Introduction
Neuroblastoma (NB) is the most common extracranial
solid tumor in children. One of the major biological factors
related to poor patient survival is amplification of the
N-myc gene, which is seen in ~30% of cases (1). This proto-oncogene encodes a basic
helix-loop-helix transcription factor that binds to E-box elements
(2) in the numerous genes it
regulates. Since the majority of neuroblastoma patients with
overexpression of N-myc have a lower survival rate, defining novel
pathways that regulate its expression is critical for developing
new therapeutic approaches to better treat this enigmatic
cancer.
MicroRNAs (miRNAs) are small (~21 nucleotides)
non-coding regulatory RNAs that downregulate the expression of
mRNAs either by inhibiting their translation and/or facilitating
the degradation of the cognate mRNA (3). Several recent studies have reported a
role for miRNAs in targeting or being targets of N-myc in
neuroblastoma (4–16). Of particular interest has been the
relationship between N-myc expression and members of the
miR-17-92 miRNA cluster, a polycistronic cluster on chromosome
13q31.32 that gives rise to 6 mature miRNAs (miR-17, miR-18a,
miR-19a, miR-20a and miR-92-1). Several previous studies in human
neuroblastoma cell lines have shown that the expression levels of
the miR-17-92 cluster members are directly correlated with
N-myc amplification and that N-myc binds to E-boxes to
increase the transcription of this gene cluster (5,16).
Likewise, miR-17-92 overexpression is associated with increased
tumorigenicity in numerous types of cancer, including neuroblastoma
(17).
Our previous studies have shown that another
RNA-binding element also plays an important role in the
amplification and overexpression of N-myc: the
neuronal-specific RNA binding protein HuD (ELAVL4). This protein is
involved in the splicing and stabilization of N-myc mRNA
(18–20), and its decreased expression
(consequent to loss of the short arm of chromosome #1) leads to
N-myc amplification in neuroblastoma. Conversely,
re-introduction of HuD genes removes the selection pressure,
resulting in loss of N-myc genes (20).
In the present study, we demonstrate that miR-17, a
member of the miR-17-92 cluster, is induced by N-myc and
reciprocally binds to the N-myc 3-untranslated region
(3′-UTR) to downregulate its expression, creating a
negative-feedback loop in N-myc-amplified neuroblastoma cell
lines. Moreover, the 3′-UTR region to which miR-17 binds contains
an overlapping binding site for HuD and the miRNA and this
RNA-binding protein compete for binding to this site. This
competition may be exacerbated in N-myc-amplified
neuroblastoma cells, which lack one copy of the HuD gene. A
possible consequence of this imbalance is a heightened selection
pressure resulting in the extremely high levels of N-myc
gene amplification seen in neuroblastoma as well as the increased
malignancy and decreased differentiation in N-myc-amplified
neuroblastoma cells.
Materials and methods
Cell culture
All cell lines were maintained as previously
described (21). Thirteen human
neuroblastoma clonal cell lines or enriched populations were used
in these studies: N-myc-amplified (SK-N-BE(1)n, LA1-55n, KCN-83n, BE(2)-M17V, SK-N-LD, SK-N-HM, BE(2)-C and LA1-5s), and N-myc
non-amplified (SH-SY5Y, SMS-LHN, CB-JMN, SH-EP1 and SMS-KCNs).
miRNA microarray
miRNAs were isolated using mirVana™ miRNA Isolation
kits (Ambion, Austin, TX, USA). Processing and initial analysis of
miRNA expression levels were carried out by LC Sciences (Houston,
TX, USA). The correlation between N-myc RNA and miRNA
expression levels was assessed by linear regression analysis.
Quantitative real-time RT-PCR
cDNA for miRNAs were synthesized using the
TaqMan® MicroRNA Reverse Transcription kit.
miRNA-specific primers and miRNAs were quantified using TaqMan
assays (Applied Biosystems, Foster City, CA, USA) by the
comparative ΔΔCt method. Expression levels of miRNAs
were normalized to U6 and expressed as a fold-change compared to
the levels of the same sample of SH-SY5Y cells.
Western blot analysis
Western blot analysis of proteins was performed as
previously described (21). Primary
antibodies used were rabbit anti-N-myc [(C-19), (SC-791)] (Santa
Cruz Biotechnology Inc., Santa Cruz, CA, USA) and mouse anti-actin
[(AC-74), (076K4762)] (Invitrogen Corp., Carlsbad, CA, USA).
Transfections and infections
miRNA inhibitors for miR-17 and control oligos (100
nM) (Ambion) were transiently transfected into SK-N-LD cells using
Lipofectamine 2000 (Invitrogen Corp.) according to the
manufacturers instructions. Lentivirus constructs containing the
miR-17-92 cluster and controls were purchased from SBI Biosciences
(Mountain View, CA, USA) and used to infect SK-N-LD and CB-JMN
cells at a multiplicity of infection of 10, according to the
manufacturer's instructions. Resulting populations were either used
directly or cloned using cloning cylinders.
Luciferase and β-galactosidase
assays
The 3′-UTR of N-myc (880 bp) was
PCR-amplified (primers available upon request) and cloned into
pMIR-Luciferase reporter plasmid (Ambion) using T4-ligase (Promega,
Madison, WI, USA). The sequences of the inserts were confirmed
(GENEWIZ, South Plainfield, NJ, USA).
SK-N-LD cells were co-transfected in quadruplicate
with either pMIR-Luciferase-N-myc-UTR or
pMIR-REPORT-Luciferase (Luciferase vector) and pMIR-REPORT-β-gal
(Ambion) using Lipofectamine 2000. After 24 h, half of each of the
co-transfected populations were treated with 100 nM of either the
miR-17 inhibitor or a non-specific control oligo (both from
Ambion). Twenty-four hours thereafter luciferase and β-gal
activities were assessed (Promega). Luciferase activity was
normalized to β-gal and expressed as a fold-change in the
miR-17-inhibitor-treated cells compared to the control-treated
cells for both the pMIR-Luciferase-full-N-myc 3′-UTR and the
vector transfectants.
Colony-forming efficiency
Colony-forming efficiencies in soft agar were
assessed as previously described (21). Mean colony-forming efficiency (CFE;
the number of colonies divided by the cell inoculum × 100) was
determined in quadruplicate in 3 independent experiments.
Gel mobility shift assays (GMSAs)
The pCRII-SNM plasmid, containing 399 nt at the
3′-end of the N-myc 3′-UTR (19)
was linearized and used to generate radioactive probes for gel
shift assays using the MAXIscript T7 in vitro transcription
kit (Ambion) and 32P-UTP (PerkinElmer, Inc., Waltham,
MA, USA). The probes generated were treated with DNase and purified
using a MEGAclear kit (both from Ambion).
HuD and HuC proteins were expressed and purified as
previously described (19).
Bacterial lysates containing either HuD or HuC protein were mixed
with the N-myc 3′-UTR probe (~10,000 cpm) and incubated for
20 min at room temperature. In competition experiments, the probe
was pre-mixed in annealing buffer (19) with either mature miR-17 RNA oligo
(8, 40 or 80 pmol) or non-specific RNA oligo (40 pmol) (sequences
available upon request) (Dharmacon Inc., Chicago, IL, USA),
denatured at 95°C for 5 min and cooled to room temperature before
use. RNA-protein complexes were resolved by electrophoresis through
3.2% non-denaturing polyacrylamide gels and exposed to Kodak XAR
film overnight at −80°C.
Results
Inverse relationship between N-myc
mRNA and miR-17-92 cluster expression
To identify miRNAs that regulate N-myc, we analyzed
the expression of miRNAs in 4 human neuroblastoma cell lines with
varying degrees of N-myc amplification using an miRNA
microarray. The 4 cell lines had N-myc relative mRNA levels
(compared to the non-amplified SH-SY5Y neuroblastoma cell line)
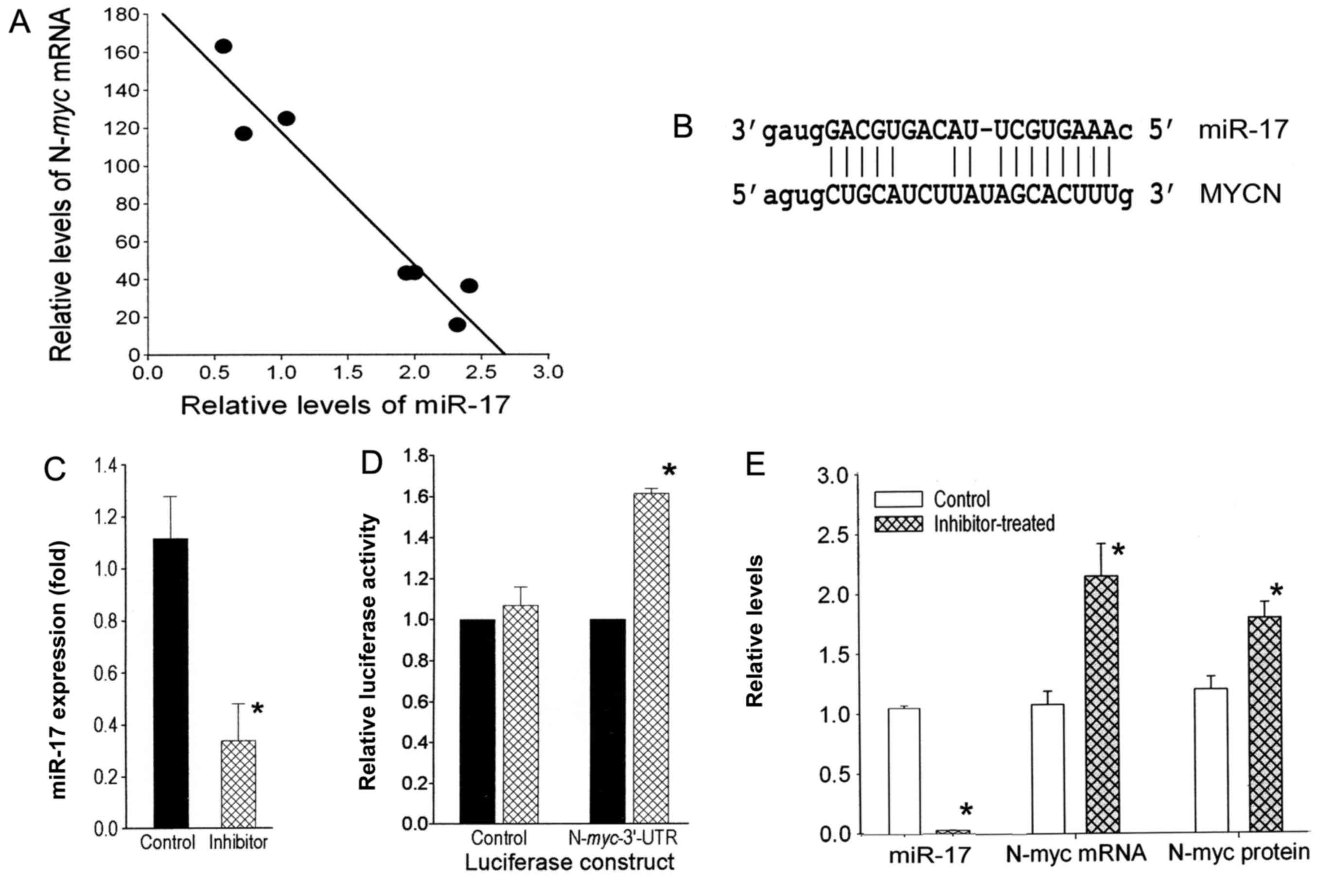
ranging from 16- to 183-fold. The levels of each miRNA in the 4
lines were compared to the N-myc levels by linear regression
analysis, and ranked in descending order according to the
R2 value. Notably, from this analysis, we found a highly
inverse correlation between the expression level of N-myc
and most of the miR-17-92 cluster members (Table I). Additional qRT-PCR analysis for 3
of the miRNAs (miR-17, miR-20a and miR-18a) with a larger set
(n=13) of neuroblastoma cell lines clearly revealed that all 3
miRNAs were significantly higher in the N-myc-amplified
group (n=8) compared to the N-myc-non-amplified group (n=6)
(data not shown) consistent with previous findings. Notably, as
predicted by the microarray data (Table
I), the qRT-PCR data ascertained that the inverse relationship
between N-myc expression and miR-17 levels was highly
significant (R2=0.98; P<0.001) (Fig. 1A). However, the inverse relationship
was not significant for miR-20a and miR-18a in the
N-myc-amplified cells. Further studies were undertaken to
investigate whether the inverse correlation between miR-17 and
N-myc levels is due to the downregulation of N-myc by
miR-17.
 | Table I.Inverse correlation between
N-myc overexpression and levels of miR-17-92 cluster
members. |
Table I.
Inverse correlation between
N-myc overexpression and levels of miR-17-92 cluster
members.
|
| Cell lines |
|
|---|
|
|
|
|
|---|
| mRNA/miRNA | BE(2)-M17 | BE(2)-C | SK-N-BE(1)n | SK-N-LD | R2 |
|---|
| N-myc | 183 | 117 | 47 | 16 |
|
|
hsa-miR-20a | 7,044 | 11,238 | 12,395 | 16,020 | 0.922 |
|
hsa-miR-17 | 7,749 | 10,688 | 12,137 | 16,145 | 0.902 |
|
hsa-miR-92 | 10,643 | 11,483 | 13,651 | 17,298 | 0.834 |
|
hsa-miR-19b | 4,865 | 5,201 | 8,840 | 13,332 | 0.815 |
|
hsa-miR-19a | 640 | 578 | 1,745 | 4,475 | 0.680 |
|
hsa-miR-18a | 1,022 | 1,381 | 1,172 | 3,173 | 0.496 |
N-myc 3′-UTR has miR-17 regulatory
sites
Sequence analysis revealed a complementary binding
site for miR-17 within the 3′-UTR of N-myc mRNA (Fig. 1B), making the oncogene a predicted
target of miR-17 (www.microrna.org and www.mirbase.org). To determine whether N-myc
mRNA is a target of miR-17, the N-myc full length 3′-UTR was
cloned into a luciferase reporter gene and this construct
(Luciferase-N-myc-UTR) or the Luciferase vector (control)
was transfected into the SK-N-LD N-myc-amplified cell line.
When each of these transfectants was treated with the miR-17
inhibitor, the miR-17 levels decreased significantly (3.3-fold;
P<0.05) (Fig. 1C) compared to
the control oligo transfectants. This decrease in miR-17 resulted
in a significant 1.6-fold (P<0.01) increase in luciferase
activity in cells expressing the Luciferase-N-myc-UTR
construct, but had no effect on the luciferase activity in the
Luciferase vector-transfectants (Fig.
1D). Thus, the binding site of miR-17 in N-myc appears
to be functional.
Alteration of miR-17 expression
inversely affects N-myc expression
To confirm the direct regulation of N-myc
expression by miR-17, a specific inhibitor for miR-17 was
transiently transfected into N-myc-amplified SK-N-LD cells.
miR-17 levels were decreased 33.3-fold (P<0.01) (Fig. 1E). Consequently, N-myc mRNA
and protein levels in the inhibitor-transfectants were
significantly increased 2.1-fold (P<0.01) and 1.5-fold
(P<0.02), respectively, compared to the control cells (Fig. 1E). Since the expression levels of
the homolog miRNAs miR-18a and miR-20a remained unaltered (data not
shown), these decreases in N-myc appear to be the result of
the specific decrease of miR-17. However, transfection of the
miR-17 inhibitor into an N-myc-non-amplified cell line
SH-SY5Y did not alter the levels of N-myc mRNA or the
protein, suggesting this regulation is not seen in
N-myc-non-amplified cells (data not shown).
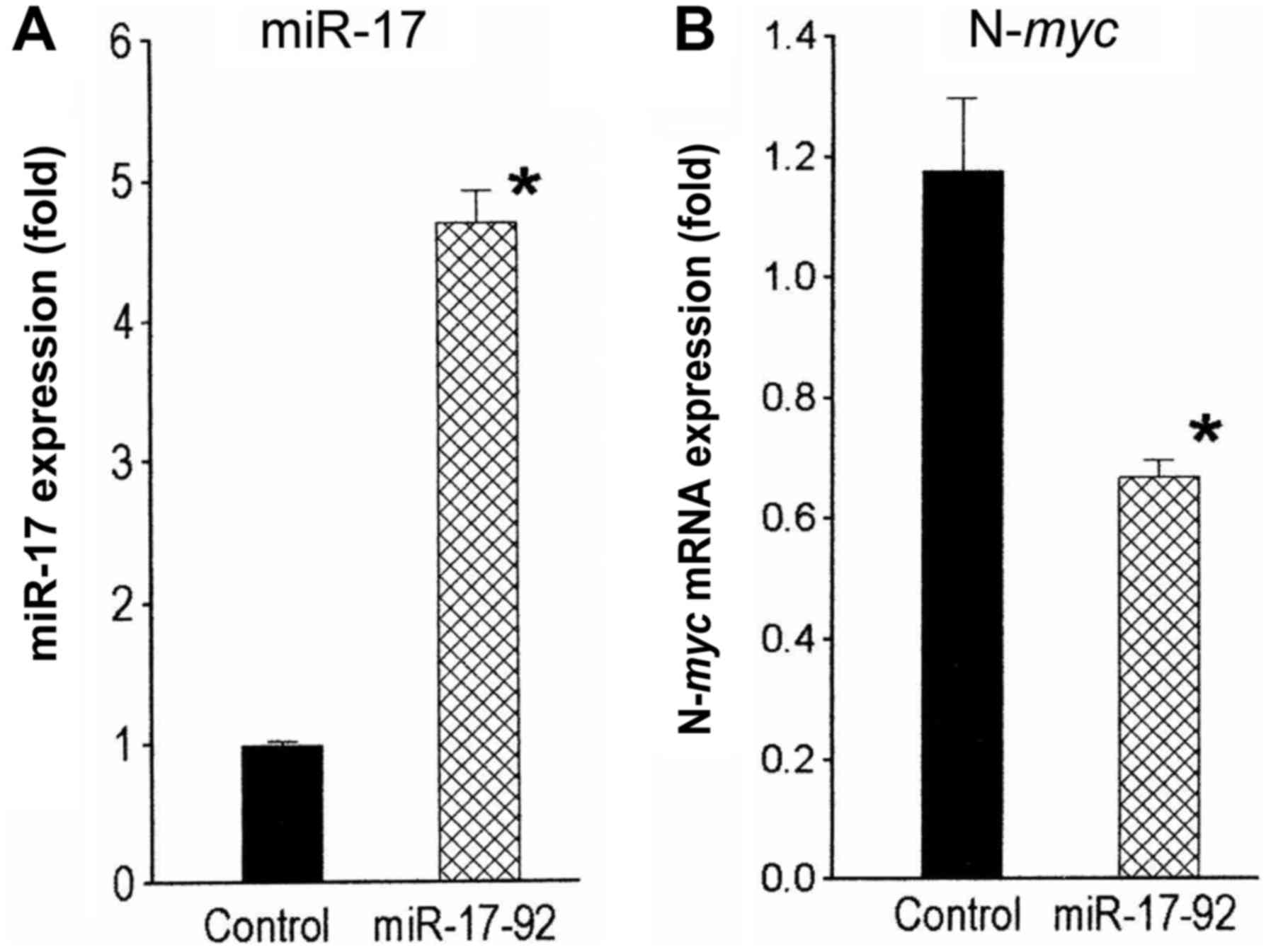
In converse experiments, levels of miR-17 were
increased in SK-N-LD cells by stable infection with a
miR-17-92-overexpressing cassette in a lentiviral vector. These
infected cell populations had 4.8-fold (P<0.03) higher miR-17
levels compared to the vector-infected control cells (Fig. 2A). N-myc mRNA levels in the
miR-17-92-overexpressing cells decreased significantly 1.7-fold
(P<0.02) compared to the control (Fig. 2B). Collectively, these observations
suggest that miR-17 regulates N-myc expression in
N-myc-amplified human neuroblastoma cells.
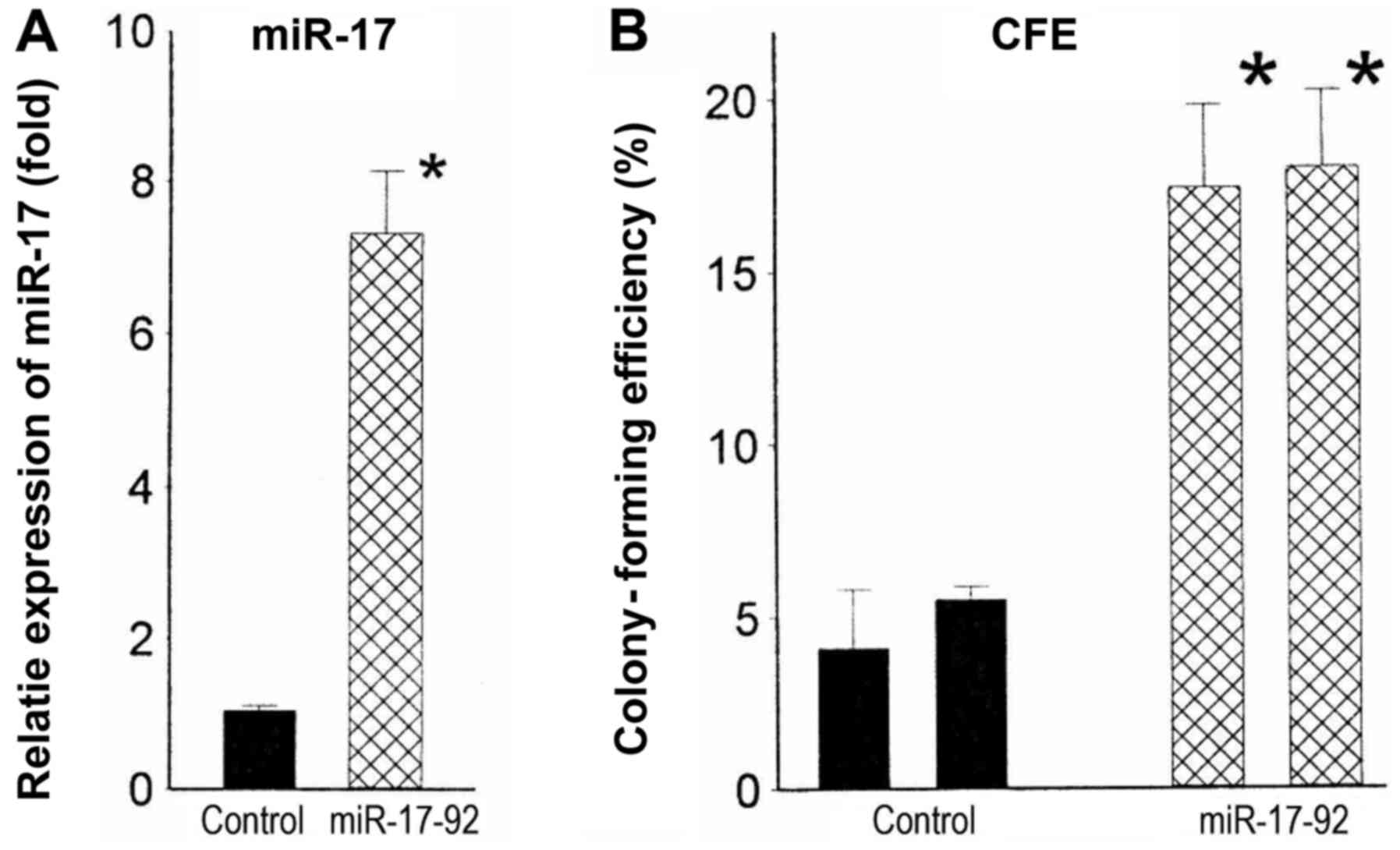
Overexpression of miR-17-92 increases
tumorigenicity
miR-17-92 overexpression is associated with
increased tumorigenicity in numerous types of cancer. To assess
whether elevated levels of miR-17-92 likewise contributed to
increased tumorigenicity of N-myc-amplified cell lines, we
stably infected the lentiviral miR-17-92 cassette into a
non-amplified cell line, CB-JMN. Two miR-17-92-infected clones with
average increases in miR-17 of 7.0-fold (P<0.001) (Fig. 3A) were tested for malignant
potential. These infectants had colony-forming efficiencies in soft
agar of 17.4 and 18.0%. By contrast, those of the control clones
were 4.4 and 5.1% (Fig. 3B). Thus,
increased expression of miR-17-92 appears to be responsible, at
least in part, for the increased malignant potential and/or
aggressiveness of N-myc-amplified tumors.
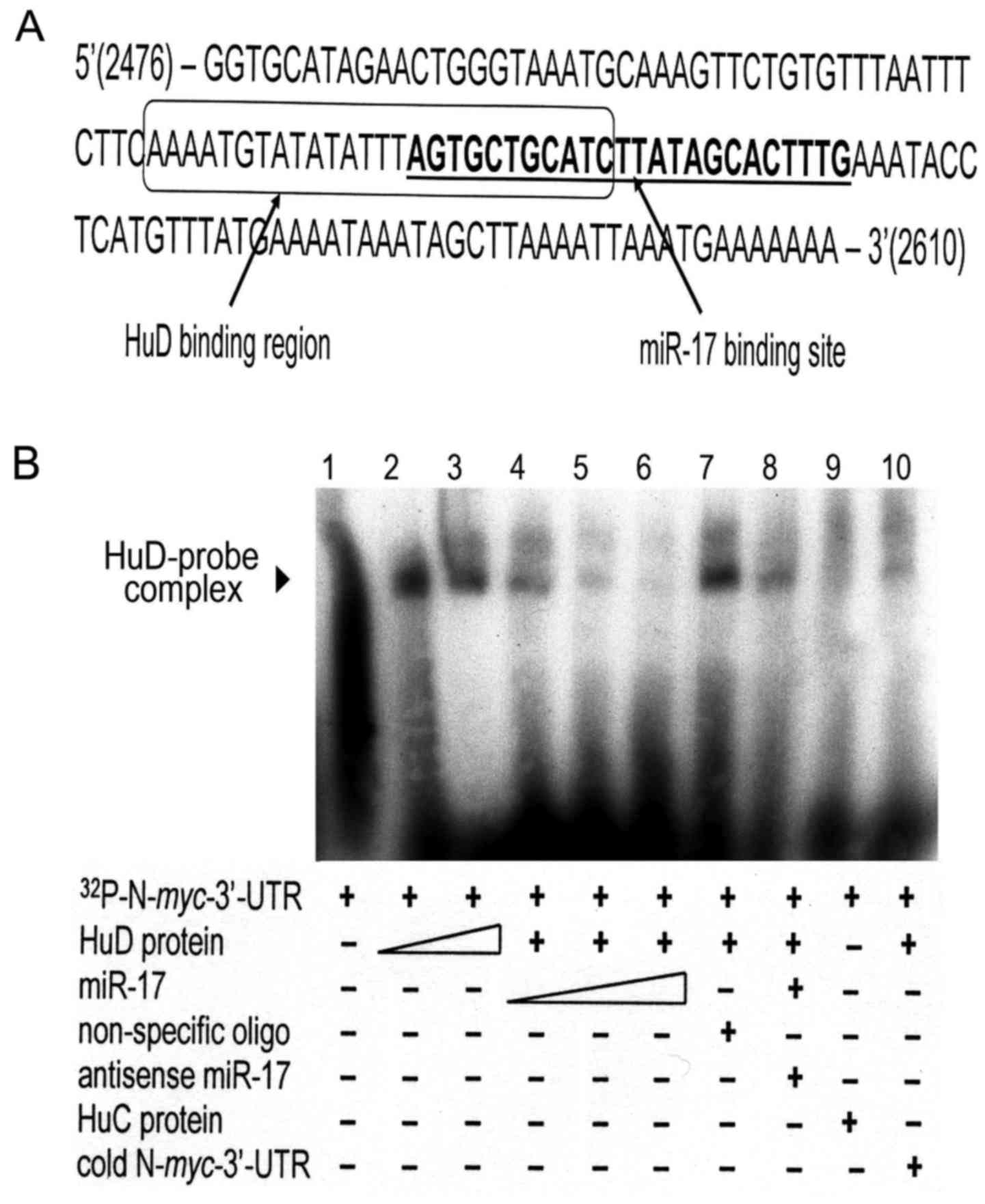
Regulation of N-myc by HuD and
miR-17
Previous studies in our laboratory and others have
shown that the neuronal-specific RNA-binding protein HuD (ELAVL4)
is involved in stabilizing the N-myc mRNA transcript by
binding to the AU-rich elements in its 3′-UTR (18–20).
Since miR-17 binds to the 3′-UTR, but destabilizes the mRNA, we
examined the location of the proposed binding sites of both
regulators. Notably, the binding sites overlap (Fig. 4A). To assess whether these 2
regulators compete for binding to the N-myc 3′-UTR, gel
shift assays were performed. As shown in Fig. 4B, binding of the recombinant HuD
protein to a radio-labeled 399-bp region from the N-myc
3′-UTR containing the 2 putative binding sites protects it from
degradation by endogenous bacterial RNases, resulting in an
RNA-protein complex that migrates more slowly through the gel
(Fig. 4B, lanes 2 and 3). Notably,
prior incubation of the N-myc probe with increasing amounts
of miR-17 decreased in a dose-dependent manner the amount of
HuD-N-myc complex (Fig. 4B,
lanes 4–6). Formation of the HuD-mRNA complex was not abrogated by
preincubation with non-specific RNA oligonucleotides (Fig. 4B, lane 7) or with miR-17
pre-annealed to its antisense sequence (Fig. 4B, lane 8) and was not noted after
substitution of HuC, a closely related Hu protein, for HuD
(Fig. 4B, lane 9) or pre-incubation
of the HuD lysate with cold N-myc 3′-UTR prior to the
addition of labeled N-myc 3′-UTR (Fig. 4B, lane 10). This experiment was
repeated with a second, shorter piece (124 bp) of the N-myc
3′-UTR with the same results (data not shown). Thus, the
neuronal-specific RNA-binding protein HuD and miR-17 appear to
compete for adjacent overlapping binding sites in the 3′-UTR of
N-myc, exerting antagonistic effects on N-myc mRNA
stability and subsequent protein expression.
Discussion
In the present study, we identified an interaction
between miR-17 and HuD, two molecules involved in the regulation of
N-myc expression. Several studies have identified miRNAs that
correlate with N-myc expression in neuroblastoma cell lines
and tumors (4–15,22).
Many of these studies revealed that the expression of the miR-17-92
cluster is: i) higher in N-myc-amplified cells (5,16,22);
and ii) regulated by N-myc (5,16), as
we also observed in our panel of cells. This cluster, also known as
oncomiR-1, has been shown to influence tumorigenicity in many forms
of cancer (17). Schulte et
al (22) noted an association
between high miR-17-92 levels and an unfavorable outcome in
children with neuroblastoma. Moreover, in the present study,
increased expression of miR-17-92 increased the tumorigenic
potential of an N-myc-non-amplified cell line. Therefore,
the aggressive behavior of neuroblastoma tumors with N-myc
amplification could be mediated, at least in part, by
downregulation of target tumor
suppressor/antiproliferative/apoptotic genes (17) consequent to increased levels of
miR-17-92.
The negative correlation of N-myc and miR-17
levels in N-myc-amplified neuroblastoma cell lines revealed
the existence of a negative feedback loop between N-myc and
miR-17. Notably, this relationship was not observed in
N-myc-non-amplified cell lines. Why is this negative
regulation observed only in N-myc-amplified cell lines? Our
previous studies have shown that HuD binds and stabilizes the
N-myc transcript (19).
N-myc-amplified cells have lower levels of HuD compared to
N-myc-non-amplified cells as a consequence of the loss of
one HuD allele with the 1p deletion invariably accompanying
N-myc amplification (20).
In non-amplified cells with 2 HuD alleles, HuD protein
amounts appear sufficient to block the common binding site and
ensure adequate levels of N-myc translation. By contrast,
lower levels of HuD may allow increased access to and promote
N-myc mRNA degradation by miR-17, leading to N-myc levels
too low to sustain viability. This effect may in turn select for
cells that have amplified N-myc, as proposed by Grandinetti
et al (20).
Overcompensation, leading to excess N-myc RNA and protein,
may further increase the amount of miR-17, leading subsequently to
both a greater imbalance between the 2 regulators and to greater
malignancy. Thus, our findings concerning the interactions between
miR-17, HuD and N-myc provided one explanation for the very
high levels of N-myc gene amplification found in
neuroblastoma.
Increased levels of the oncogenic miR-17-92 cluster,
and in particular miR-17, in N-myc-amplified tumors could be
a major mediator of their aggressive nature. In addition, the
negative feedback regulatory mechanism between N-myc and
miR-17 could be an important regulator of cell fate (proliferation
vs. apoptosis) in normal as well as in malignant neuroblasts. A
similar negative feedback regulatory mechanism has been observed
between c-myc and miR-17-92, in which upregulation of the
miR-17-92 cluster results in downregulation of E2F (23). Greater knowledge of the complex
interactions between miR-17-92, HuD and N-myc could lead to
better understanding of the regulation of N-myc expression and to
the discovery of additional drugs or therapeutic approaches for the
more effective treatment of N-myc-amplified cancers.
Acknowledgements
The present study was supported in part by a grant
(CA 77593) from the National Institutes of Health (Bethesda, MD,
USA).
References
|
1
|
Park JR, Bagatell R, London WB, Maris JM,
Cohn SL, Mattay KK and Hogarty M: COG Neuroblastoma Committee:
Children's Oncology Group's 2013 blueprint for research:
Neuroblastoma. Pediatr Blood Cancer. 60:985–993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bell E, Chen L, Liu T, Marshall GM, Lunec
J and Tweddle DA: MYCN oncoprotein targets and their therapeutic
potential. Cancer Lett. 293:144–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schulte JH, Schowe B, Mestdagh P, Kaderali
L, Kalaghatgi P, Schlierf S, Vermeulen J, Brockmeyer B, Pajtler K,
Thor T, et al: Accurate prediction of neuroblastoma outcome based
on miRNA expression profiles. Int J Cancer. 127:2374–2385. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fontana L, Fiori ME, Albini S, Cifaldi L,
Giovinazzi S, Forloni M, Boldrini R, Donfrancesco A, Federici V,
Giacomini P, et al: Antagomir-17-5p abolishes the growth of
therapy-resistant neuroblastoma through p21 and BIM. PLoS One.
3:e22362008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu H, Du L, Nagabayashi G, Seeger RC and
Gatti RA: ATM is down-regulated by N-Myc-regulated microRNA-421.
Proc Natl Acad Sci USA. 107:1506–1511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Buckley PG, Alcock L, Bryan K, Bray I,
Schulte JH, Schramm A, Eggert A, Mestdagh P, De Preter K,
Vandesompele J, et al: Chromosomal and microRNA expression patterns
reveal biologically distinct subgroups of 11q- neuroblastoma. Clin
Cancer Res. 16:2971–2978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen Y and Stallings RL: Differential
patterns of microRNA expression in neuroblastoma are correlated
with prognosis, differentiation, and apoptosis. Cancer Res.
67:976–983. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shohet JM, Ghosh R, Coarfa C, Ludwig A,
Benham AL, Chen Z, Patterson DM, Barbieri E, Mestdagh P, Sikorski
DN, et al: A genome-wide search for promoters that respond to
increased MYCN reveals both new oncogenic and tumor suppressor
microRNAs associated with aggressive neuroblastoma. Cancer Res.
71:3841–3851. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma L, Young J, Prabhala H, Pan E, Mestdagh
P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S,
et al: miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin
and cancer metastasis. Nat Cell Biol. 12:247–256. 2010.PubMed/NCBI
|
|
11
|
Wei JS, Song YK, Durinck S, Chen QR, Cheuk
AT, Tsang P, Zhang Q, Thiele CJ, Slack A, Shohet J, et al: The MYCN
oncogene is a direct target of miR-34a. Oncogene. 27:5204–5213.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lovén J, Zinin N, Wahlström T, Müller I,
Brodin P, Fredlund E, Ribacke U, Pivarcsi A, Påhlman S and
Henriksson M: MYCN-regulated microRNAs repress estrogen receptor-α
(ESR1) expression and neuronal differentiation in human
neuroblastoma. Proc Natl Acad Sci USA. 107:1553–1558. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haug BH, Henriksen JR, Buechner J, Geerts
D, Tømte E, Kogner P, Martinsson T, Flægstad T, Sveinbjørnsson B
and Einvik C: MYCN-regulated miRNA-92 inhibits secretion of the
tumor suppressor DICKKOPF-3DKK3) in neuroblastoma. Carcinogenesis.
32:1005–1012. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Buechner J, Tømte E, Haug BH, Henriksen
JR, Løkke C, Flægstad T and Einvik C: Tumour-suppressor microRNAs
let-7mir-101 target the proto-oncogene MYCN and inhibit cell
proliferation in MYCN-amplified neuroblastoma. Br J Cancer.
105:296–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bray I, Bryan K, Prenter S, Buckley PG,
Foley NH, Murphy DM, Alcock L, Mestdagh P, Vandesompele J, Speleman
F, et al: Widespread dysregulation of MiRNAs by MYCN amplification
and chromosomal imbalances in neuroblastoma: Association of miRNA
expression with survival. PLoS One. 4:e78502009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schulte JH, Horn S, Otto T, Samans B,
Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L,
Buettner R, et al: MYCN regulates oncogenic MicroRNAs in
neuroblastoma. Int J Cancer. 122:699–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Olive V, Jiang I and He L: mir-17-92, a
cluster of miRNAs in the midst of the cancer network. Int J Biochem
Cell Biol. 42:1348–1354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chagnovich D, Fayos BE and Cohn SL:
Differential activity of ELAV-like RNA-binding proteins in human
neuroblastoma. J Biol Chem. 271:33587–33591. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lazarova DL, Spengler BA, Biedler JL and
Ross RA: HuD, a neuronal-specific RNA-binding protein, is a
putative regulator of N-myc pre-mRNA processing/stability in
malignant human neuroblasts. Oncogene. 18:2703–2710. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grandinetti KB, Spengler BA, Biedler JL
and Ross RA: Loss of one HuD allele on chromosome #1p selects for
amplification of the N-myc proto-oncogene in human neuroblastoma
cells. Oncogene. 25:706–712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walton JD, Kattan DR, Thomas SK, Spengler
BA, Guo HF, Biedler JL, Cheung NK and Ross RA: Characteristics of
stem cells from human neuroblastoma cell lines and in tumors.
Neoplasia. 6:838–845. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schulte JH, Marschall T, Martin M,
Rosenstiel P, Mestdagh P, Schlierf S, Thor T, Vandesompele J,
Eggert A, Schreiber S, et al: Deep sequencing reveals differential
expression of microRNAs in favorable versus unfavorable
neuroblastoma. Nucleic Acids Res. 38:5919–5928. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005. View Article : Google Scholar : PubMed/NCBI
|