Introduction
The tumor-suppressor LKB1 is an evolutionarily
conserved and ubiquitously expressed serine/threonine kinase, which
can be inactivated either by germline mutations resulting in
Peutz-Jeghers syndrome or by somatic mutations causing
predisposition to multiple sporadic cancers (1–3).
Termed a ‘master kinase’ LKB1 directly phosphorylates and activates
a set of 14 kinases from the adenosine monophosphate
(AMP)-activated protein kinase (AMPK) family, which includes AMPK
(AMPK-α1 and AMPK-α2), brain-specific kinases (BRSKs; BRSK1 and
BRSK2), microtubule affinity-regulating kinases (MARKs; MARK1,
MARK2, MARK3 and MARK4), salt-inducible kinases (SIKs; SIK1, SIK2
and SIK3), nua (novel)/SNF1-like kinases (NUAK1 and NUAK2), and the
Snf-related serine/threonine kinase (SNRK) (4,5).
Through these downstream kinases, LKB1 regulates multiple cellular
processes contributing to tumor suppression, including cell cycle
regulation, cell polarity establishment, energy metabolic balance
and apoptosis control (6–8). However, the underlying molecular
mechanisms are still not completely understood.
The majority of the known functions of LKB1 kinase
are mediated by its ability to activate AMPK, a central conserved
regulator of energy metabolism and cell growth (9). When cellular AMP:ATP ratios rise, LKB1
directly phosphorylates the threonine 172 (Thr172) in the
activation loop of AMPK and results in an increase in ATP-producing
activities and a decrease in ATP-consuming processes including
essentially all biosynthetic pathways required for cell growth
(10,11). Previous investigations have shown
that activation of the LKB1-AMPK pathway is able to suppress cell
growth by arresting the cell cycle in the G1 phase (12,13).
In a similar manner, we confirmed and extended these observations
by demonstrating that exogenous activation of LKB1-AMPK signaling
induces downregulation of cyclins (cyclin D1 and D3) and
upregulation of the cyclin-dependent kinase (CDK) inhibitors (p53,
p21 and p16), and thus, inhibits G1/S cell cycle transition, even
in cells with endogenous expression of LKB1 (14).
Disruption of polarity, as uncontrolled cell growth,
is a quintessential characteristic of epithelial-derived cancer
cells, and is critical in epithelial-mesenchymal transition (EMT),
a crucial step during tumor invasiveness, metastasis and fibrosis
(15,16). LKB1 has been firmly established as a
highly conserved regulator of cell polarization, and there is
growing evidence that MARKs may be primary mediators of LKB1 action
in that process (17,18). In epithelial cells, the polarizing
activity of LKB1 kinase is relayed through MARKs, which
phosphorylate microtubule-associated proteins including tau, and
increase the dynamic instability of microtubules (19,20).
According to current knowledge, LKB1 signaling predominantly
regulates cell growth via AMPK and cell polarity via MARKs.
However, Müller et al suggested that MARK2 also plays a role
in the regulation of cell proliferation by phosphorylating the cell
cycle regulatory phosphatase Cdc25 resulting in a complex of Cdc25
and 14-3-3 and cell cycle blockage (21), which indicates that LKB1-mediated
regulation of cell growth and polarity signaling are not separate,
but connected to each other. However, how these two pathways are
orchestrated to maintain cellular homeostasis has not been fully
investigated. To address this question and to better understand
MARK signaling, we enforced MARK expression using a lentiviral
system in adherent HeLa cells, and revealed a dual role of MARK2 in
the regulation of AMPK-mediated G1/S transition and the actin-based
cytoskeletal system, indicating that cell proliferation and cell
polarity may be broadly integrated under the control of MARK2
signaling.
Materials and methods
Materials
Cell culture media and supplements were purchased
from Invitrogen Life Technologies (Carlsbad, CA, USA). Antibodies
targeting MARK1, MARK2, MARK3, MARK4, AMP-activated protein kinase
α (AMPKα), phospho-AMPKα (Thr172), p21, E-cadherin, N-cadherin and
vimentin were purchased from Cell Signaling Technology (Beverly,
MA, USA). Antibodies against p53, p16 and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Horseradish
peroxidase (HRP)-conjugated secondary antibodies were purchased
from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Phalloidin-tetramethylrhodamine-conjugated (phalloidin-TRITC) and
compound C were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Unless otherwise noted, chemicals and organic solvents were
obtained from Sigma-Aldrich and were of the highest grade.
Cell culture and viral infection
Human cervical cancer cell line HeLa cells were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai). Cells were maintained as frozen stocks.
After viral infections, only pooled clones were used to avoid
clonal variations. Culturing of cells was performed as previously
described (22).
Lentivirus production and
transduction
Expression plasmids of MARKs were obtained from
Professor Dario Alessi (University of Dundee). LV5-GFP-Puro
lentiviral vectors expressing MARKs (LV-MARK1, LV-MARK2, LV-MARK3
and LV-MARK4) were constructed by GenePharma Co., Ltd. (Shanghai,
China). HeLa cells were infected with LV-MARKs or LV-GFP mock
vector at multiplicities of infection (MOI) of 20 for 48 h in the
presence of 8 µg/ml Polybrene (Sigma, St. Louis, MO, USA) and
maintained in puromycin.
Cell proliferation assay
Cell proliferation was examined using a
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfonphenyl)-2H-tetrazolium
(MTS) assay (CellTiter 96 AQueous One Solution Cell Proliferation
Assay; Promega, Madison, WI, USA), according to the manufacturer's
instructions. Cells (1,000/well) were seeded onto 96-well plates in
triplicates and incubated in culture medium containing MTS every
day for 4 days. Absorbance was measured at 490 nm in a SpectraMax
M5 (Molecular Devices, Sunnyvale, CA, USA). Experiments were
repeated 3 times and representative results are shown.
Soft agar colony formation assay
Cells were seeded at a density of 1×103
cells in 60-mm dishes in complete media with a bottom layer of 0.6%
low melting point (LMP) agar and a top layer of 0.35% low LMP agar.
The plates were incubated at 37°C in a humidified incubator for
10–14 days. Cells were fed twice a week by adding fresh medium.
Colonies were stained with crystal violet, photographed and
manually counted.
Flow cytometric analysis
Exponentially growing cells were infected with the
indicated lentiviral vectors for 48 h, detached and fixed with 70%
ethanol overnight. Propidium iodide/RNase A was used to stain the
nuclei. Cell cycle distribution was determined by flow cytometry
using a FACSVantage SE cell sorter (BD Biosciences, Franklin Lakes,
NJ, USA). Percentage of cells at different phases of the cell cycle
was analyzed using the CellQuest™ Pro software (BD
Biosciences).
Western blot analysis
Total proteins were analyzed and blotted as
previously described (22,23). Compound C (CC; 20 µM) or dimethyl
sulfoxide (DMSO) was added 24 h post infection. Specific primary
antibodies recognizing MARK1 (1:1,000), MARK2 (1:1,000), MARK3
(1:1,000), MARK4 (1:1,000), AMPKα (1:1,000), phospho-AMPKα (Thr172)
(1:1,000), p53 (1:1,000), p21 (1:500), p16 (1:500), E-cadherin
(1:1,000), N-cadherin (1:1,000), vimentin (1:1,000) and GAPDH
(1:1,000) were used at the indicated dilutions. HRP-conjugated
secondary antibodies were used at 1:10,000 dilutions. Signals were
detected by Substrate SuperSignal West Pico Chemiluminescent
Substrate kit (Pierce Biotechnology, Rockford, IL, USA), and
quantified using Quantity One software (Bio-Rad, Hercules, CA,
USA).
Cell staining and confocal
imaging
Cells grown on glass coverslips were fixed with 4%
paraformaldehyde and permeabilized in 0.1% Triton X-100. F-actin
was visualized using TRITC-conjugated phalloidin
(phalloidin-TRITC). Nuclei were counterstained with
4,6-diamidino-2-phenylindole (DAPI). Digital images of 10 random
fields (>500 cells) were used to calculate the percentage of
elongated cells. Cells were classified as elongated when the length
of the protrusion was 2-fold longer than the width of the cell body
(24). Representative images were
captured using an inverted confocal microscope (FluoView FV1000;
Olympus, Tokyo, Japan) at a magnification of ×800.
Wound-healing assay
HeLa cells were seeded into 6-well plates and
infected with MARK2 or green fluorescence protein (GFP) mock
lentiviruses. Confluent cell monolayers were scratched with
micropipette tips, and images were captured at each time courses
for 48 h. The wound healing capacities were calculated by measuring
the distance of the migrating edge. Mean values were obtained from
3 separate experiments.
Transwell invasion assay
Cell invasion assays were performed using Transwell
chambers with 8-µm pores (Corning Costar Corp., Cambridge, MA, USA)
according to the manufacturer's instructions. Infected cells were
seeded on top of the Matrigel in the upper chamber, and the bottom
chamber was filled with culture medium containing 10% fetal bovine
serum (FBS), as the chemoattractant for 24 h. The invasive cells on
the underside of the filter, were fixed with paraformaldehyde,
stained with crystal violet and counted. Experiments were performed
in triplicate.
Statistical analysis
Data are presented as mean ± standard deviation
(SD). Unless otherwise indicated, cell culture experiments were
reproduced 3 or more times. Paired t-tests were used to determine
statistical significance. p-values <0.05 were considered
statistically significant.
Results
Lentiviral transduction of MARKs into
HeLa cells
MARKs have been identified as important components
of LKB1-related signal transduction pathways in a variety of
species (4,5). In the present study, LKB1-deficient
HeLa cells were transduced with lentiviral vectors expressing 4
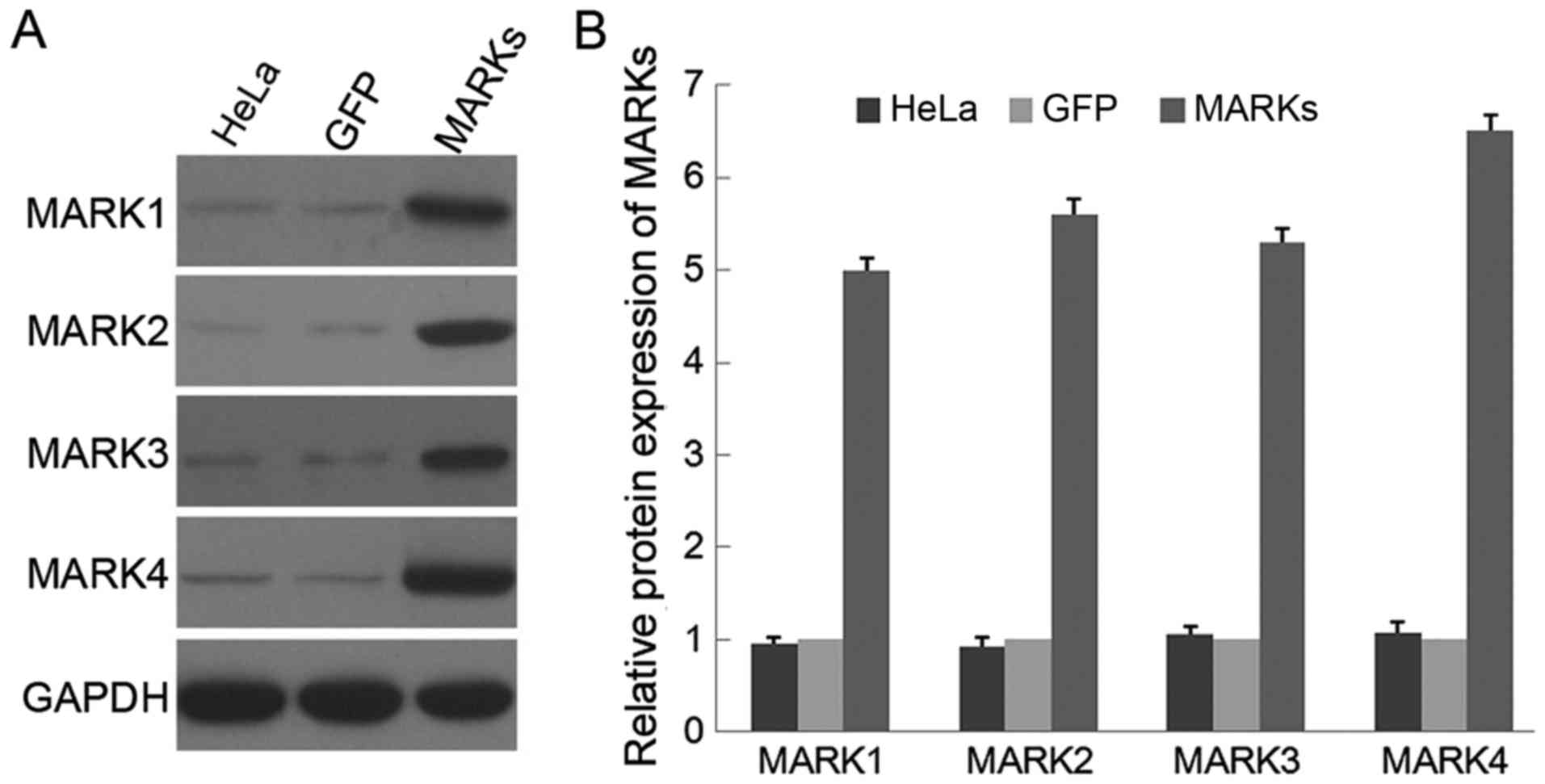
isoforms of MARKs respectively. Western blot analysis confirmed a
5- to 6-fold increase in protein expression of 4 MARK kinases in
the HeLa cells infected with the corresponding MARK lentivirus
compared with that in cells infected with a GFP mock vector
(Fig. 1). Uninfected HeLa cells
were also included, and no difference was observed in the
expression of MARK protein between the parental HeLa and HeLa cells
infected with the GFP vector. Thus, an efficient overexpression of
MARKs was successfully achieved in the LKB1-null HeLa cells.
Overexpression of MARK2 inhibits
proliferation of HeLa cells
It is known that reintroducing LKB1 into HeLa cells
restores LKB1 activity and induces growth suppression (25,26).
As important downstream kinases regulated by LKB1, MARKs may also
be involved in growth inhibition. To investigate this possibility,
we performed MTS assay, which is used as colorimetric method for
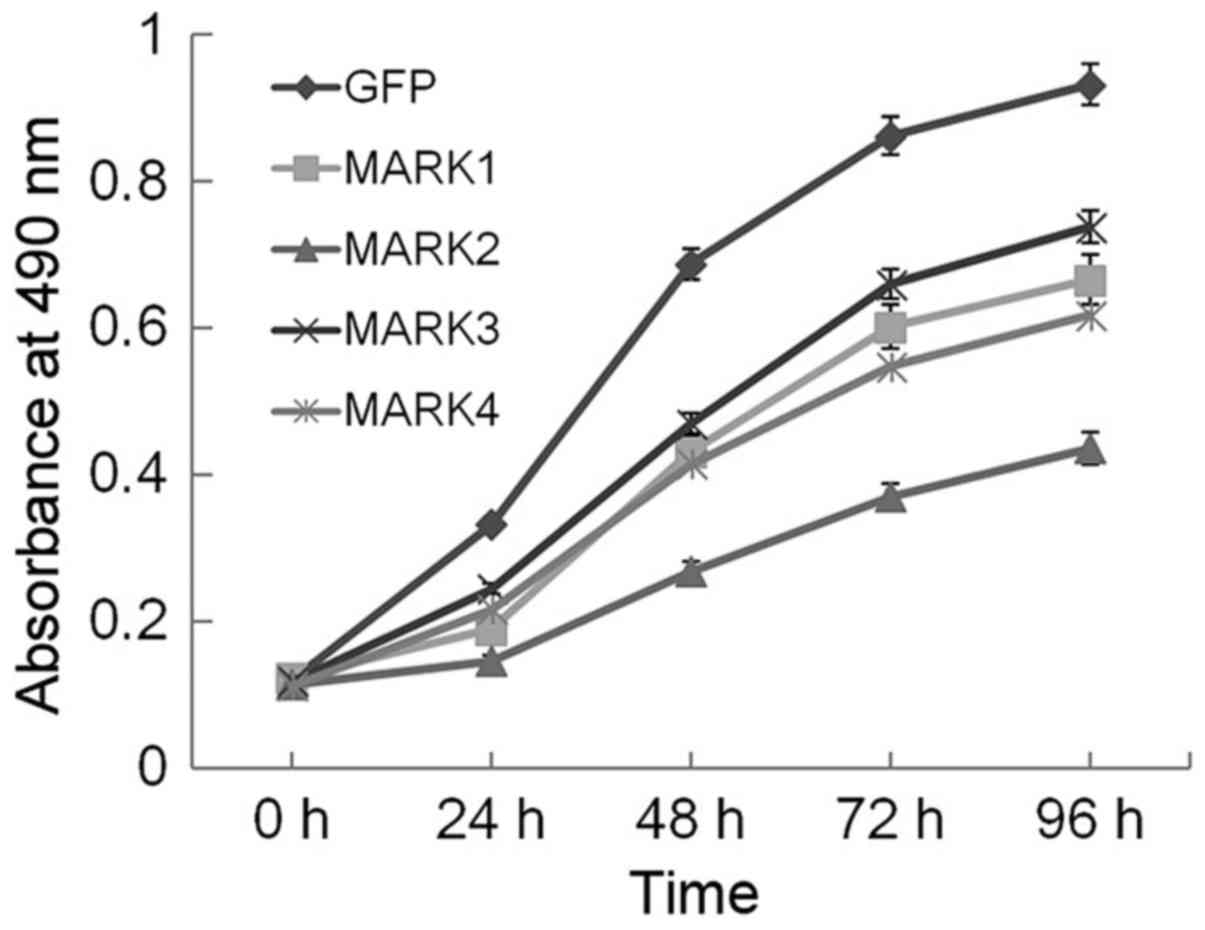
sensitive quantification of viable cells in proliferation (27). Over the 96-h interval examined
(Fig. 2), growth curve experiments
clearly demonstrated that enforced expression of MARKs conferred a
proliferative disadvantage to HeLa cells, which was most evident in
the MARK2-transduced cells and suggests that overexpression of
MARKs, particularly MARK2, strongly inhibited the proliferation of
HeLa cells.
Overexpression of MARK2 inhibits
anchorage-independent growth of HeLa cells
Anchorage-independent growth of cells in soft agar
is one of the hallmark characteristics of cellular transformation
and uncontrolled cell growth (28).
The LKB1-deficient HeLa cell line is one widely used human cervical
cancer line, and forced LKB1 expression reverses the oncogenesis of
HeLa cells. To explore whether enhanced expression of MARKs had a
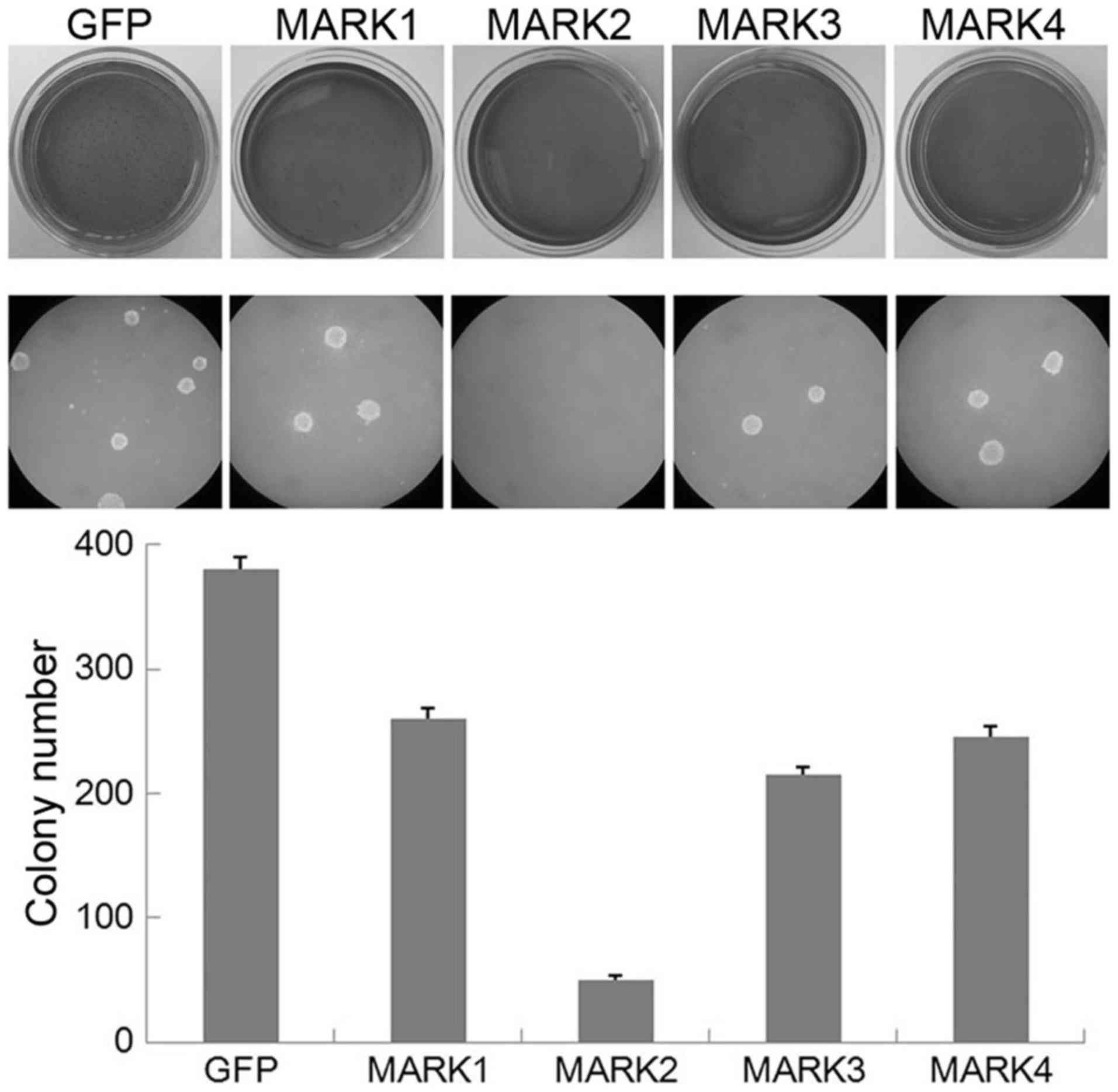
similar effect, we performed a soft agar colony formation assay.
Notably, the HeLa cells infected with the GFP control vector grew
efficiently in soft agar and formed numerous colonies, whereas the
MARK2-overexpressing cells exhibited a significant reduction in
anchorage-independent growth on soft agar (Fig. 3). Overexpression of MARK1, MARK3 or
MARK4 led to minor attenuation of colony formation. These results
suggest that overexpression of MARK2 inhibits the
anchorage-independent growth of HeLa cells, which mimics the effect
of the upstream kinase LKB1.
Overexpression of MARK2 arrests the
cell cycle at G1 phase
In mammalian cells, proliferation control is
primarily achieved in the G1 phase of the cell cycle (29). Previous studies have provided
compelling evidence that ectopic LKB1 expression in LKB1-deficient
cancer cells inhibits G1/S transition and arrests cells in the G1
phase (25,30,31).
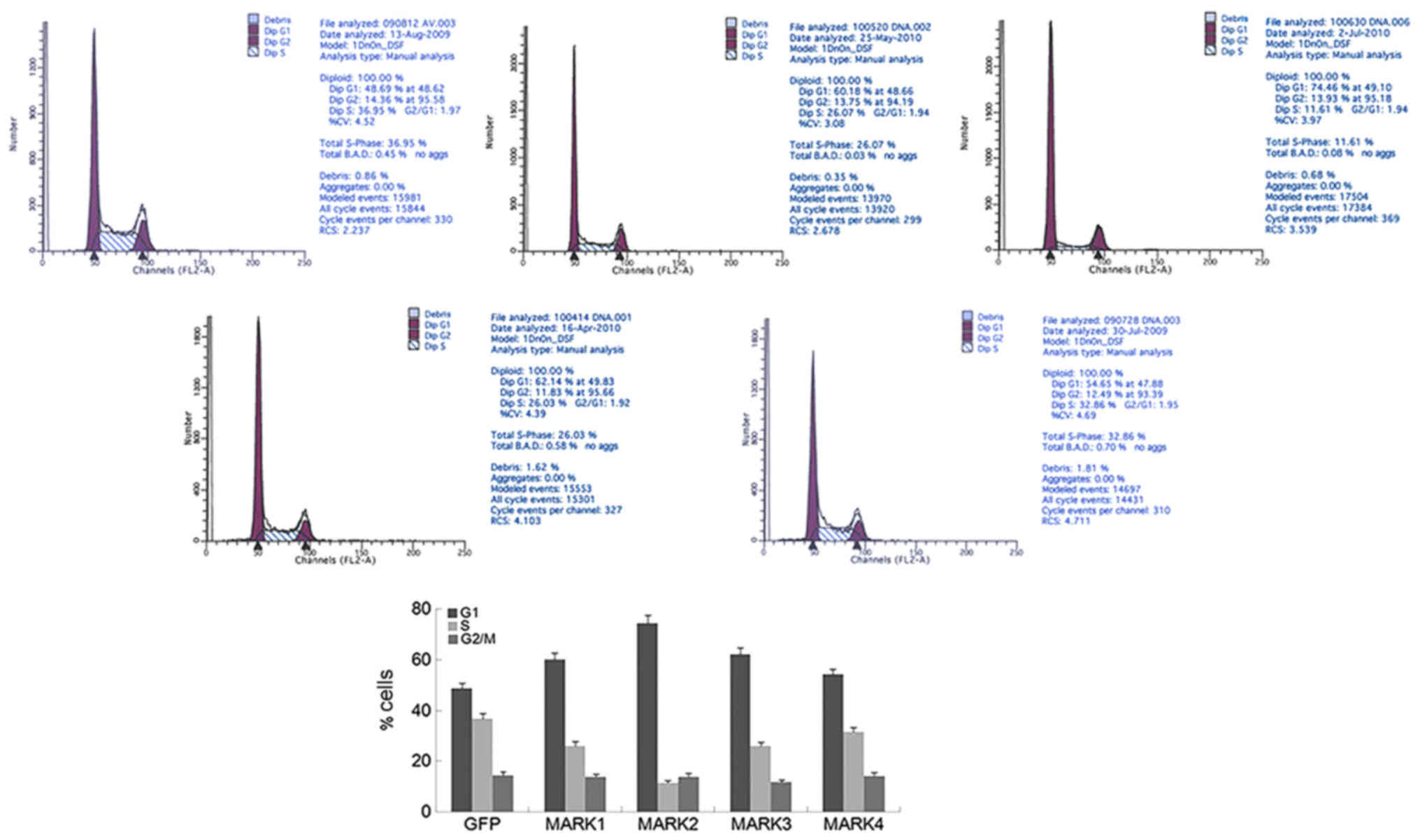
Transduction of MARK2 into HeLa cells led to increased accumulation
of cells in G1 and a reduced proportion of cells in S phase,
compared with that in cells infected with the GFP vector (Fig. 4). Similar but less pronounced
changes in cell cycle profile were observed for the other 3 MARKs.
These results indicate that exogenous expression of MARK2 is
sufficient to induce G1 arrest in cells with endogenous LKB1
expression deficiency.
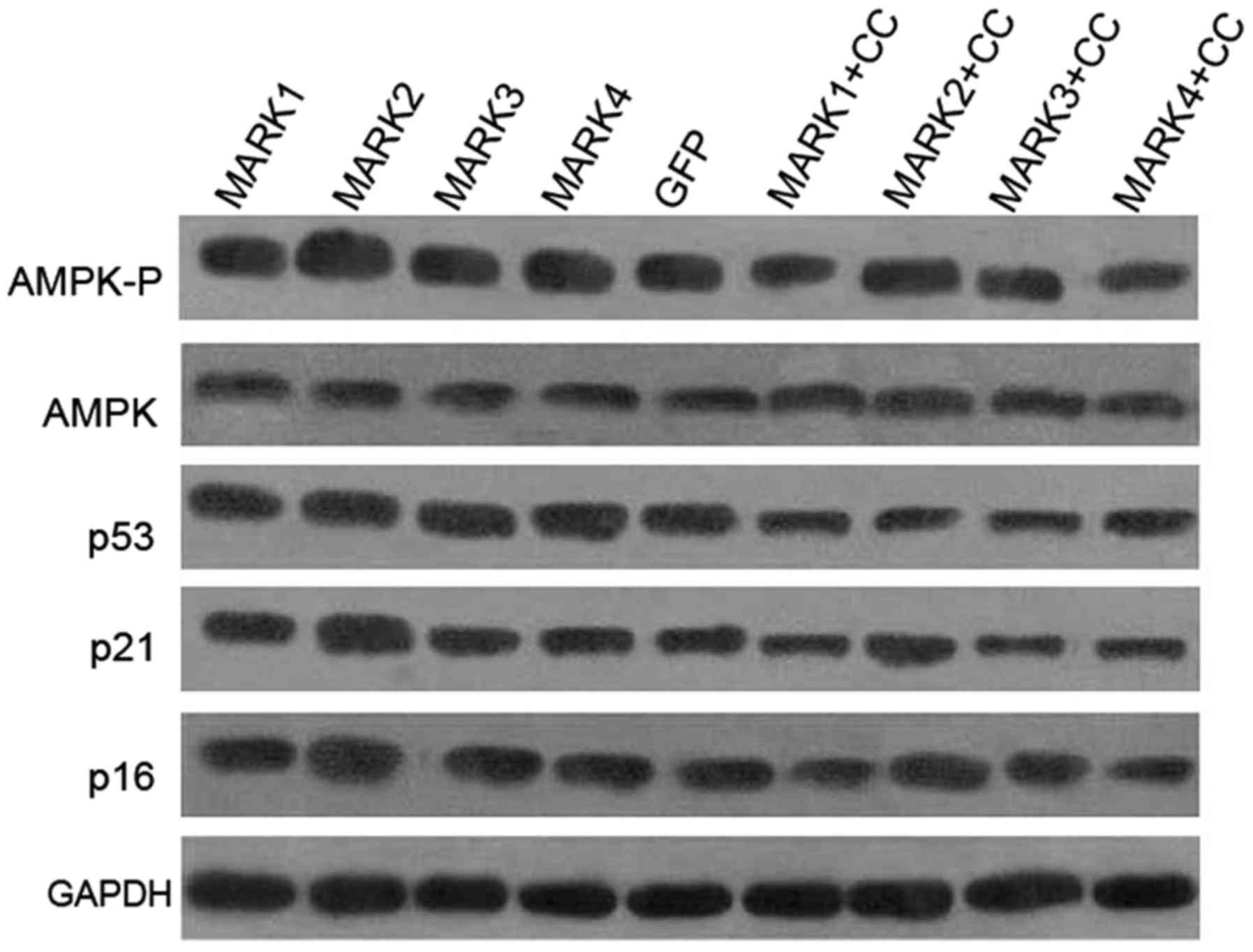
Overexpression of MARK2 activates AMPK
and increases expression of p21 and p16, but not p53
The role of AMPK as a key mediator in LKB1-related
signaling cascades raises the possibility that it may play a role
in the observed intracellular cell cycle modulation by MARK2.
Overexpression of MARK2 promoted phosphorylation of AMPKα at Thr172
in the HeLa cells, while the protein level of AMPKα was unaffected
(Fig. 5), suggesting that inducible
expression of MARK2 activated AMPK in the HeLa cells. No observable
differences were detected in AMPK phosphorylation in the HeLa cells
transduced with MARK1, MARK3 or MARK4 lentiviral vectors.
Induction of CDK inhibitors is a well-characterized
mechanism through which to inhibit G1/S transition (32). To investigate whether MARKs mediate
cell cycle arrest by regulating expression of CDK inhibitors, we
analyzed the expression levels of p21, p16 and p53 protein in the
HeLa cells. The results presented in Fig. 5 indicate a specific increase in p21
and p16 levels in the MARK2-transduced cells, whereas the levels of
p53 were not changed. Furthermore, to test whether AMPK mediates
the cell cycle-controlling functions of MARK2, we used compound C,
a potent and selective inhibitor of AMPK. As expected, treatment
with compound C (CC) was able to repress AMPKα phosphorylation in
all groups (Fig. 5), further
confirming that compound C is an effective inhibitor of AMPK
activity. Induction of p21 and p16 expression in MARK2 transduced
cells was markedly suppressed by compound C (Fig. 5), suggesting that in HeLa cells AMPK
is a critical downstream effector of MARK2, and AMPK kinase
activity is required for p21 and p16 induction.
No differences were observed in the expression of
p21, p16 or p53 protein after transduction with MARK1, MARK3 or
MARK4 lentivirus. Additionally, inactivation of AMPK using
pharmacologic inhibitor, compound C, attenuated the expression of
p53, p21 and p16 in all groups, which is in good agreement with our
previous study that activation of AMPK signaling inhibits G1/S
progression by upregulating the p53, p21 and p16 pathways (14).
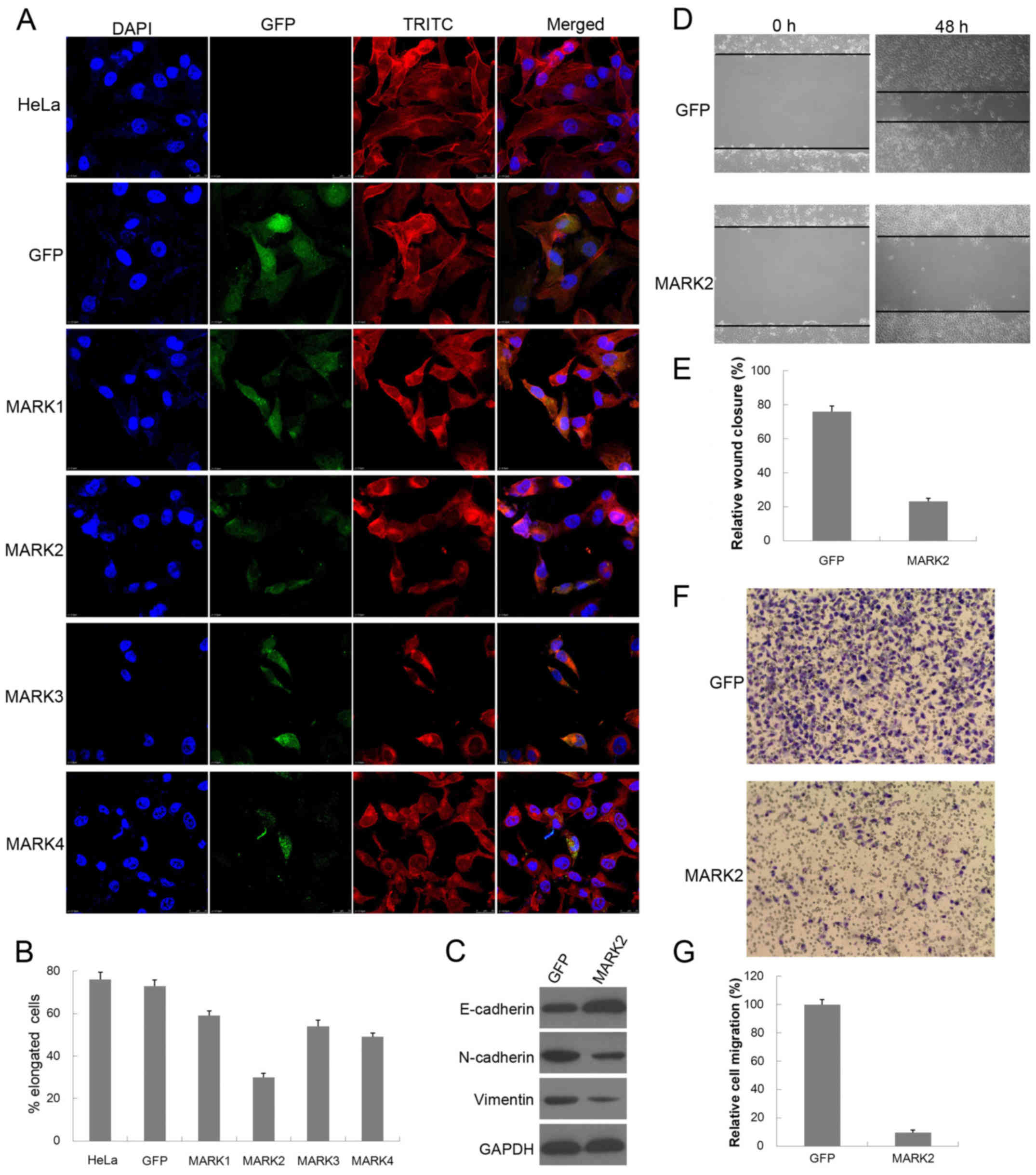
Overexpression of MARK2 reverses the
EMT phenotype of HeLa cells
MARKs are thought to function as regulators of cell
polarity since their activation is associated with cytoskeletal
modification (19,20). Next, we examined whether
overexpression of MARKs was able to reorganize the actin
cytoskeleton and restore HeLa cells to an epithelial phenotype.
Analysis of the actin cytoskeleton was performed by fluorescence
microscopy following phalloidin staining. As shown in Fig. 6A, parental HeLa and HeLa cells
infected with the GFP vector display elongated and spindle-shaped
phenotypic characteristics of mesenchymal cells, with transcellular
filamentous actin (F-actin) stress fiber formation, whereas
overexpression of MARK2 induced the typical cobblestone-like
epithelial characteristic phenotype, with cortical actin staining
and actin filament bundles below the plasma membrane. MARK2
overexpression significantly decreased the elongated
(mesenchymal-like) cell population of HeLa cells in comparison to
the GFP control vector (73 vs. 30%) (Fig. 6B). Additionally, overexpression of
MARK1, MARK3 or MARK4 induced a morphological switch to create an
epithelial cell population, in a similar, but less profound way.
Western blot analysis further showed upregulation of the epithelial
marker E-cadherin, and downregulation of the mesenchymal marker
N-cadherin and vimentin in MARK2-overexpressing HeLa cells
(Fig. 6C). These results indicate
that enforced expression of MARK2 may remodel the actin
cytoskeleton and inhibit the development of a mesenchymal-like
phenotype of HeLa cells, uncovering the MARK2-negative role in
regulating the EMT process.
Overexpression of MARK2 suppresses
cell migration and invasion of HeLa cells
The finding that MARK2 appears to influence EMT
prompted us to more closely examine its effects on cell migration
and invasion. To assay collective cell migration, we performed
scratch wound assay of cell monolayers. As expected, we observed
delayed closure upon overexpression of MARK2 compared with the
control cells (Fig. 6D).
Statistical analysis indicated that the migratory activity of
control HeLa cells was ~3-fold higher than that of the
MARK2-infected cells (Fig. 6E). We
also used Matrigel-coated Transwell chambers to assess cell
invasiveness and yielded similar results (Fig. 6F). The invasive capacity of the
MARK2-transduced cells was decreased to ~10% of the control cells
(Fig. 6G). These results indicate
that enhanced expression of MARK2 led to inhibited cell migration
and invasion, which is a characteristic feature of EMT.
Discussion
Among the numerous molecular mechanisms altered in
human cancers, those involving cell division cycle control are
believed fundamental for oncogenesis (33). Originally discovered as polarity
proteins, the importance of MARKs has been greatly limited to the
regulation of cell polarization in a context-dependent manner.
However, by utilizing lentiviral systems in LKB1-deficient HeLa
cells, we revealed that overexpression of MARK2 led to retarded
cell growth, decreased colony formation and cell cycle arrest,
which suggests that MARK2 may be an important regulator of cell
proliferation and provides further insights into the roles of MARK2
in anti-tumorigenesis. The suppressive mechanism of HeLa cell
proliferation by MARK2 appeared to be cell cycle arrest, rather
than apoptosis, since the cell population of the G1 phase, but not
G0 phase was significantly increased by MARK2 overexpression. In
addition, the HeLa cell line is severely impaired in LKB1 activity
(30). The present study showed
that inducible expression of MARK2 in HeLa cells was sufficient to
mimic LKB1 signaling activation as previously reported (12–14),
and further confirms that MARK2 is a critical downstream target of
LKB1.
MARKs and AMPK belong to the same protein kinase
family, referred as AMPK-related kinase family, and are
well-established substrates of LKB1 signaling (4,5). In
the present study, we found that enforced expression of MARK2
induced phosphorylation of AMPK, and led to the upregulation of p21
and p16 in an AMPK-dependent manner. The present study demonstrated
that MARK2 upregulation could be responsible for the AMPK-mediated
increase of p21 and p16 protein expression; this increased
expression is of critical importance in the induction of G1 arrest
and cell growth inhibition, as widely reported in the literature
(34,35). To the best of our knowledge, this is
the first study to report that AMPK is a novel and essential
downstream target of MARK2 signaling and acts as an important
factor during MARK2-induced G1 arrest, although additional analyses
are required to precisely define how MARK2 induces AMPK activation.
Our findings showcase the complexity of MARK2 biology and indicate
that cell growth and polarity pathways mediated by LKB1 are
intimately connected to each other.
Induction of CDK inhibitors is a well-characterized
mechanism to inhibit the activity of cyclin/CDK complexes and
prevent cell cycle progression (32). Notably, in the present study,
overexpression of MARK2 activates AMPK and induces the expression
of p21 and p16, but not p53. These results suggest that MARK2-AMPK
activation triggers cell cycle blockage in a p53-independent mode,
which is not in complete agreement with our earlier study that
LKB1-AMPK mediates G1 cell cycle arrest by inducing the expression
of p21, p16 and p53 (14). One
plausible explanation is that HeLa cells constitutively express
human papilloma virus E6 protein, which targets p53 for
proteasome-mediated degradation and abrogates its tumor-suppressor
function (36,37). This idea is sustained by a previous
study which showed that LKB1 forms a complex with transcription
regulatory factors LMO4, GATA-6 and Ldb1, and induces GATA-mediated
p21 expression and G1 cell cycle arrest through p53-independent
mechanism in HeLa cells (38).
Further studies are warranted to validate the GATA-related model in
the MARK2-AMPK pathway.
In this context, we stained F-actin cytoskeleton
with phalloidin and observed that overexpression of MARK2 in HeLa
cells led to rearrangement of actin cytoskeleton and formation of
stress fibers, supporting a role of MARK2 in the regulation of the
actin cytoskeleton in HeLa cells and raising the possibility that
MARK2 may interact with the Rho family of small GTPases, which are
regarded as central regulators of the actin cytoskeletal system
(39,40). Further investigations may elucidate
these issues more clearly. Loss of cell polarity, along with the
acquisition of motility and invasiveness, are regarded as typical
phenomena during the epithelial-mesenchymal transition (EMT)
process (41,42). Enforced expression of MARK2
inhibited the mesenchymal and stimulated an epithelial-like
morphology with concomitant alterations in EMT markers expression,
and reduced migration and invasion, thus causing the shift of the
EMT balance in favor of an epithelial state. Our experiments
provide new insights into the function of MARK2 in suppressing EMT
and suggest that modulation of MARK2 expression is a potential
approach to inhibit the invasiveness and migration of cervical
cancer cells and the attendant pathologic processes including
metastasis.
In summary, our results reveal that MARK2 plays a
role in synergistically activating AMPK and reorganizing the actin
cytoskeleton, and functions as an intracellular inhibitor for cell
cycle progression and EMT in HeLa cells. The present study has
provided a fascinating link between LKB1-mediated control of cell
proliferation and cell polarity, which may benefit cancer research,
but may also shed light on basic principles of epithelial
biology.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81201544).
Glossary
Abbreviations
Abbreviations:
|
AMPK
|
adenosine monophosphate-activated
protein kinase
|
|
MARK
|
microtubule affinity-regulating
kinase
|
|
CDK
|
cyclin-dependent kinase
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
McGarrity TJ and Amos C: Peutz-Jeghers
syndrome: Clinicopathology and molecular alterations. Cell Mol Life
Sci. 63:2135–2144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hearle N, Schumacher V, Menko FH,
Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott
RJ, Lim W, et al: Frequency and spectrum of cancers in the
Peutz-Jeghers syndrome. Clin Cancer Res. 12:3209–3215. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara
K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al:
LKB1 modulates lung cancer differentiation and metastasis. Nature.
448:807–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lizcano JM, Göransson O, Toth R, Deak M,
Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, et
al: LKB1 is a master kinase that activates 13 kinases of the AMPK
subfamily, including MARK/PAR-1. EMBO J. 23:833–843. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alessi DR, Sakamoto K and Bayascas JR:
LKB1-dependent signaling pathways. Annu Rev Biochem. 75:137–163.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ylikorkala A, Rossi DJ, Korsisaari N,
Luukko K, Alitalo K, Henkemeyer M and Mäkelä TP: Vascular
abnormalities and deregulation of VEGF in Lkb1-deficient mice.
Science. 293:1323–1326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jishage K, Nezu J, Kawase Y, Iwata T,
Watanabe M, Miyoshi A, Ose A, Habu K, Kake T, Kamada N, et al: Role
of Lkb1, the causative gene of Peutz-Jegher's syndrome, in
embryogenesis and polyposis. Proc Natl Acad Sci USA. 99:8903–8908.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Williams T and Brenman JE: LKB1 and AMPK
in cell polarity and division. Trends Cell Biol. 18:193–198. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hardie DG: AMPK - sensing energy while
talking to other signaling pathways. Cell Metab. 20:939–952. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sebbagh M, Olschwang S, Santoni MJ and
Borg JP: The LKB1 complex-AMPK pathway: The tree that hides the
forest. Fam Cancer. 10:415–424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dasgupta B and Chhipa RR: Evolving lessons
on the complex role of AMPK in normal physiology and cancer. Trends
Pharmacol Sci. 37:192–206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fogarty S, Ross FA, Ciruelos D Vara, Gray
A, Gowans GJ and Hardie DG: AMPK causes cell cycle arrest in
LKB1-deficient cells via activation of CAMKK2. Mol Cancer Res.
14:683–695. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang HW, Lee YS, Nam HY, Han MW, Kim HJ,
Moon SY, Jeon H, Park JJ, Carey TE, Chang SE, et al: Knockdown of
β-catenin controls both apoptotic and autophagic cell death through
LKB1/AMPK signaling in head and neck squamous cell carcinoma cell
lines. Cell Signal. 25:839–847. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang X, Wang P, Gao Q and Tao X:
Exogenous activation of LKB1/AMPK signaling induces G1 arrest in
cells with endogenous LKB1 expression. Mol Med Rep. 9:1019–1024.
2014.PubMed/NCBI
|
|
15
|
Okada T, Sinha S, Esposito I, Schiavon G,
López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M,
et al: The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT
by restraining Ras-MAPK signalling. Nat Cell Biol. 17:81–94. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen N, Balasenthil S, Reuther J and
Killary AM: DEAR1, a novel tumor suppressor that regulates cell
polarity and epithelial plasticity. Cancer Res. 74:5683–5689. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mohseni M, Sun J, Lau A, Curtis S,
Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong KK, et al: A
genetic screen identifies an LKB1-MARK signalling axis controlling
the Hippo-YAP pathway. Nat Cell Biol. 16:108–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shelly M and Poo MM: Role of LKB1-SAD/MARK
pathway in neuronal polarization. Dev Neurobiol. 71:508–527. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee S, Wang JW, Yu W and Lu B:
Phospho-dependent ubiquitination and degradation of PAR-1 regulates
synaptic morphology and tau-mediated Aβ toxicity in Drosophila. Nat
Commun. 3:13122012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matenia D and Mandelkow EM: The tau of
MARK: A polarized view of the cytoskeleton. Trends Biochem Sci.
34:332–342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Müller J, Ritt DA, Copeland TD and
Morrison DK: Functional analysis of C-TAK1 substrate binding and
identification of PKP2 as a new C-TAK1 substrate. EMBO J.
22:4431–4442. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liang X, Xu G, Gao Q and Tao X: LKB1
expression reverses the tumorigenicity of L02 cells. Oncol Rep.
36:1055–1061. 2016.PubMed/NCBI
|
|
23
|
Liang X, Wang P, Gao Q, Xiang T and Tao X:
Endogenous LKB1 knockdown accelerates G1/S transition through p53
and p16 pathways. Cancer Biol Ther. 9:156–160. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ladhani O, Sánchez-Martinez C, Orgaz JL,
Jimenez B and Volpert OV: Pigment epithelium-derived factor blocks
tumor extravasation by suppressing amoeboid morphology and
mesenchymal proteolysis. Neoplasia. 13:633–642. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tiainen M, Vaahtomeri K, Ylikorkala A and
Mäkelä TP: Growth arrest by the LKB1 tumor suppressor: Induction of
p21WAF1/CIP1. Hum Mol Genet. 11:1497–1504. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fogarty S and Hardie DG: C-terminal
phosphorylation of LKB1 is not required for regulation of
AMP-activated protein kinase, BRSK1, BRSK2, or cell cycle arrest. J
Biol Chem. 284:77–84. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gopinath P and Ghosh SS: Apoptotic
induction with bifunctional E.coli cytosine deaminase-uracil
phosphoribosyltransferase mediated suicide gene therapy is
synergized by curcumin treatment in vitro. Mol Biotechnol.
39:39–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee CJ, Jang JH, Lee JY, Lee MH, Li Y, Ryu
HW, Choi KI, Dong Z, Lee HS, Oh SR, et al: Aschantin targeting on
the kinase domain of mammalian target of rapamycin suppresses
epidermal growth factor-induced neoplastic cell transformation.
Carcinogenesis. 36:1223–1234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sclafani RA and Holzen TM: Cell cycle
regulation of DNA replication. Annu Rev Genet. 41:237–280. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tiainen M, Ylikorkala A and Mäkelä TP:
Growth suppression by Lkb1 is mediated by a G1 cell cycle arrest.
Proc Natl Acad Sci USA. 96:9248–9251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gurumurthy S, Hezel AF, Sahin E, Berger
JH, Bosenberg MW and Bardeesy N: LKB1 deficiency sensitizes mice to
carcinogen-induced tumorigenesis. Cancer Res. 68:55–63. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cánepa ET, Scassa ME, Ceruti JM, Marazita
MC, Carcagno AL, Sirkin PF and Ogara MF: INK4 proteins, a family of
mammalian CDK inhibitors with novel biological functions. IUBMB
Life. 59:419–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Manning AL and Dyson NJ: pRB, a tumor
suppressor with a stabilizing presence. Trends Cell Biol.
21:433–441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pei XH and Xiong Y: Biochemical and
cellular mechanisms of mammalian CDK inhibitors: A few unresolved
issues. Oncogene. 24:2787–2795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pei XH and Xiong Y: Biochemical and
cellular mechanisms of mammalian CDK inhibitors: A few unresolved
issues. Oncogene. 24:2787–2795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oei AL, van Leeuwen CM, ten Cate R,
Rodermond HM, Buist MR, Stalpers LJ, Crezee J, Kok HP, Medema JP
and Franken NA: Hyperthermia selectively targets human
papillomavirus in cervical tumors via p53-dependent apoptosis.
Cancer Res. 75:5120–5129. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Setogawa T, Shinozaki-Yabana S, Masuda T,
Matsuura K and Akiyama T: The tumor suppressor LKB1 induces p21
expression in collaboration with LMO4, GATA-6, and Ldb1. Biochem
Biophys Res Commun. 343:1186–1190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aktories K: Bacterial protein toxins that
modify host regulatory GTPases. Nat Rev Microbiol. 9:487–498. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mokady D and Meiri D: RhoGTPases - A novel
link between cytoskeleton organization and cisplatin resistance.
Drug Resist Updat. 19:22–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Savagner P: The epithelial-mesenchymal
transition (EMT) phenomenon. Ann Oncol. 21 Suppl 7:vii89–vii92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gandalovičová A, Vomastek T, Rosel D and
Brábek J: Cell polarity signaling in the plasticity of cancer cell
invasiveness. Oncotarget. 7:25022–25049. 2016.PubMed/NCBI
|