Introduction
Ovarian cancer is the leading cause of death among
all gynecologic malignancies, and the mortality rate of ovarian
cancer continues to increase while the incidence rate remains high
in recent decades according to recently published cancer statistics
in China, 2015 (1). Therefore,
efficient targets for early detection and treatment of ovarian
cancers are urgently needed. Normal ovarian epithelial cells have a
limited ability to proliferate and migrate for wound healing after
ovulation or rupture of the mature follicle. However,
epithelium-originated ovarian cancer cells are able to spread
through the abdominal cavity forming multiple implants on the
peritoneal surface. While early-stage epithelial ovarian cancers
can be cured when they are still confined to the ovary upon
diagnosis, a majority of epithelial ovarian cancers are diagnosed
at the advanced stage after peritoneal dissemination has occurred,
which is often too late for efficient treatment (2). In this context, it is critical to
understand the molecular mechanisms driving epithelial ovarian
cancer progression, especially the development of metastasis, for
the identification of valuable drug targets and development of
effective therapeutic strategies.
Previous studies have shed light on the multiple
signaling pathways involved in epithelial ovarian cancer, including
the PI3K/AKT pathway that commonly participates in the
proliferation and survival of tumor cells (3–5). In
addition to PI3K, other players in the phosphoinositide signaling
pathway are also implicated in regulating cancer cells (6,7). For
example, type Iγ phosphatidylinositol phosphate kinase (PIPKIγ)
generates phosphatidylinositol 4,5-biphosphate [PtdIns(4,5)P2] as the substrate of PI3K
to activate AKT and downstream signaling cascades, which then
promote proliferation and survival (8). Moreover, PtdIns(4,5)P2 is an important secondary
messenger that regulates various cellular events including protein
trafficking, actin reorganization, cell adhesion and migration
(9). We recently observed that
PIPKIγ is engaged in the metastasis of breast cancer by regulating
the proliferation, migration and invasion of breast cancer cells
(6,10,11).
In this context, we proposed that PIPKIγ may also contribute to
epithelial ovarian cancer metastasis as both cancers share similar
pathways. Interestingly, we found that PIPKIγ was highly expressed
in the epithelial ovarian cancer cells. This lipid kinase was
necessary for the activation of the PI3K/AKT pathway and regulated
the migration and invasion of these cells. Furthermore, loss of
PIPKIγ impaired signal transducer and activator of transcription 3
(STAT3) activation that is closely associated with the poor
prognosis of ovarian carcinomas (12). Our data strongly suggest that PIPKIγ
may have profound influence on facilitating the progression of
epithelial ovarian cancer. These results endorse the potential of
this lipid kinase as a novel therapeutic target for epithelial
ovarian cancer treatment and call for further investigation.
Materials and methods
Cell culture
All five human epithelial ovarian cancer cell lines
(OVCAR-7, OVCAR-8, PEO-1, PEO-4 and SKOV-3) were kindly provided by
Dr William A. Cliby (Mayo Clinic, Rochester, MN, USA), and the
immortalized OSE (OSE hTERT) cells were obtained from Dr
Vijayalakshmi Shridhar (Mayo Clinic). All cancer cell lines were
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin
(Invitrogen, Carlsbad, CA, USA), while OSE hTERT cells were
maintained in NOE complete media consisting of 50% v/v Medium 199
and 50% v/v MCDB (Sigma-Aldrich, St. Louis, MO, USA) supplemented
with 15% FBS, 1% penicillin/streptomycin and 0.1% epithelial growth
factor (EGF; Sigma-Aldrich). Cell cultures were maintained at 37°C
with 5% CO2.
Antibodies
The rabbit polyclonal PIPKIγ antibody was generated
and purified as described previously (6). Antibodies against phosphorylated AKT
(pSer473), total AKT, phosphorylated ERK1/2 (pThr202/Tyr204), total
ERK1/2, phosphorylated STAT3 (pTyr705), total STAT3, phosphorylated
JAK2 (pTyr1007/1008) and total JAK2 were procured from Cell
Signaling Technology (Danvers, MA, USA). Antibody for β-actin was
purchased from Sigma-Aldrich.
Constructs and transfection
Two distinct siRNA sequences specifically targeting
human PIPKIγ were: PIPKIγ-siRNA1 (ATCCGCGTCGTGGTCATGAACAACA) and
PIPKIγ- siRNA2 (GCGTGGTCAAGATGCACCTCAAGTT). PIPKIγ siRNAs and
control siRNA were synthesized at Invitrogen (Stealth RNAi). Mouse
PIPKIγ constructs encoding isoform 1 and isoform 2 that are
resistant to human PIPKIγ siRNAs were constructed as described
previously (6).
For transient transfection of siRNAs, OVCAR-8 and
SKOV-3 cells were plated in 6-well culture plates at
4×105 cells/well, and then reversely transfected using
Lipofectamine RNAiMAX (Invitrogen) for 48 h before further
analyses. For the rescue experiments, SKOV-3 cells were transiently
transfected with DNA plasmids using X-tremeGENE 9 (Roche) for 24 h,
and then lifted and reversely transfected with siRNAs as described
above.
Immunoblotting
The transfected OVCAR-8 and SKOV-3 cells were
collected in 200 µl of 1X SDS lysis buffer (40 mM Tris-HCl, 1 mM
EDTA, 150 mM KCl, 100 mM NaVO3, 1% Triton X-100, 1 mM
PMSF, pH 7.5) on ice. Proteins in the lysates were separated by
electroporation using 10% SDS-PAGE gels and then transferred onto
PVDF membranes. After being blocked with 5% non-fat milk in TBS-T
at room temperature for 1 h, membranes were incubated with primary
antibodies overnight in blocking buffer at 4°C, followed by
HRP-conjugated secondary antibody for 1 h at room temperature. Then
membranes were incubated with Supersignal Chemiluminescent
Substrate (Thermo Fischer Scientific, Waltham, MA, USA) and imaged
using ChemiDOC imaging system (Bio-Rad, Hercules, CA, USA).
Cell viability assay
OVCAR-8 and SKOV-3 cells transfected with
PIPKIγ-siRNA1, PIPKIγ-siRNA2 or control siRNA for 48 h were plated
(1×105 cells/well) as triplicates in 96-well plates and
cultured for 48 h at 37°C in tissue culture incubator. Then 10 µl
of 12 mM 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) was added to each well and incubated for another 4 h.
After replacing the culture medium with Stop Solution (40 mM HCL in
isopropanol; 100 µl/well), the absorbance was measured at 590 nm on
a plate reader.
Flow cytometry
For cell cycle analysis, the cells transfected with
PIPKIγ-siRNA1, PIPKIγ-siRNA2, or control siRNA for 48 h were washed
twice with phosphate-buffered saline (PBS) and then fixed with
prechilled 70% ethanol at 4°C overnight. Fixed cells were washed
twice with PBS and then stained in 500 µl propidium iodide solution
containing 50 µg/ml propidium iodide (eBioscience, USA), 0.1 mg/ml
RNase A and 0.05% Triton X-100 at 37°C for 1 h. The DNA content was
determined by a flow cytometer (BD Biosciences, San Jose, CA, USA).
Data were then analyzed using ModFit software (Verity Software,
Topsham, ME, USA). For apoptosis analyses, cells were stained with
7-AAD (BioLegend, USA) at 4°C for 15 min or fixed with fixation
buffer (BioLegend) followed by caspase-3 staining (BD Biosciences).
The cells were then anayzed by flow cytometer (BD Biosciences).
Cell migration assay
The migration assay was performed using modified
Boyden chambers (Neuroprobe, Gaithersburg, MD, USA) according to
the manufacturers instructions (6).
The polycarbonate membranes (8-µm pore size; Neuroprobe) were
precoated with 10 µg/ml type I collagen for 3 h at 37°C and placed
on the top of the lower chamber filled with DMEM containing 10%
FBS. Serum-starved cells (3×105) in serum-free medium
were added to the upper chamber and incubated at 37°C in a tissue
culture incubator for 8 h. After the removal of non-migrated cells
on top of the membrane with a cotton swab, the membranes were fixed
with methanol and stained with 0.2% crystal violet. The number of
cells that migrated to the lower side of the membrane were counted
under a ×20 magnification lens using a Nikon microscope (TE-2000)
and averaged from at least 5 randomly selected fields. We set
duplicates for each sample.
Cell invasion assay
Cell invasion assay was carried out in
Matrigel-coated Transwells (BD Biosciences, USA) as previously
described (11). Transwells were
incubated with DMEM for 4 h and the lower compartment was filled
with DMEM containing 10% FBS. OVCAR-8 or SKOV-3 cells
(4×104) were plated in the upper compartment and
incubated at 37°C in a tissue culture incubator for 24 h. The cells
that invaded the Matrigel and reached the lower surface of the
membrane were fixed with 4% polyformaldehyde and stained with 0.2%
crystal violet. The number of invaded cells was counted under
microscope as described above.
Quantitative RT-PCR
OVCAR-8 and SKOV-3 cells transfected with
PIPKIγ-siRNA1, PIPKIγ-siRNA2or control siRNAfor 48 h were collected
and washed in PBS, and then used to prepare total RNA using RNA kit
(Invitrogen, Life Technologies). RNAs (2 mg) were reversely
transcripted into cDNAs using M-MLV (Invitrogen, Life
Technologies), followed by quantitative PCR using the following
primers (BGI, USA). Matrix metalloproteinase (MMP)-2 forward,
5′-GTTCATTTGGCGGACTGT-3′ and reverse, 5′-AGGGTGCTGGCTGAGTAG-3′;
MMP-9 forward, 5′-AATCTCACCGACAGGCAGCT-3′ and reverse,
5′-CCAAACTGGATGACGATGTC-3′; and GAPDH forward,
5′-GAAGGTGAAGGTCGGAGT-3′ and reverse, 5′-CATGGGTGGAATCATATTGGAA-3′.
PCR program consisted of 50°C for 2 min, 95°C for 5 min, followed
by 49 cycles at 95°C for 20 sec, 60°C for 20 sec, 72°C for 25 sec.
The samples were then heated at 95°C for 10 sec and 65°C for 30 sec
followed by gradual heating to 95°C for 15 sec. The results were
analyzed by CFX Manager software (13).
Statistical analysis
All experiments were repeated at least three times.
Results were analyzed with Prism 6 and are presented as mean ±
standard deviation (SD). The significance of group differences was
identified as P<0.01 or P<0.05.
Results
PIPKIγ is upregulated in ovarian
cancer cells
Abnormally altered expression of a protein in cancer
cells often indicates a correlation between this protein and the
development and/or progression of cancer (6). Thus, we firstly examined the
expression of PIPKIγ in epithelial ovarian cancer cells. According
to the Human Protein Atlas, both RNA and protein levels of PIPKIγ
are relatively low in normal ovarian tissues, however higher PIPKIγ
expression is detected in some epithelial ovarian carcinomas. To
verify this, we examine PIPKIγ expression in five most commonly
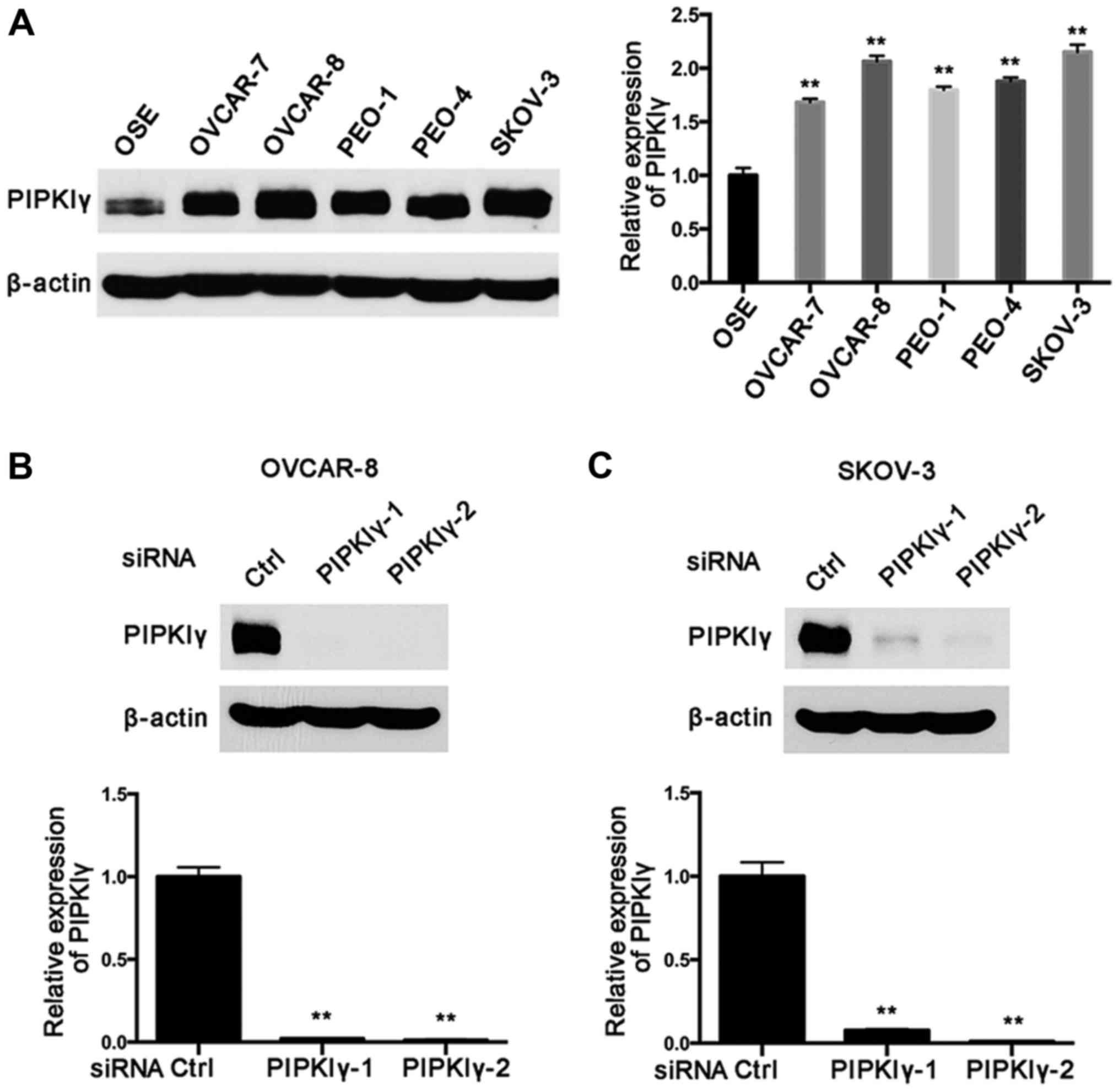
used human epithelial ovarian cancer cell lines, including OVCAR-7,
OVCAR-8, PEO-1, PEO-4 and SKOV-3. As shown in Fig. 1A, PIPKIγ was found to be highly
expressed in all the tested cell lines compared to that in the
normal epithelial cell line OSE-hTERT. Notably, OVCAR-8 and SKOV-3
cells exhibited substantially elevated PIPKIγ expression compared
to that in the normal epithelial cells (Fig. 1A). These data suggest that
upregulation of PIPKIγ may correlate with the development and/or
progression of epithelial ovarian cancers.
Silencing of PIPKIγ impairs the
viability of human epithelial ovarian cancer cells and promotes
apoptosis
To investigate the role of PIPKIγ in epithelial
ovarian cancer cells, we utilized two distinct PIPKIγ-specific
siRNAs (PIPKIγ-1 and PIPKIγ-2) to knock down endogenous PIPKIγ in
OVCAR-8 and SKOV-3 cells, as these two lines showed the highest
expression of PIPKIγ and share similar characteristics. After
confirming the knockdown efficiency of these siRNAs (Fig. 1B and C), we examined the aggressive
behaviors of PIPKIγ-depleted cells in comparison with the control
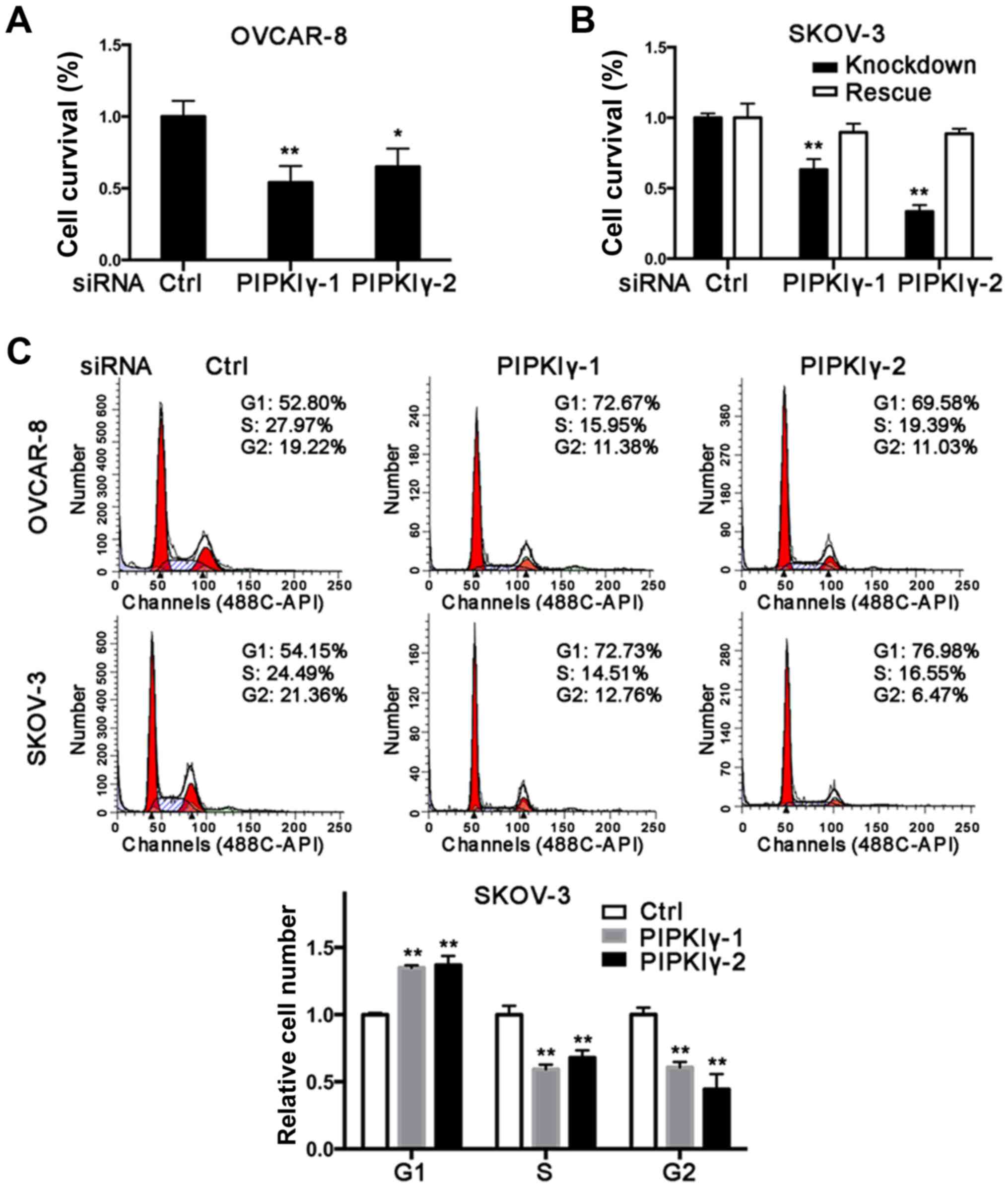
cells treated with the scrambled siRNA. As shown in Fig. 2A and B, reduction of PIPKIγ in the
OVCAR-8 and SKOV-3 cells led to a significant decrease in cell
survival as determined by MTT assay. Importantly, the decreased
viability was rescued by introducing the expression of
RNAi-resistant PIPKIγ back in PIPKIγ-depleted SKOV-3 cells
(Fig. 2B), further demonstrating
that PIPKIγ is indeed required for the viability of ovarian cancer
cells. To delineate how PIPKIγ affects cell viability, we
determined both cell cycle progression and apoptosis in the control
and PIPKIγ-deficient cells. As shown in Fig. 2C, the cells treated with PIPKIγ RNAi
exhibited a significant increase in G1 phase and less cells were
observed in the S phase, indicating a delayed G1-to-S transition in
both OVCAR-8 and SKOV-3 cells (Fig.
2C). Furthermore, we found that loss of PIPKIγ also induced
higher apoptosis when we examined 7-AAD (Fig. 2D) and caspase-3 (Fig. 2E). These results indicate that
PIPKIγ likely regulates multiple pathways and contributes to the
growth of ovarian cancer cells by both promoting cell proliferation
and inhibiting apoptosis.
Cell migration and invasion are
suppressed in PIPKIγ-deficient human epithelial ovarian cancer
cells
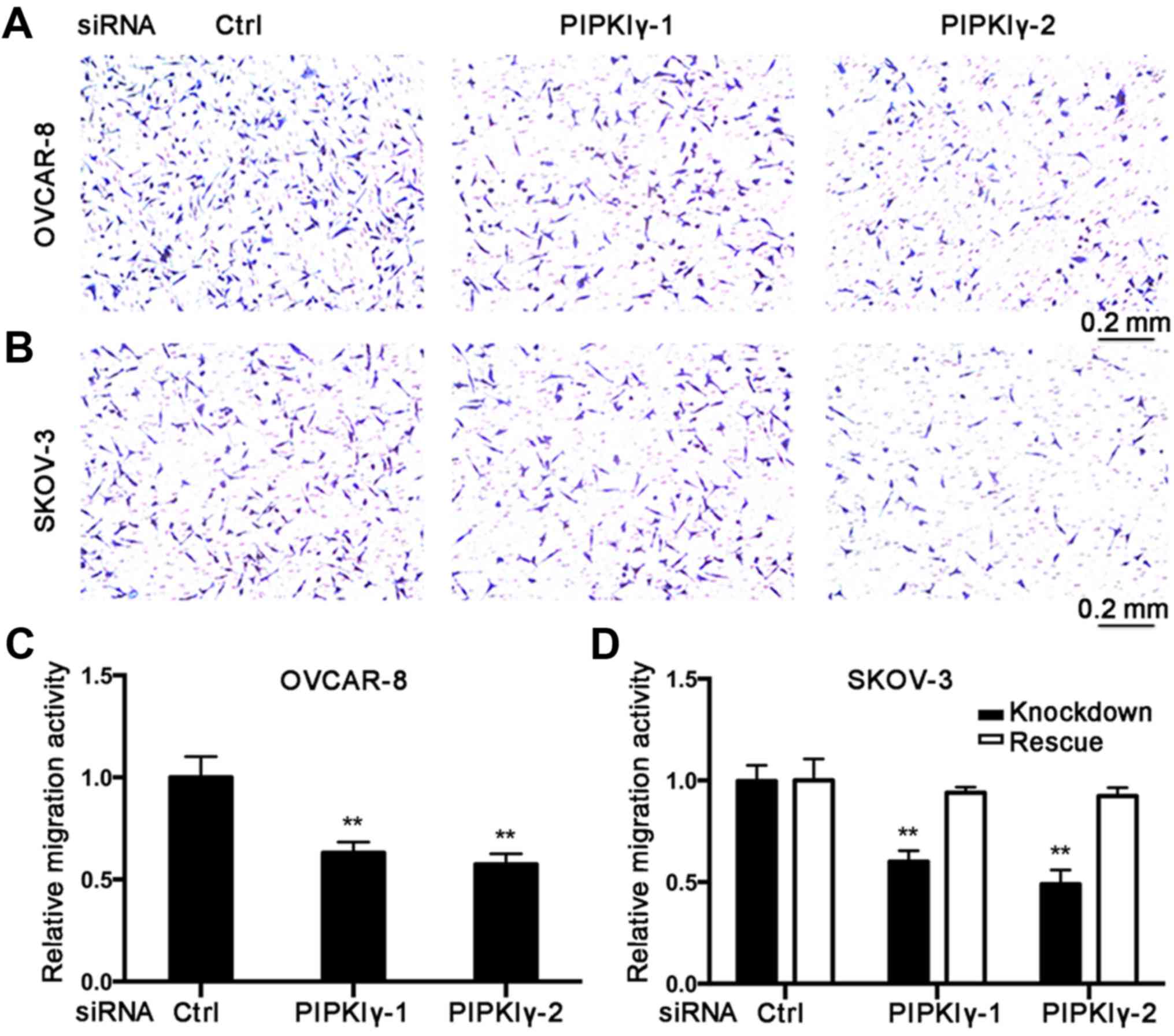
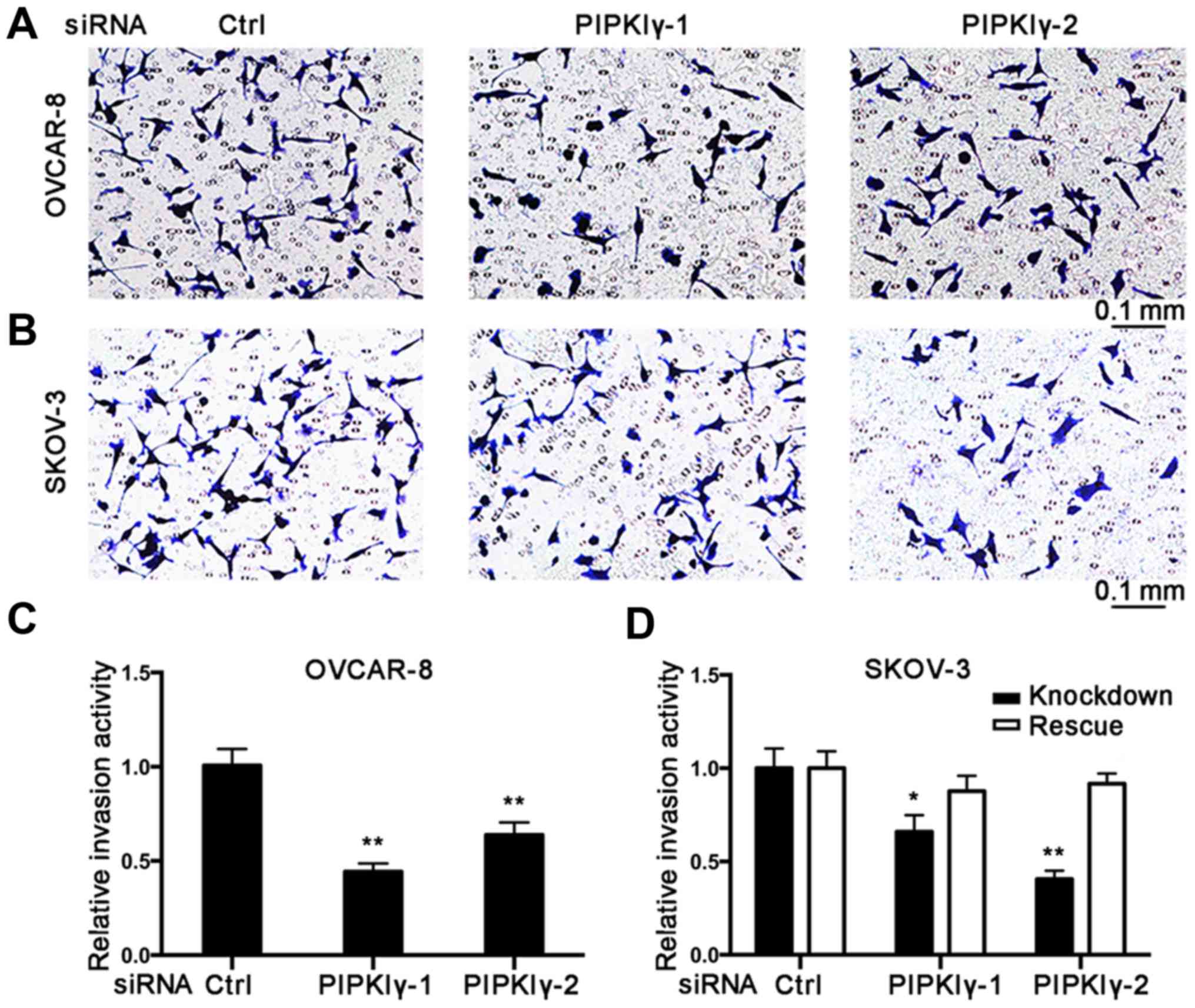
To further elucidate the role of PIPKIγ in
regulating the malignant behaviors of epithelial ovarian cancer
cells, we conducted in vitro cell migration and invasion
assays. Using the Boyden chamber system, we found that the
PIPKIγ-depleted cells migrated significantly slower responding to
serum when compared to the control cells (Fig. 3). Results from the Transwell
invasion assay showed that knockdown of PIPKIγ led to a
substantially impaired invasive ability (Fig. 4). Furthermore, both migration and
invasion capacities were almost completely rescued when the
expression of PIPKIγ was recovered in the SKOV-3 cells (Figs. 3 and 4). Taken together, these results
demonstrate that PIPKIγ indeed is required for the malignant
behavior of epithelial ovarian tumor cells, indicating that
inhibition of PIPKIγ may suppress the development of metastasis in
epithelial ovarian cancer.
PIPKIγ is required for the activation
of the PI3K/AKT pathway in human epithelial ovarian cancer
cells
Since our results indicated that PIPKIγ regulates
the proliferation and migration of epithelial ovarian cancer cells,
we then tested whether this is through PI3K/AKT and/or MAPK/ERK
pathways that often participate in ovarian carcinogenesis (14,15).
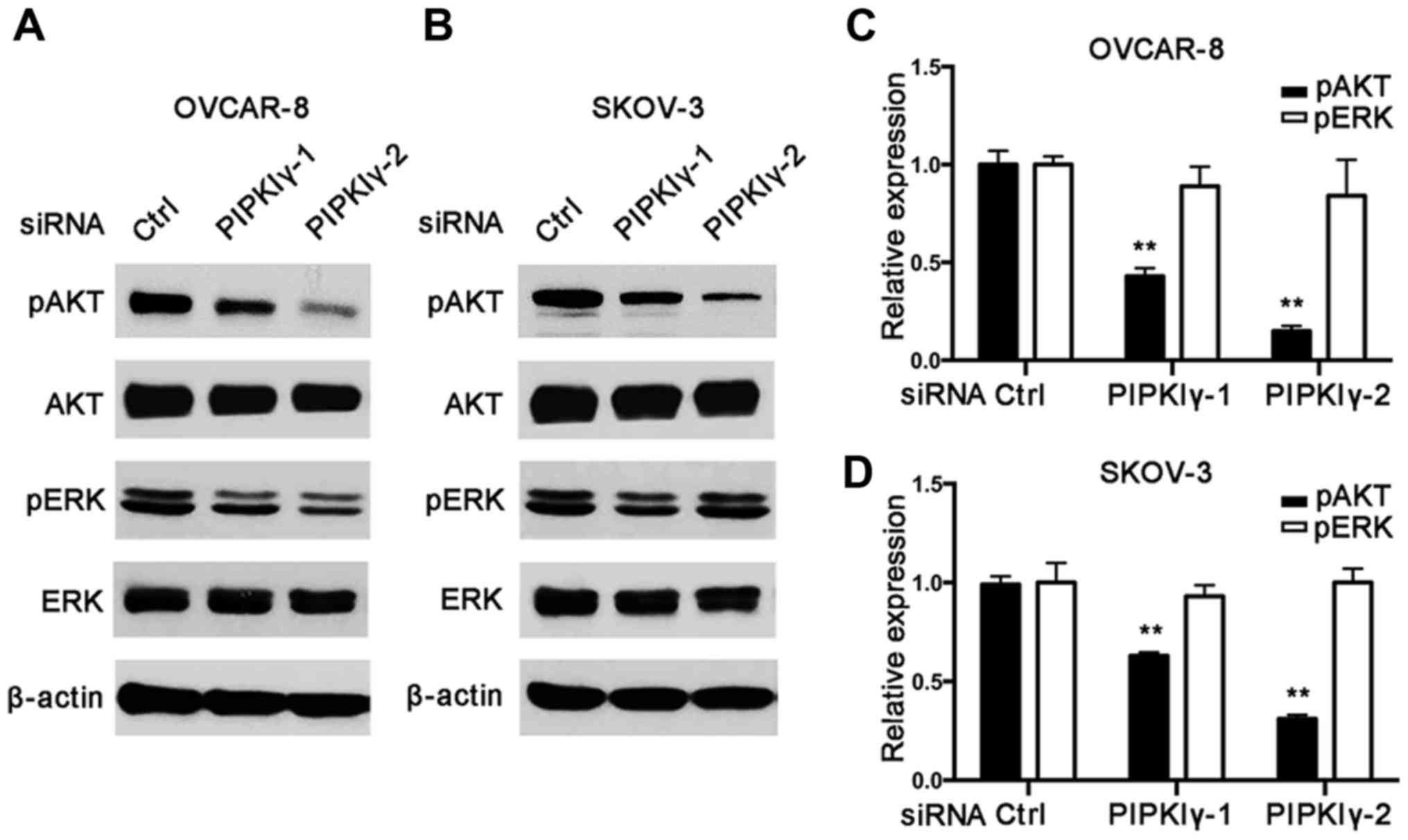
As shown in Fig. 5, PIPKIγ-depleted
cells exhibited much less activated AKT than the control cells;
however, activation of the MAPK pathway appeared similar in the
control and PIPKIγ-depleted cells. These results indicate that
PIPKIγ is necessary for the activation of the PI3K/AKT pathway but
not the MAPK pathway, although MAPK is known to be closely related
to migration in epithelial ovarian cancers (14,15).
Our data suggest that inhibition of PIPKIγ blocks ovarian tumor
cell proliferation and migration by downregulating the PI3K/AKT
pathway, which may subsequently interrupt the metastasis of
epithelial ovarian cancer.
Expression of MMP-2 and MMP-9 is
regulated by PIPKIγ
MMPs are known as a family of proteolytic enzymes
that remodel the extracellular matrix to promote tumor metastasis.
Among all 23 members in the MMP family, MMP-2, MMP-7 and MMP-9 are
most closely associated with ovarian cancer tumor metastasis
(16), and MMP-2 and MMP-9 are
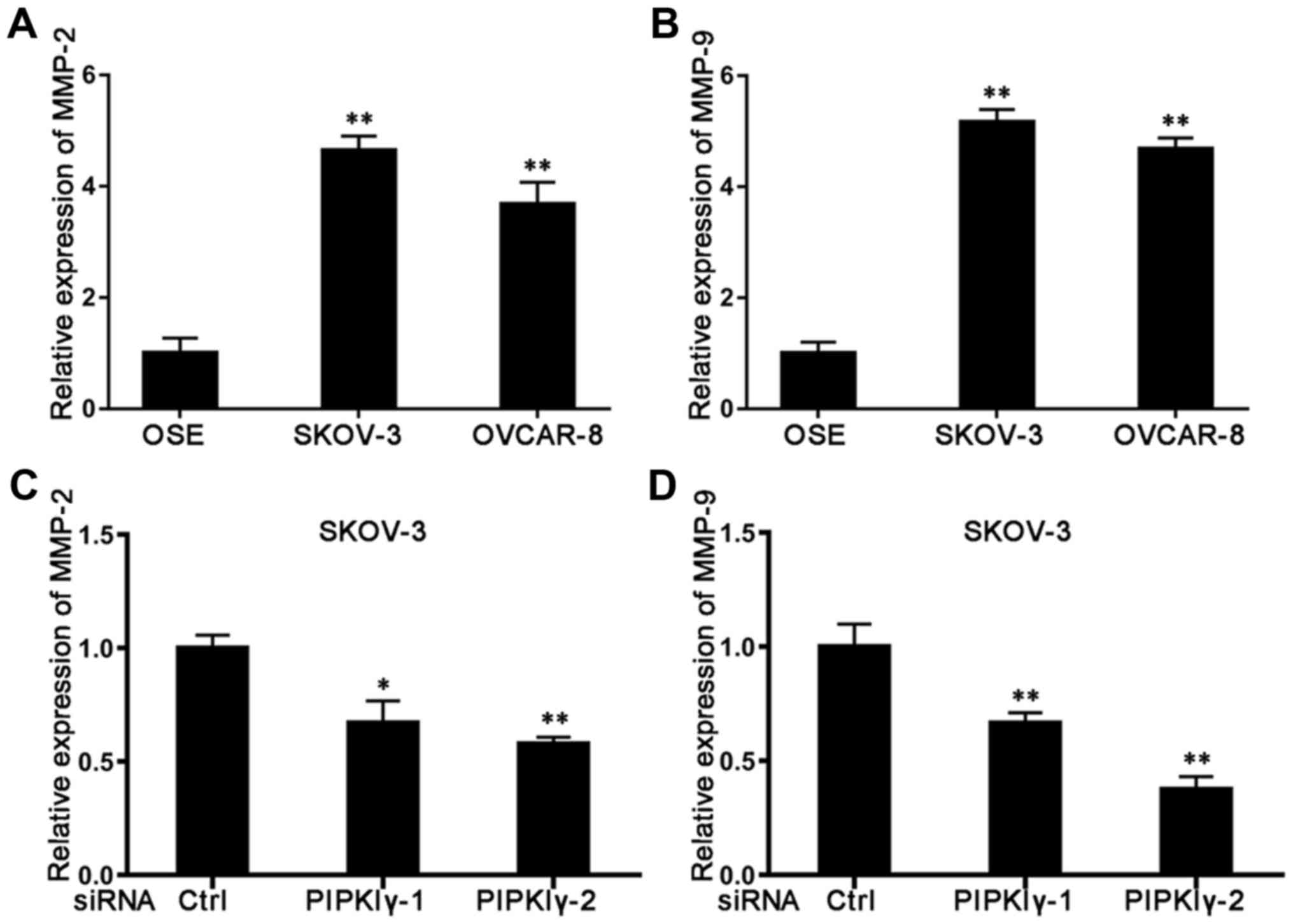
highly expressed in ovarian cancer ascites and tissues (17). When we utilized qRT-PCR to measure
the expression of these MMPs, we found that both MMP-2 and MMP-9
were expressed at significantly higher levels in the epithelial
ovarian cancer cells than levels noted in the normal OSE cells
(Fig. 6A and B). Compared to normal
SKOV-3 cells, depletion of PIPKIγ led to a reduction in both MMP-2
(Fig. 6C) and MMP-9 (Fig. 6D). However, levels of these MMPs
remained the same in the control and PIPKIγ-depleted OVCAR-8 cells
(data not shown), suggesting that PIPKIγ mediates a cell
type-specific regulation in the expression of MMP-2 and MMP-9.
Considering the role of MMP-2 and MMP-9 in metastasis formation,
our results suggest that PIPKIγ may promote cell invasion by
regulating the expression of these MMPs, and therefore may
potentially facilitate metastasis in certain epithelial ovarian
cancers.
PIPKIγ activates the STAT3 pathway in
ovarian cancer cells
STAT3 is activated by phosphorylation of a tyrosine
residue by activated EGFR, JAK or Src (18). Constitutively phosphorylated and
activated in 70% of ovarian cancers, aberrant STAT3 signaling is
significantly correlated with the development of ovarian cancer
(18,19). Currently, targeting the STAT3
pathway has become a major focus of drug development, and numerous
STAT3 inhibitors have been shown to be effective in suppressing
tumor cell migration (18–20). Importantly, it has been recently
reported that elevated STAT3 expression in ovarian cancer ascites
promotes tumor invasion and metastasis (21). In this context, we explored whether
PIPKIγ, which is required for migration and invasion of epithelial
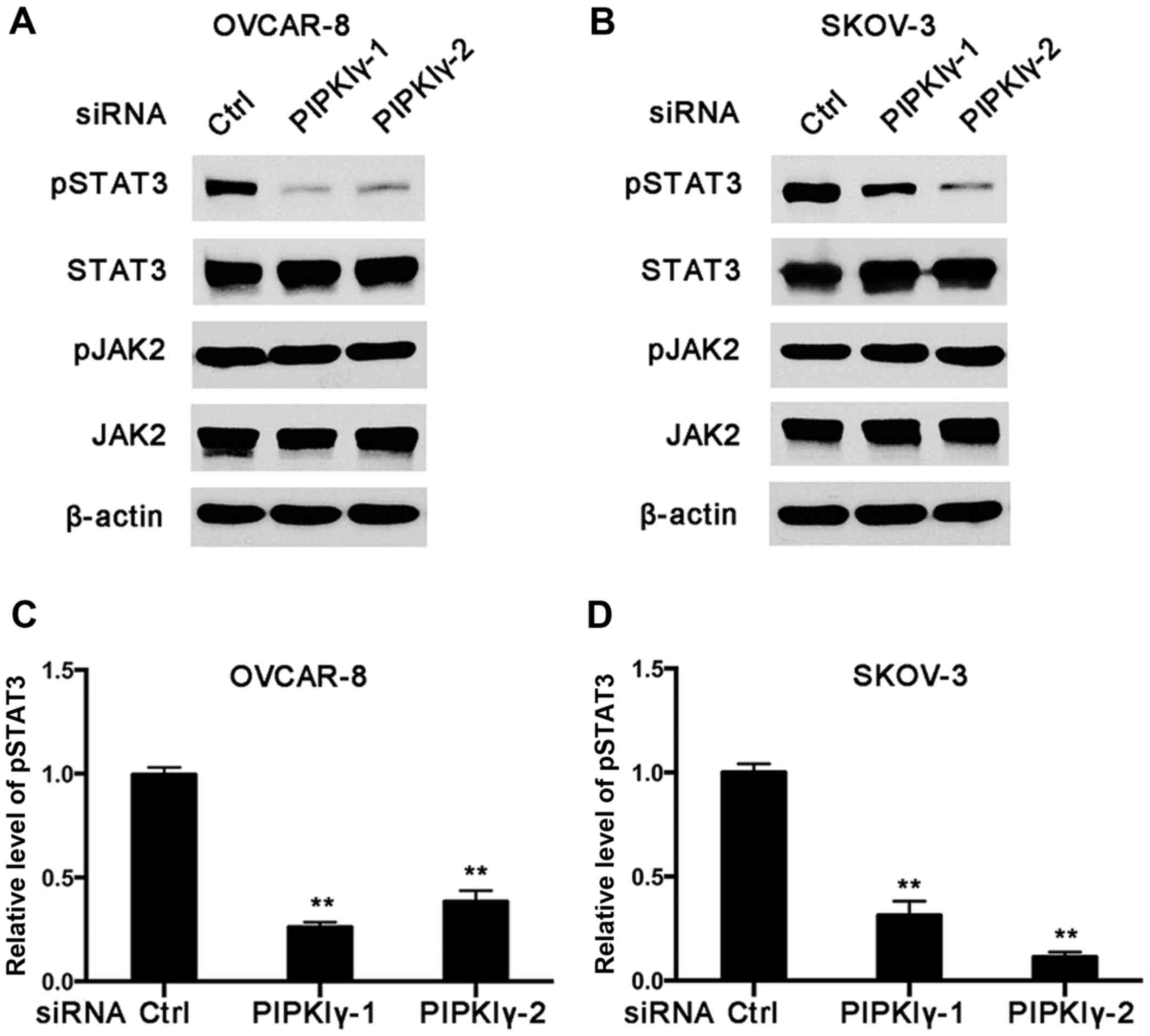
ovarian cancer cells, regulates the activation of STAT3. As shown
in Fig. 7, the level of
Tyr705-phosphorylated STAT3 was notably decreased in the
PIPKIγ-depleted cells compared to that noted in the control cells
in both the OVCAR-8 and SKOV-3 cell lines. However, the levels of
phosphorylated JAK2 and total JAK2 appeared comparable between the
control and PIPKIγ-depleted cells (Fig.
7A and B). These results indicate that PIPKIγ is required for
the basal activity of STAT3 in non-stimulated ovarian cancer cells,
which is independent of JAK2. Considering the important role of the
STAT3 pathway in the progression of ovarian cancer, our findings
reveal PIPKIγ as a novel JAK2-independent regulator of STAT3, which
may be an important mechanism underlying PIPKIγ-dependent survival
and migration/invasion of ovarian cancer cells.
Discussion
In recent years, lipid kinases have been intensively
explored in the tumor metastasis of ovarian cancer. Among them, the
PI3K pathway, which is deregulated in epithelial ovarian cancers,
has been extensively studied (4,22,23).
Recent genomic analyses have revealed that components of the PI3K
pathway are often mutated or altered in many human cancers
(24–30), which supports PI3K as one of the
most prospecting targets for therapeutic intervention in cancers
(31). Correspondingly, a number of
PI3K inhibitors have shown antitumor activities when applied alone
or combined with chemotherapies (24,25).
However, only a small portion of patients benefit from each single
PI3K inhibitor, as distinct PI3K isoforms play different roles in
cellular signaling and oncogenic transformation (32–36).
In addition, side effects resulting from inhibition of other
pathways such as RAF/MAPK (37) and
the development of acquired resistance (38) further increase the complexity of the
clinical application of PI3K inhibitors. These limitations call for
new drug targets and novel therapeutic strategies, such as the
combination of multiple targeted therapies. As reported previously,
PIPKIγ that functions upstream of PI3K targets focal adhesion and
regulates growth factor-induced cell migration and invasion of
breast cancer cells (39,40). Here we report for the first time
that PIPKIγ is also required for ovarian cancer cells to
proliferate, survive, migrate and invade in vitro. Our
results suggest that activation of the PI3K/AKT and STAT3 pathways
both require PIPKIγ in epithelial ovarian cancer cells, indicating
a molecular connection between PIPKIγ and conventional survival and
metastasis pathways.
Among all of the four subtypes of ovarian cancers,
the majority of tumors are derived from the ovarian surface
epithelium. Utilizing several epithelial ovarian cancer cell lines,
we found that epithelial ovarian cancer cells displayed elevated
expression of PIPKIγ, indicating a correlation between PIPKIγ and
malignancy of these cells. Indeed, proliferation, migration and
invasion of these cells were significantly suppressed when PIPKIγ
was depleted, accompanied by enhanced apoptosis. Since the PI3K/AKT
pathway has been implicated in the survival and metastasis of
epithelial ovarian cancers (41),
the effects following the knockdown of PIPKIγ likely resulted from
the inhibition of AKT activation, for AKT activity was repressed by
depleting PIPKIγ in these cells.
Moreover, our results suggest that other signaling
cascades important for ovarian cancer progression could be
regulated by PIPKIγ, such as STAT3. In addition to a transcription
factor, phosphorylated STAT3 can also localize to focal adhesions
and promote ovarian cancer cell motility (12). Importantly, a recent study revealed
that STAT3 expression is elevated in ovarian cancer ascites and
promotes the progression/metastasis of ovarian cancer in
vivo (21). This study
demonstrated that phosphorylation of Tyr705 in STAT3, which
indicates the constitutive activation of STAT3, is directly
correlated with the extent and severity of ovarian cancer. In this
context, our finding that deficiency of PIPKIγ severely impaired
Tyr705 phosphorylation in STAT3 in both OVCAR-8 and SKOV-3 cells
reveals STAT3 as a novel regulator in ovarian cancer cells.
Notably, this PIPKIγ-dependent STAT3 activation was independent of
JAK1/2. Therefore, we reason that the PIPKIγ-dependent Tyr705
phosphorylation of STAT3 is likely mediated by EGFR or Src kinases;
both are important for PIPKIγ functions (11,40).
Since STAT3, upon phosphorylated by Src, targets focal adhesions
(12), it is important to explore
whether this requires PIPKIγ in future research.
STAT3 is an important transcription factor
regulating the expression of a wide variety of proteins including
MMPs, which are frequently upregulated in malignant tumor cells
(42,43). In ovarian cancers, MMP-2 and MMP-9
are two of the most commonly elevated MMPs and contribute to the
development of tumor metastasis and poor prognosis (44–48).
We previously reported the substantial downregulation of MMP-9 in
PIPKIγ-depleted breast cancer cells (45). Here we further showed that
PIPKIγ-deficient SKOV-3 cells indeed exhibited lower mRNA levels of
both MMP-2 and MMP-9. Although it has been suggested that the
expression of MMP-2 and MMP-9 in ovarian epithelial cells can be
regulated by STAT3 (49,50) and PIPKIγ depletion inhibits STAT3
activity similarly in OVCAR-8 cells as in SKOV-3 cells, no
significant MMP-2 or MMP-9 reduction was detected in OVCAR-8 cells
after PIPKIγ depletion. This suggests some complexity in the
regulation of MMP-2 and MMP-9 in different types of epithelial
ovarian cancer cells. Nevertheless, loss of PIPKIγ undeniably
caused substantially reduced invasion in the OVCAR-8 and SKOV-3
cells. We reason that in SKOV-3 cells, PIPKIγ likely promotes
invasion via the STAT3/MMPs axis; whereas in OVCAR-8 cells, other
signaling cascades regulated by PI3K/AKT and/or STAT3 contribute
more to cell invasion. Considering the structural similarity
between invadopodia and focal adhesions and the role of STAT3 in
focal adhesion assembly (12), it
should be tested whether invadopodium assembly may be disturbed by
the inhibited STAT3 phosphorylation in PIPKIγ-depleted cells. The
underlying mechanism should be explored in the future.
In the present study, we mainly focused on the role
of PIPKIγ in epithelial ovarian cancer cells and provide initial
evidence supporting the contribution of PIPKIγ in epithelial
ovarian cancer cell proliferation, migration and invasion, which is
consistent with previous studies that revealed the role of PIPKIγ
in oncogenic growth and cancer metastasis (6,11,51).
Importantly, our results establish a solid base for further in
vivo and translational studies to confirm whether PIPKIγ could
be a valuable drug target alone or combined with other therapeutic
strategies targeting ovarian cancer.
Acknowledgements
We appreciate Dr Vijayalakshmi Shridhar (Mayo
Clinic, Rochester, MN, USA) for providing the immortalized OSE (OSE
hTert) cells, and Dr William A. Cliby (Mayo Clinic) for sharing
ovarian cancer cell lines (OVCAR-7, OVCAR-8, PEO-1, PEO-4 and
SKOV-3). This research was supported by the National Cancer
Institute R01 grant to K.L. (1R01CA149039) and by National Natural
Science Foundation of China (no.81472428).
Glossary
Abbreviations
Abbreviations:
|
PIPKIγ
|
type Iγ phosphatidylinositol phosphate
kinase
|
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bunney TD and Katan M: Phosphoinositide
signalling in cancer: Beyond PI3K and PTEN. Nat Rev Cancer.
10:342–352. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Samuels Y, Diaz LA Jr, Schmidt-Kittler O,
Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C,
Kinzler KW, et al: Mutant PIK3CA promotes cell growth and invasion
of human cancer cells. Cancer Cell. 7:561–573. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen C, Wang X, Xiong X, Liu Q, Huang Y,
Xu Q, Hu J, Ge G and Ling K: Targeting type Iγ phosphatidylinositol
phosphate kinase inhibits breast cancer metastasis. Oncogene.
34:4635–4646. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Toker A: Phosphoinositides and signal
transduction. Cell Mol Life Sci. 59:761–779. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thapa N and Anderson RA: PIP2 signaling,
an integrator of cell polarity and vesicle trafficking in
directionally migrating cells. Cell Adhes Migr. 6:409–412. 2012.
View Article : Google Scholar
|
|
10
|
Ling K, Schill NJ, Wagoner MP, Sun Y and
Anderson RA: Movin' on up: The role of PtdIns(4,5)P(2) in cell
migration. Trends Cell Biol. 16:276–284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun Y, Turbin DA, Ling K, Thapa N, Leung
S, Huntsman DG and Anderson RA: Type I gamma phosphatidylinositol
phosphate kinase modulates invasion and proliferation and its
expression correlates with poor prognosis in breast cancer. Breast
Cancer Res. 12:R62010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Silver DL, Naora H, Liu J, Cheng W and
Montell DJ: Activated signal transducer and activator of
transcription (STAT) 3: Localization in focal adhesions and
function in ovarian cancer cell motility. Cancer Res. 64:3550–3558.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Du Y, Feng J, Wang R, Zhang H and Liu J:
Effects of flavonoids from Potamogeton crispus L on proliferation,
migration, and invasion of human ovarian cancer cells. PLoS One.
10:e01306852015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller CR, Oliver KE and Farley JH: MEK1/2
inhibitors in the treatment of gynecologic malignancies. Gynecol
Oncol. 133:128–137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fresno Vara JA, Casado E, De Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Al-Alem L and Curry TE Jr: Ovarian cancer:
Involvement of the matrix metalloproteinases. Reproduction.
150:R55–R64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davidson B, Goldberg I, Gotlieb WH,
Kopolovic J, Ben-Baruch G, Nesland JM and Reich R: The prognostic
value of metalloproteinases and angiogenic factors in ovarian
carcinoma. Mol Cell Endocrinol. 187:39–45. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grandis JR, Drenning SD, Chakraborty A,
Zhou MY, Zeng Q, Pitt AS and Tweardy DJ: Requirement of Stat3 but
not Stat1 activation for epidermal growth factor receptor- mediated
cell growth In vitro. J Clin Invest. 102:1385–1392. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johnston PA and Grandis JR: STAT3
signaling: Anticancer strategies and challenges. Mol Interv.
11:18–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saini U, Naidu S, ElNaggar AC, Bid HK,
Wallbillich JJ, Bixel K, Bolyard C, Suarez AA, Kaur B, Kuppusamy P,
et al: Elevated STAT3 expression in ovarian cancer ascites promotes
invasion and metastasis: A potential therapeutic target. Oncogene.
36:168–181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang D, Li C, Zhang Y, Wang M, Jiang N,
Xiang L, Li T, Roberts TM, Zhao JJ, Cheng H, et al: Combined
inhibition of PI3K and PARP is effective in the treatment of
ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol.
142:548–556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Banerjee S and Kaye SB: New strategies in
the treatment of ovarian cancer: Current clinical perspectives and
future potential. Clin Cancer Res. 19:961–968. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carden CP, Stewart A, Thavasu P, Kipps E,
Pope L, Crespo M, Miranda S, Attard G, Garrett MD, Clarke PA, et
al: The association of PI3 kinase signaling and chemoresistance in
advanced ovarian cancer. Mol Cancer Ther. 11:1609–1617. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thomas RK, Baker AC, Debiasi RM, Winckler
W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L,
et al: High-throughput oncogene mutation profiling in human cancer.
Nat Genet. 39:347–351. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cancer Genome Atlas Research Network.
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Samuels Y, Wang Z, Bardelli A, Silliman N,
Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al:
High frequency of mutations of the PIK3CA gene in human cancers.
Science. 304:554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee
SH, Zhang J, Signoretti S, Loda M, Roberts TM, et al: Essential
roles of PI(3)K-p110β in cell growth, metabolism and tumorigenesis.
Nature. 454:776–779. 2008.PubMed/NCBI
|
|
33
|
Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup
OV, Mikami A and Roberts TM: The p110α isoform of PI3K is essential
for proper growth factor signaling and oncogenic transformation.
Proc Natl Acad Sci USA. 103:16296–16300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ciraolo E, Iezzi M, Marone R, Marengo S,
Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, et al:
Phosphoinositide 3-kinase p110β activity: Key role in metabolism
and mammary gland cancer but not development. Sci Signal.
1:ra32008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guillermet-Guibert J, Bjorklof K, Salpekar
A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJ, Okkenhaug K
and Vanhaesebroeck B: The p110β isoform of phosphoinositide
3-kinase signals downstream of G protein-coupled receptors and is
functionally redundant with p110γ. Proc Natl Acad Sci USA.
105:8292–8297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Graupera M, Guillermet-Guibert J, Foukas
LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J,
Cutillas PR, et al: Angiogenesis selectively requires the p110α
isoform of PI3K to control endothelial cell migration. Nature.
453:662–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jun T, Gjoerup O and Roberts TM: Tangled
webs: Evidence of cross-talk between c-Raf-1 and Akt. Sci STKE.
1999:PE11999.PubMed/NCBI
|
|
38
|
Zhang J, Yang PL and Gray NS: Targeting
cancer with small molecule kinase inhibitors. Nat Rev Cancer.
9:28–39. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Di Paolo G, Pellegrini L, Letinic K,
Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR and De
Camilli P: Recruitment and regulation of phosphatidylinositol
phosphate kinase type 1 γ by the FERM domain of talin. Nature.
420:85–89. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ling K, Doughman RL, Firestone AJ, Bunce
MW and Anderson RA: Type I γ phosphatidylinositol phosphate kinase
targets and regulates focal adhesions. Nature. 420:89–93. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wulfkuhle JD, Aquino JA, Calvert VS,
Fishman DA, Coukos G, Liotta LA and Petricoin EF III: Signal
pathway profiling of ovarian cancer from human tissue specimens
using reverse-phase protein microarrays. Proteomics. 3:2085–2090.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Coticchia CM, Curatolo AS, Zurakowski D,
Yang J, Daniels KE, Matulonis UA and Moses MA: Urinary MMP-2 and
MMP-9 predict the presence of ovarian cancer in women with normal
CA125 levels. Gynecol Oncol. 123:295–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chakraborti S, Mandal M, Das S, Mandal A
and Chakraborti T: Regulation of matrix metalloproteinases: An
overview. Mol Cell Biochem. 253:269–285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Delassus GS, Cho H, Park J and Eliceiri
GL: New pathway links from cancer-progression determinants to gene
expression of matrix metalloproteinases in breast cancer cells. J
Cell Physiol. 217:739–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Deryugina EI and Quigley JP: Matrix
metalloproteinases and tumor metastasis. Cancer Metastasis Rev.
25:9–34. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liotta LA, Steeg PS and Stetler-Stevenson
WG: Cancer metastasis and angiogenesis: An imbalance of positive
and negative regulation. Cell. 64:327–336. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Stetler-Stevenson WG: The role of matrix
metalloproteinases in tumor invasion, metastasis, and angiogenesis.
Surg Oncol Clin N Am. 10:383–392. 2001.PubMed/NCBI
|
|
49
|
Zou M, Zhang X and Xu C: IL6-induced
metastasis modulators p-STAT3, MMP-2 and MMP-9 are targets of
3,3-diindolylmethane in ovarian cancer cells. Cell Oncol (Dordr).
39:47–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang X, Liu P, Zhang B, Mao H, Shen L and
Ma Y: Inhibitory effects of STAT3 decoy oligodeoxynucleotides on
human epithelial ovarian cancer cell growth in vivo. Int J Mol Med.
32:623–628. 2013.PubMed/NCBI
|
|
51
|
Thapa N, Choi S, Hedman A, Tan X and
Anderson RA: Phosphatidylinositol phosphate 5-kinase Iγi2 in
association with Src controls anchorage-independent growth of tumor
cells. J Biol Chem. 288:34707–34718. 2013. View Article : Google Scholar : PubMed/NCBI
|