencing at
the Norwegian Sequencing Centre, Ullevål Hospital (http://www.sequencing.uio.no/). Detailed information
about the high-throughput paired-end RNA-sequencing was given
elsewhere (18). The software
FusionCatcher (19) (https://github.com/ndaniel/fusioncatcher) was used for
discovery of fusion transcripts.
Molecular genetic analysis
Total RNA (1 µg) was reverse-transcribed in a 20 µl
reaction volume using iScript Advanced cDNA Synthesis kit for
RT-qPCR according to the manufacturer's instructions (Bio-Rad
Laboratories). The 25 µl PCR volume contained 12.5 µl Premix Ex Taq
DNA Polymerase Hot Start version (Takara Bio Europe/SAS,
Saint-Germain-en-Laye, France), 2 µl of cDNA, and 0.4 µM of each of
the forward primer SETD2-7227F1 (5′-CCTCCCAACTGGAAGACAGCTCGA-3′)
and reverse primer NF1-020-452R1 (5′-AGCTTTCCAACCCAGGACTGTGGTC-3′).
The PCR was run on a C-1000 Thermal cycler (Bio-Rad Laboratories).
The PCR conditions for amplification were: initial denaturation at
94°C for 30 sec followed by 35 cycles of 7 sec at 98°C and 2 min at
68°C, and a final extension for 5 min at 68°C. PCR products (3 µg)
were stained with GelRed (Biotium), analyzed by electrophoresis
through 1.0% agarose gel, and photographed. The remaining 22 µl PCR
products were purified using the MinElute PCR purification kit
(Qiagen Nordic) and direct sequenced using the light run sequencing
service of GATC Biotech (http://www.gatc-biotech.com/en/sanger-services/lightrun-sequencing.html).
The BLAST software (http://www.ncbi.nlm.nih.gov/BLAST/) was used for
computer analysis of sequence data.
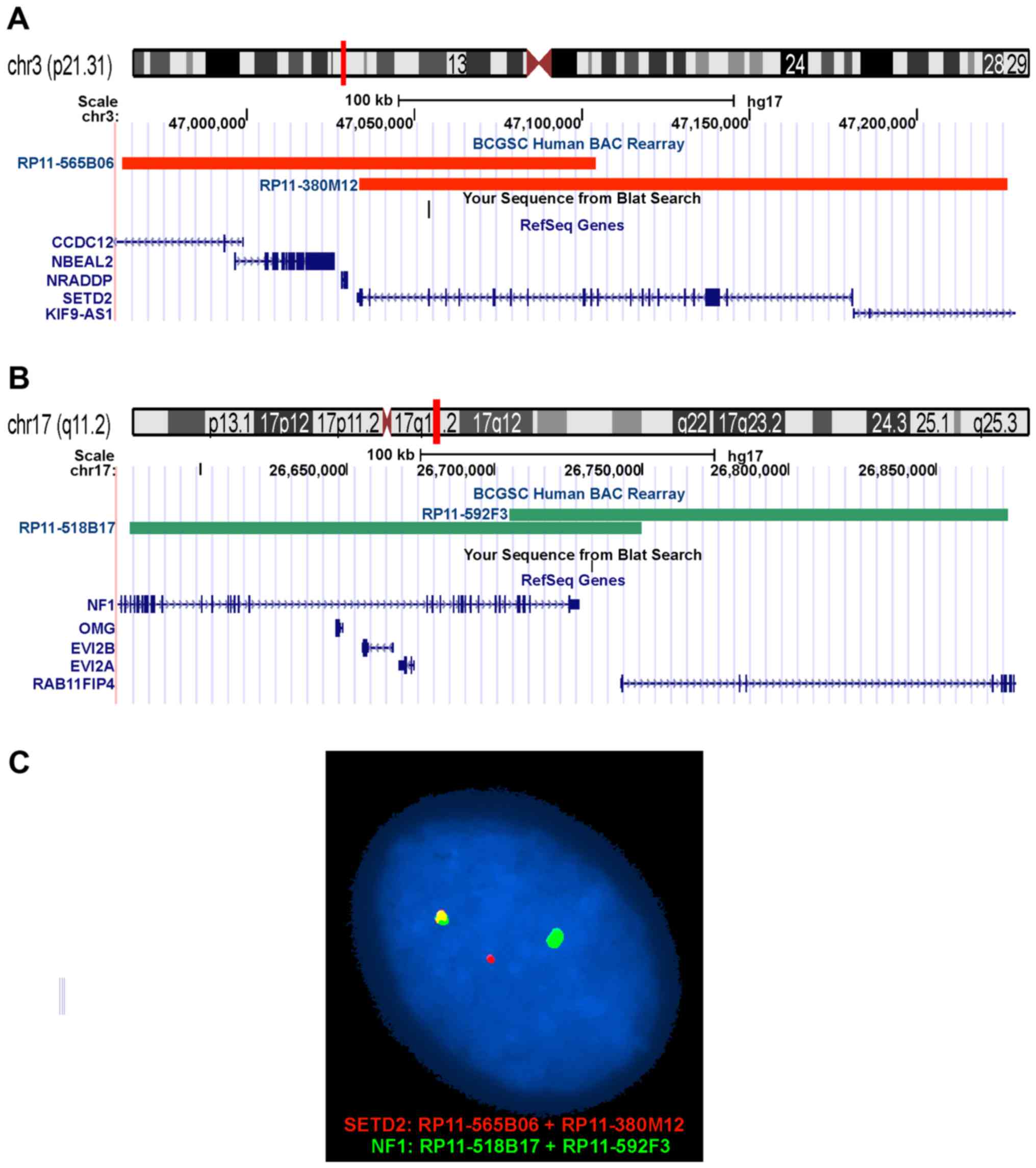
Fluorescence in situ hybridization
(FISH)
BAC probes were retrieved from the Human ‘32K’ BAC
Re-Array library (BACPAC Resources, http://bacpac.chori.org/home.htm). They were selected
according to physical and genetic mapping data on chromosomes 3 and
17 (see below) as reported on the Human Genome Browser at the
University of California, Santa Cruz website (May 2004, http://genome.ucsc.edu/). FISH mapping of the clones
on normal controls was performed to confirm their chromosomal
location. The clones were RP11-565B06 (chr3:46962826-47104129) and
RP11-380M12 (chr3:47033474-47226748) mapping to chromosome subband
3p21.31 which contains the SETD2 gene (red signal), and
RP11-518B17 (chr17:26576215-26749754) and RP11-592F3
(chr17:26705272-26874157) mapping to chromosome subband 17q11.2
which contains the NF1 gene (green signal). FISH was
performed as described elsewhere (18). Fluorescent signals were captured and
analyzed using the CytoVision system (Leica Biosystems, Newcastle,
UK).
Results
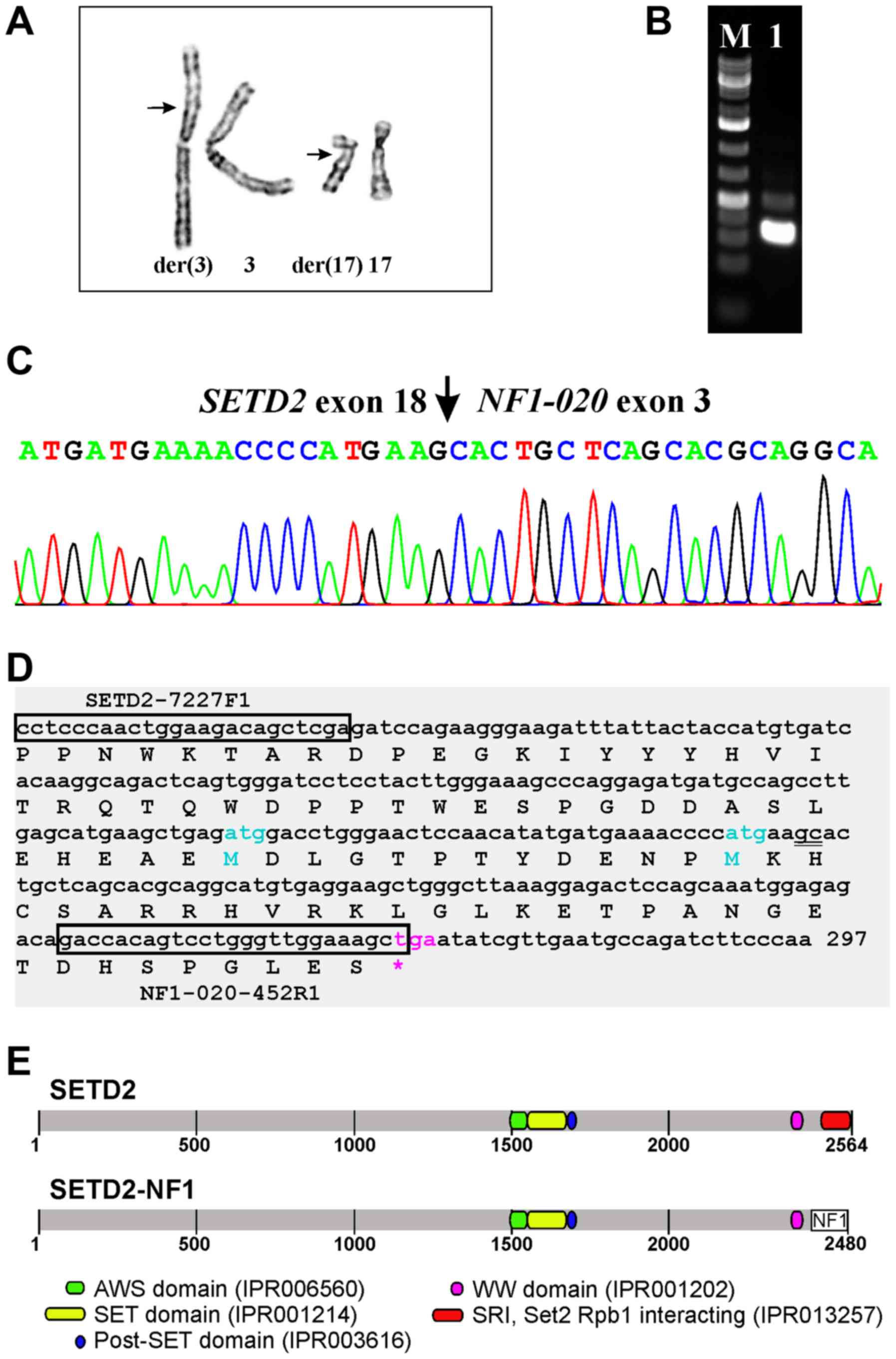
G-banding
The G-banding analysis of short-term cultured cells
from both the core needle biopsy and surgical specimen yielded the
karyotype 46,XY,t(3;17)(p21;q12),del(10)(q24)[11] (Fig. 2A).
RNA-sequencing, molecular genetic
analysis, and FISH confirmation of SETD2-NF1 fusion
Using the FusionCatcher software with the fastq
files obtained from the Norwegian Sequencing Centre, 31 potential
fusion transcripts were found (Table
I), among them SETD2-NF1. Taking into consideration that
SETD2 and NF1 map to chromosome bands 3p21.31 and
17q11.2, respectively (http://genome-euro.ucsc.edu/index.html), the bands
identified by G-banding analysis as being recombined by the
3;17-translocation, we decided to investigate further the
SETD2-NF1 fusion transcript using molecular techniques. No
other fusions were examined.
 | Table I.Fusion transcripts detected using
FusionCatcher. |
Table I.
Fusion transcripts detected using
FusionCatcher.
| 5′-Chr | 3′-Chr | 5′-Partner
gene | 3′-Partner
gene | Fusion
description | Fusion
sequence |
|---|
| 17 | 3 | COL1A1 | APOD |
|
GCTCCCTCCCACCCAACCAACTTTC*cccccccccataaagacaaaccaat |
|
3 | 17 |
SETD2 |
NF1 |
|
AACATATGATGAAAACCCCATGAAG*cactgctcagcacgcaggcatgtga |
| 7 | 3 | COL1A2 | APOD |
|
CTGGCAACATTGGATTCCCTGGACC*catcggcaccgtactggatcctggc |
| 19 | 19 | ADCK4 | NUMBL | readthrough |
TCCAGCCTCTCAGTGTGTTGGAGAG*acggggcgggcaccatgaacaagtt |
| 1 | 1 | PEAR1 | LRRC71 | readthrough |
TCCAGGGCCCTGTGTACATAAACTG*aggagtaccagtgctccggggtcct |
| 2 | 3 | IGFBP5 | APOD |
|
CAGGACTGACCCTCCTTCCTCCAGC*cacccagccccaagatggtgatgct |
| 19 | 19 | CYP4F12 |
CYP4F24P | pseudogene |
ACCGCGATCCTAAAGAGATTGAATG*gcattatctgcatcatcaacattat |
| 17 | X | COL1A1 | TIMP1 |
|
CTTTCCACCCTCTCTCCACCTGCCT*ctggcttctggcatcctgttgttgc |
| 3 | 3 | DVL3 | AP2M1 | readthrough |
TTCCGCATGGCCATGGGAAACCCCA*gtctgttctcagagcgatgggccgc |
| 3 | 3 | COL7A1 | UCN2 | readthrough |
CGGGTGGTCCAGAGCCAGGGGACAG*cctgacctcacgatgaccaggtgtg |
| 11 | 11 | CTSC | RAB38 | readthrough |
TTTGTCAGTCCTGTTCGAAACCAAG*gtcaagaaagatttggaaacatgac |
| 10 | 10 | MTG1 |
RP11-108K14.4 | readthrough |
CCCTCAACAAACACCAGCGCTTTGG*gtggaccaggtgctctgaggctggc |
| 11 | 11 | LSP1 | TNNT3 | short distance |
CCGGCTCCCTAGGCGTCCCATCTCG*aaaccacccaccttcaccatgtctg |
| 1 | 1 | CTBS | GNG5 | readthrough |
GCGGGCTCCTTATTATAACTATAAA*gtttcccaggcagctgcagacttga |
| 19 | 19 | GRAMD1A | SCN1B | readthrough |
CCTCGGCGGCCACTGCTGGCACGGT*tgtcctcagcctgcgggggctgcgt |
| 10 | 10 | SYNPO2L | MYOZ1 | readthrough |
CTAAGCGGCAGAGCCGTGCGGACAG*tgctgcccccacgcctgcccagctc |
| 19 | 19 | XRCC1 | ETHE1 | readthrough |
TATGGGGTGGTGCCGCAAGCCTGAA*gcgttggagaccagggccagccctg |
| 11 | 11 | HPX | APBB1 | readthrough |
TCTTCTCGGCTCCATATCATGGCAG*gagctgccaaggccatgtctgttcc |
| 16 | 16 | LCAT | PSMB10 | readthrough |
TGAATAAAGACCTTCCTTTGCTACC*agtacccagtgagcagcacagaggg |
| 2 | 2 | SOCS5 |
LINC01119 | short distance |
GAGGCCGCCCCGCGCGCCCCAAACG*atgattccaatgtacagccaatgat |
| 3 | 3 | TBC1D23 | NIT2 | readthrough |
GAATAGAAATCTTGGCAATCGAAAG*ctttccgcttggccctcatccagct |
| 2 | 2 | ADCY3 | PTRHD1 | readthrough |
CACGGGGGTCATGGGCAACATTCAG*gccccagatgagaccaccctaaagg |
| 19 | 19 | CADM4 | ZNF428 | readthrough |
GCGCTCTACGTACTTGTGGTCTACG*catccctctctacctgccaacatcc |
| 20 | 20 | CCM2L | HCK | short distance |
CCGACTTCAGCTGCTGCAGCTCCTT*gatggggtgcatgaagtccaagttc |
| 17 | 20 | COL1A1 | CPXM1 |
|
CCACCCAACCAACTTTCCCCCCAAC*catcacctgccattgcccacttact |
| 16 | 16 | COQ9 | POLR2C | readthrough |
TGAGACAAGTGCCTGCTGGACAGAG*gaggccgcagaagacgctcggcagt |
| 1 | 1 | EIF4G3 | HP1BP3 | readthrough |
AAAAAGGTGATCATGGAGGAAAAAG*gttgattcttcaccacactgaaacc |
| X | X |
MORF4L2-AS1 | TMEM31 | readthrough |
AGAAGAGTGCAGGAAAGCAAACCAA*gtgatcactttactgtagaagaaat |
| 7 | 7 | RHBDD2 | POR | readthrough |
AGAGGAGGGCAGCCCAGAGCCGGAA*tttcatgatcaacatgggagactcc |
| 14 | 3 |
SERPINA3 | APOD |
|
CAACAGGCCCTTCCTGATGATCATT*gtcccttctccagccacccagcccc |
| 1 | 1 | VPS45 | PLEKHO1 | readthrough |
GCACCACAGTGCACAACACGAAAAG*ggacctcaggatggaaaccagcagc |
RT-PCR with the SETD2-7227F1 and NF1-020-452R1
primer combination amplified a 268 bp cDNA fragment (Fig. 2B). Sanger sequencing showed that it
was a SETD2-NF1 chimeric cDNA fragment with the fusion point
identical to that found using FusionCatcher (Fig. 2C and D; Table I). In this fusion transcript, the
sequence of SETD2 coding for the last 114 amino acids of the
SETD2 protein are replaced by the NF1 sequence coding for 30
amino acids (Fig. 2D and E).
Interphase FISH analysis confirmed the
SETD2-NF1 fusion. All 100 counted nuclei showed a red signal
corresponding to the SETD2 (Fig.
3A and C), a green signal corresponding to NF1 (Fig. 3B and C), and a yellow fusion signal
corresponding to the SETD2-NF1 (Fig. 3C).
Discussion
Fusion transcripts of both NF1 and
SETD2 with various partners have been described in
hematologic malignancies as well as solid tumors (20–22).
However, this is the first time that a fusion between SETD2
and NF1 was found.
The SETD2 gene is ubiquitously expressed and
codes for a protein which belongs to a class of huntingtin
interacting proteins characterized by WW motifs (23,24).
SETD2 is also a DNA-binding factor that binds the proximal
E1A promoter of adenovirus serotype 12 (24). In addition, SETD2 was shown to
possess histone H3 lysine 36 (H3-K36) specific HMTase activity,
auto-methylation activity, a novel transcriptional activation
domain, and association with hyperphosphorylated RNA polymerase II
(25). The SETD2 protein is solely
responsible for all H3K36 trimethylation in humans (26). SETD2 interacts with the Ser2/Ser5
hyperphosphorylated RNA polymerase II during transcriptional
elongation via its SRI (Set2 Rpb1 interacting) domain, which
explains why H3K36 trimethylation is found in the body of actively
transcribed genes (27). Deletion
of the SRI domain in yeast Set2 abolishes H3K36 methylation, which
in turn prevents elongation by RNA polymerase II. This suggests
that the SRI domain is responsible for coupling transcription to
histone methylation by Set2 (28).
It seems that SETD2 serves as a linker between histone H3-K36
methylation and transcriptional regulation in yeast and mammals
(25,28).
Involvement of the SETD2 gene has been
reported in many types of malignancy (29). Inactivation of SETD is common
in clear cell renal carcinoma with loss or decrease of H3K36me3
mark (30), when it is associated
with worse prognosis and development of recurrent and/or metastatic
disease (31). Downregulation of
SETD2 at transcriptional and protein levels was observed in
breast cancer (32,33). The expression of SETD2 was
lower in malignant samples, decreased with increasing tumor stage,
and was lower in samples from patients who developed metastasis,
local recurrence, or died from breast cancer compared to those who
were disease-free for >10 years (32). SETD2 mutations were also
described in high-grade gliomas and in leukemias (34–36).
The mutations are either nonsense or frameshift mutations that
truncate a portion of the C terminus domain of SETD2. Truncating
mutations result in loss of the C terminus SRI domain which is
responsible for the recruitment of SETD2 to its target gene locus
through binding to the phosphor-C-terminal repeat domain (PCTD) of
elongating RNA polymerase II (36).
Recently, genomic disruption of SETD2 was reported in
chronic lymphocytic leukemia and the data suggested that
SETD2 aberrations may be clinically relevant (37). Patients with SETD2
abnormalities and wild-type TP53 and ATM had
significantly shorter progression-free and overall survival
compared with cases with wild-type for all three genes (37). In malignant mesotheliomas, a
combination of the methods array comparative genomic hybridization
and targeted next-generation sequencing revealed biallelic
SETD2 inactivation in 9 out of 33 examined tumors (38). Gene fusions and splice alterations
were also reported to be frequent mechanisms for SETD2
inactivation (39).
SETD2 was found to be the most significantly
and recurrently mutated gene in type II enteropathy-associated
T-cell lymphoma (EATL-II); 86% (13/15) of EATL-II tumors with 20
distinctive mutations (40).
Fourteen of these mutations consisted of premature stop codon,
nonsense, frameshift indels or splicing mutations expected to
confer critical changes in protein structure. The other six
missense mutations occurred in highly conserved residues of
functional domains and were predicted to be deleterious with a
damaging effect on the protein (40).
The NF1 gene spans approximately 280 kbp, has
58 exons (mRNA transcript variant 1, NM_001042492.2) and codes for
the cytoplasmatic and multidomain protein neurofibromin (41). Neurofibromin is a negative regulator
of the RAS cellular proliferation pathway (42–45).
Several other functions of neurofibromin were also reported, among
them positive regulation of adenyl cyclase, regulation of cell
adhesion and motility, and suppression of epithelial mesenchymal
transition (42–45). The NF1 gene is a classical
tumor suppressor gene whose inactivation is responsible for the
neurofibromatosis type 1 (NF1) tumor predisposition syndrome
(http://omim.org/entry/613113). Mutations
of NF1 are also linked to juvenile myelomonocytic leukemia
(http://omim.org/entry/607785) and Watson
syndrome (http://omim.org/entry/193520). The NF1 syndrome is
characterized by the development of multiple neurofibromas,
café-au-lait spots, and Lisch nodules (42,45).
Patients with NF1 syndrome are at increased risk to develop
malignant peripheral nerve sheath tumors, phaeochromocytoma,
leukemia, glioma, rhabdomyosarcoma, and breast cancer (42,45).
Both alleles of NF1 are inactivated in the tumors in NF1
patients. Mutations in the NF1 gene may also result in
cardiovascular, musculoskeletal, and nervous system anomalies
(45,46).
Splicing in the NF1 gene is complex and
several alternative transcripts were found (47); altogether 23 according to the
ensemble genome browser (http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000196712;r=17:31378891-31382106;t=ENST00000422121).
The transcript NF1-020 (ENST00000422121) has three exons and is
thought to undergo nonsense mediated decay, a process which detects
nonsense mutations and prevents the expression of truncated or
erroneous proteins (48). Thus, the
functional significance, if any, of the NF1-020 transcript is
unclear.
In the present case, the t(3;17) resulted in a
SETD2-NF1 fusion transcript in which the first 18 exons of
SETD2 (sequence with accession number NM_014159 version 6)
are fused to exon 3 of the transcript NF1-020
(ENST00000422121) (Fig. 2C and D).
The fusion transcript would code for a protein in which the last
114 amino acids of SETD2, in other words the entire SRI domain, are
replaced by 30 amino acids encoded by the NF1 sequence
(Fig. 2D and E). The result would
be similar to that seen with the truncating SETD2 mutations
found in leukemias (36). Absence
of the SRI domain would result in inability to recruit SETD2 to its
target gene locus through binding to the phosphor-C-terminal repeat
domain of elongating RNA polymerase II and may affect H3K36
methylation. Alternatively, loss of one of two functional
SETD2 alleles might be the crucial factor in tumorigenesis.
Whether aberrations of SETD2 are recurrent and define a
specific subgroup of spindle cell tumors, remains to be seen.
Acknowledgements
The authors thank Hege Kilen Andersen and Nina Øino
for excellent technical assistance. This work was supported by
grants from the Norwegian Radium Hospital Foundation.
References
|
1
|
Collini P, Sorensen PH, Patel S, Blay JY,
Issels RD, Maki RG, Eriksson M and del Muro XG: Sarcomas with
spindle cell morphology. Semin Oncol. 36:324–337. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fisher C: Spindle cell sarcomas. Surg
Pathol Clin. 4:721–744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heim S and Mitelman F: Cancer
Cytogenetics: Chromosomal and Molecular Genetic Abberations of
Tumor Cells. Fourth. Wiley-Blackwell; 2015, doi:
10.1002/9781118795569. View Article : Google Scholar
|
|
4
|
Sandberg AA and Bridge JA: Updates on the
cytogenetics and molecular genetics of bone and soft tissue tumors:
Congenital (infantile) fibrosarcoma and mesoblastic nephroma.
Cancer Genet Cytogenet. 132:1–13. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Llombart B, Serra-Guillén C, Monteagudo C,
López Guerrero JA and Sanmartín O: Dermatofibrosarcoma protuberans:
A comprehensive review and update on diagnosis and management.
Semin Diagn Pathol. 30:13–28. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fletcher CDM, Bridge JA, Hogendoorn PCW
and Mertens F: WHO Classification of Tumours of Soft Tissue and
Bone. 4th. IARC; Lyon: 2013
|
|
7
|
Chmielecki J, Crago AM, Rosenberg M,
O'Connor R, Walker SR, Ambrogio L, Auclair D, McKenna A, Heinrich
MC, Frank DA, et al: Whole-exome sequencing identifies a recurrent
NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet.
45:131–132. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mohajeri A, Tayebwa J, Collin A, Nilsson
J, Magnusson L, von Steyern FV, Brosjö O, Domanski HA, Larsson O,
Sciot R, et al: Comprehensive genetic analysis identifies a
pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic
imbalances, and a characteristic gene expression profile in
solitary fibrous tumor. Genes Chromosomes Cancer. 52:873–886. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robinson DR, Wu YM, Kalyana-Sundaram S,
Cao X, Lonigro RJ, Sung YS, Chen CL, Zhang L, Wang R, Su F, et al:
Identification of recurrent NAB2-STAT6 gene fusions in solitary
fibrous tumor by integrative sequencing. Nat Genet. 45:180–185.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schweizer L, Koelsche C, Sahm F, Piro RM,
Capper D, Reuss DE, Pusch S, Habel A, Meyer J, Göck T, et al:
Meningeal hemangiopericytoma and solitary fibrous tumors carry the
NAB2-STAT6 fusion and can be diagnosed by nuclear expression of
STAT6 protein. Acta Neuropathol. 125:651–658. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fruth K, Hansen T, Katenkamp D, Mann W and
Lippert BM: Recurrence of a laryngeal spindle cell sarcoma with a
transformation into a higher grade of malignancy. Auris Nasus
Larynx. 36:491–495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alaggio R, Rosolen A, Sartori F, Leszl A,
d'Amore ES, Bisogno G, Carli M, Cecchetto G, Coffin CM and Ninfo V:
Spindle cell tumor with EWS-WT1 transcript and a favorable clinical
course: A variant of DSCT, a variant of leiomyosarcoma, or a new
entity? Report of 2 pediatric cases. Am J Surg Pathol. 31:454–459.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Panagopoulos I, Bjerkehagen B, Gorunova L,
Berner JM, Boye K and Heim S: Several fusion genes identified by
whole transcriptome sequencing in a spindle cell sarcoma with
rearrangements of chromosome arm 12q and MDM2 amplification. Int J
Oncol. 45:1829–1836. 2014.PubMed/NCBI
|
|
14
|
Nord KH, Macchia G, Tayebwa J, Nilsson J,
von Steyern F Vult, Brosjö O, Mandahl N and Mertens F: Integrative
genome and transcriptome analyses reveal two distinct types of ring
chromosome in soft tissue sarcomas. Hum Mol Genet. 23:878–888.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lestou VS, O'Connell JX, Ludkovski O,
Gosling H, Lesack D and Horsman DE: Coamplification of 12p11 and
12q13 approximately q22 in multiple ring chromosomes in a spindle
cell sarcoma resolved by novel multicolor fluorescence in situ
hybridization analysis. Cancer Genet Cytogenet. 139:44–47. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mandahl N: Methods in solid tumour
cytogeneticsHuman cytogenetics: Malignancy and Acquired
Abnormalities. Rooney DE: 3rd. Oxford University Press; New York:
pp. 165–203. 2001
|
|
17
|
Schaffer LG, McGowan-Jordan J and Schmid
M: ISCN 2013: An International System for Human Cytogenetic
Nomenclature Karger, Basel. 2013. View Article : Google Scholar
|
|
18
|
Panagopoulos I, Gorunova L, Bjerkehagen B
and Heim S: The ‘grep’ command but not FusionMap, FusionFinder or
ChimeraScan captures the CIC-DUX4 fusion gene from whole
transcriptome sequencing data on a small round cell tumor with
t(4;19)(q35;q13). PLoS One. 9:e994392014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nicorici D, Satalan H, Edgren H,
Kangaspeska S, Murumagi A, Kallioniemi O, Virtanen S and Kikku O:
FusionCatcher - a tool for finding somatic fusion genes in
paired-end RNA-sequencing data. bioRxiv. 2014.doi:
10.1101/011650.
|
|
20
|
Andersson AK, Ma J, Wang J, Chen X, Gedman
AL, Dang J, Nakitandwe J, Holmfeldt L, Parker M, Easton J, et al:
St. Jude Children's Research Hospital-Washington University
Pediatric Cancer Genome Project: The landscape of somatic mutations
in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet.
47:330–337. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen K, Navin NE, Wang Y, Schmidt HK,
Wallis JW, Niu B, Fan X, Zhao H, McLellan MD, Hoadley KA, et al:
BreakTrans: Uncovering the genomic architecture of gene fusions.
Genome Biol. 14:R872013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoshihara K, Wang Q, Torres-Garcia W,
Zheng S, Vegesna R, Kim H and Verhaak RG: The landscape and
therapeutic relevance of cancer-associated transcript fusions.
Oncogene. 34:4845–4854. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Faber PW, Barnes GT, Srinidhi J, Chen J,
Gusella JF and MacDonald ME: Huntingtin interacts with a family of
WW domain proteins. Hum Mol Genet. 7:1463–1474. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rega S, Stiewe T, Chang DI, Pollmeier B,
Esche H, Bardenheuer W, Marquitan G and Pützer BM: Identification
of the full-length huntingtin-interacting protein p231HBP/HYPB as a
DNA-binding factor. Mol Cell Neurosci. 18:68–79. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang
HH, Zhang QH, Chen SJ, Huang QH and Chen Z: Identification and
characterization of a novel human histone H3 lysine 36-specific
methyltransferase. J Biol Chem. 280:35261–35271. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Edmunds JW, Mahadevan LC and Clayton AL:
Dynamic histone H3 methylation during gene induction: HYPB/Setd2
mediates all H3K36 trimethylation. EMBO J. 27:406–420. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wagner EJ and Carpenter PB: Understanding
the language of Lys36 methylation at histone H3. Nat Rev Mol Cell
Biol. 13:115–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kizer KO, Phatnani HP, Shibata Y, Hall H,
Greenleaf AL and Strahl BD: A novel domain in Set2 mediates RNA
polymerase II interaction and couples histone H3 K36 methylation
with transcript elongation. Mol Cell Biol. 25:3305–3316. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kudithipudi S and Jeltsch A: Role of
somatic cancer mutations in human protein lysine
methyltransferases. Biochim Biophys Acta. 1846:366–379.
2014.PubMed/NCBI
|
|
30
|
Duns G, van den Berg E, van Duivenbode I,
Osinga J, Hollema H, Hofstra RM and Kok K: Histone
methyltransferase gene SETD2 is a novel tumor suppressor gene in
clear cell renal cell carcinoma. Cancer Res. 70:4287–4291. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hakimi AA, Ostrovnaya I, Reva B, Schultz
N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, et al:
Adverse outcomes in clear cell renal cell carcinoma with mutations
of 3p21 epigenetic regulators BAP1 and SETD2: A report by MSKCC and
the KIRC TCGA research network. Clin Cancer Res. 19:3259–3267.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Al Sarakbi W, Sasi W, Jiang WG, Roberts T,
Newbold RF and Mokbel K: The mRNA expression of SETD2 in human
breast cancer: Correlation with clinico-pathological parameters.
BMC Cancer. 9:2902009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Newbold RF and Mokbel K: Evidence for a
tumour suppressor function of SETD2 in human breast cancer: A new
hypothesis. Anticancer Res. 30:3309–3311. 2010.PubMed/NCBI
|
|
34
|
Fontebasso AM, Schwartzentruber J,
Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, Jones DT, Witt H,
Kool M, Albrecht S, et al: Mutations in SETD2 and genes affecting
histone H3K36 methylation target hemispheric high-grade gliomas.
Acta Neuropathol. 125:659–669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mar BG, Bullinger LB, McLean KM, Grauman
PV, Harris MH, Stevenson K, Neuberg DS, Sinha AU, Sallan SE,
Silverman LB, et al: Mutations in epigenetic regulators including
SETD2 are gained during relapse in paediatric acute lymphoblastic
leukaemia. Nat Commun. 5:34692014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu X, He F, Zeng H, Ling S, Chen A, Wang
Y, Yan X, Wei W, Pang Y, Cheng H, et al: Identification of
functional cooperative mutations of SETD2 in human acute leukemia.
Nat Genet. 46:287–293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Parker H, Rose-Zerilli MJ, Larrayoz M,
Clifford R, Edelmann J, Blakemore S, Gibson J, Wang J, Ljungström
V, Wojdacz TK, et al: Genomic disruption of the histone
methyltransferase SETD2 in chronic lymphocytic leukaemia. Leukemia.
30:2179–2186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yoshikawa Y, Emi M, Hashimoto-Tamaoki T,
Ohmuraya M, Sato A, Tsujimura T, Hasegawa S, Nakano T, Nasu M,
Pastorino S, et al: High-density array-CGH with targeted NGS unmask
multiple noncontiguous minute deletions on chromosome 3p21 in
mesothelioma. Proc Natl Acad Sci USA. 113:pp. 13432–13437. 2016;
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bueno R, Stawiski EW, Goldstein LD,
Durinck S, De Rienzo A, Modrusan Z, Gnad F, Nguyen TT, Jaiswal BS,
Chirieac LR, et al: Comprehensive genomic analysis of malignant
pleural mesothelioma identifies recurrent mutations, gene fusions
and splicing alterations. Nat Genet. 48:407–416. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Roberti A, Dobay MP, Bisig B, Vallois D,
Boéchat C, Lanitis E, Bouchindhomme B, Parrens MC, Bossard C,
Quintanilla-Martinez L, et al: Type II enteropathy-associated
T-cell lymphoma features a unique genomic profile with highly
recurrent SETD2 alterations. Nat Commun. 7:126022016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Barron VA and Lou H: Alternative splicing
of the neurofibromatosis type I pre-mRNA. Biosci Rep. 32:131–138.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kiuru M and Busam KJ: The NF1 gene in
tumor syndromes and melanoma. Lab Invest. 97:146–157. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mahalingam M: NF1 and Neurofibromin:
Emerging players in the genetic landscape of desmoplastic melanoma.
Adv Anat Pathol. 24:1–14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ratner N and Miller SJ: A RASopathy gene
commonly mutated in cancer: The neurofibromatosis type 1 tumour
suppressor. Nat Rev Cancer. 15:290–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yap YS, McPherson JR, Ong CK, Rozen SG,
Teh BT, Lee AS and Callen DF: The NF1 gene revisited - from bench
to bedside. Oncotarget. 5:5873–5892. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Larizza L, Gervasini C, Natacci F and Riva
P: Developmental abnormalities and cancer predisposition in
neurofibromatosis type 1. Curr Mol Med. 9:634–653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vandenbroucke I, Callens T, De Paepe A and
Messiaen L: Complex splicing pattern generates great diversity in
human NF1 transcripts. BMC Genomics. 3:132002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schweingruber C, Rufener SC, Zünd D,
Yamashita A and Mühlemann O: Nonsense-mediated mRNA decay -
mechanisms of substrate mRNA recognition and degradation in
mammalian cells. Biochim Biophys Acta. 1829:612–623. 2013.
View Article : Google Scholar : PubMed/NCBI
|