Introduction
Esophageal carcinoma is the sixth most common type
of cancer worldwide and has remained an aggressive cancer because
it is commonly diagnosed at later stages (1). Esophageal carcinoma is divided into
two major types [squamous cell carcinoma (SCC) and adenocarcinoma],
and the incidence of esophageal adenocarcinoma (EAC) is rapidly
increasing in Western countries (2). The overall mortality of this disease
remains high with a 5-year survival rate of less than 20%, despite
remarkable advances in the care of patients with EAC (3). A poor prognosis has been associated
with diagnosis at an advanced stage and metastasis (4,5).
Galectin-9 (Gal-9) is a tandem-repeat type galectin
with two carbohydrate recognition domains (CRDs); it was first
identified as an eosinophil chemoattractant and activation factor
(6–8). Similar to other galectins, Gal-9
regulates various cellular functions in eosinophils, including cell
aggregation, adhesion and apoptosis (9,10).
Gal-9 also enhances antitumor immunity by initiating
CRD-independent dendritic cell maturation and Th1-mediated
antitumor immunity (11). Treatment
with recombinant Gal-9 prolonged survival in a murine melanoma
model by increasing the number of CD8+ cytotoxic T cells
(CTLs), natural killer (NK) cells and macrophages (12). Furthermore, the Gal-9 receptor T
cell immunoglobulin mucin-3 (Tim-3) negatively regulated T cell
responses by promoting CD8+ T cell exhaustion and
inducing the expansion of myeloid-derived suppressor cells
(13,14).
Recombinant Gal-9 induces dose-dependent apoptosis
in various leukemic T cell lines in the presence of a functional
CRD (15,16). Additionally, Gal-9 inhibits the
growth of myeloma (17), chronic
myeloid leukemia (18) and human
melanoma both in vitro and in vivo (19,20).
Moreover, we have recently reported that recombinant Gal-9 exerts
antitumor effects on various solid malignancies by affecting the
phosphorylation of various proteins, angiogenesis and the
expression of microRNAs (miRNAs) (21–23).
However, little is known about the antitumor effects
of Gal-9 on EAC cells or the miRNAs associated with these effects.
Therefore, the present study evaluated the effects of Gal-9 on the
growth of EAC cell lines, the mechanism of action and the miRNAs
associated with its antitumor effects.
Materials and methods
Reagents and antibodies
A mutant form of Gal-9 lacking the entire linker
region was recombinantly produced and purified as described in our
previous report (24). Fetal bovine
serum (FBS) was purchased from Wako Pure Chemical Industries, Ltd.,
(Osaka, Japan), Cell Counting kit-8 (CCK-8) was purchased from
Dojindo Laboratories (Kumamoto, Japan), and all other chemicals
were obtained from Sigma Chemical Corp. (Tokyo, Japan). Z-VAD-FMK
and Z-DEVD-FMK were purchased from AdooQ Bioscience (Irvine, CA,
USA).
The primary antibodies used in the present study
included monoclonal anti-β-actin (A5441, 1:3,000; Sigma-Aldrich,
St. Louis, MO, USA), anti-cyclin D1 (RB-9041, 1:1,000; Thermo
Fisher Scientific, Waltham, MA, USA), anti-cyclin E (1:1,000;
Thermo Fisher Scientific), anti-Cdk6 (sc-177, 1:1,000; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), anti-Cdk4 (sc-749, 1:1,000;
Santa Cruz Biotechnology), anti-Cdk2 (sc-163, 1:2,000; Santa Cruz
Biotechnology) and anti-phosphorylated retinoblastoma protein
(558385, 1:1,000 Rb; BD Biosciences, San Jose, CA, USA). Antibodies
to caspase-3 (#9665), cleaved caspase-3 (#9664), caspase-7
(#12827), caspase-9 (#9508), cleaved caspase-9 (#7237), PARP
(#9542), cleaved PARP (#5625), LC3 (#12741) and SQSTM1/p62 (#8025)
were purchased from Cell Signaling Technology (Boston, MA,
USA).
The secondary antibodies used in the present study
included horseradish peroxidase (HRP)-conjugated anti-mouse and
anti-rabbit IgG antibodies purchased from Cell Signaling Technology
(1:2,000 each).
Cell culture and cell proliferation
assay
Four human EAC cell lines (OE19, OE33, SK-GT4 and
OACM5.1c) were obtained from the European Collection of
Authenticated Cell Cultures (ECACC). All cell lines were grown in
RPMI-1640 medium (Gibco/Invitrogen, Carlsbad, CA, USA) supplemented
with 10% FBS and 9100 mg/l of penicillin-streptomycin (Invitrogen)
at 37°C in a humidified atmosphere containing 5%
CO2.
Cell proliferation was assayed using a Cell Counting
kit-8 (CCK-8) according to the manufacturers instructions. Briefly,
5×103 cells were seeded into each well of a 96-well
plate and cultured in 100 µl of RPMI-1640 medium supplemented with
10% FBS. After 24 h, Gal-9 (0, 0.1, 0.3 or 1.0 µM) was added to
each well, and the cells were cultured for an additional 48 h.
Then, the CCK-8 reagent (10 µl) was added to each well, and the
plates were incubated at 37°C for 3 h. The absorbance of each well
was measured at 450 nm using a microplate reader.
ELISA to assess apoptosis
Caspase-cleaved cytokeratin 18 (CCK18) expression
was evaluated using an M30 Apoptosense ELISA kit obtained from
Peviva AB (Bromma, Sweden) according to the manufacturers
instructions (25). Cells
(5×103/well) were seeded into 96-well plates, cultured
in 100 µl of culture medium for 24 h and then treated with 0.3 µM
Gal-9. The antigen concentrations in the control and treated
samples were calculated via interpolation from a standard
curve.
Cell cycle and apoptosis analyses
The cell cycle and apoptosis analyses were performed
separately using a Cell Cycle Phase Determination kit (Cayman
Chemical, Ann Arbor, MI, USA) and an Annexin V-FITC Early apoptosis
detection kit (Cell Signaling Technology), respectively.
SK-GT4 cells (1.0×106 cells in a 100-mm
dish) were treated with 0.3 µM Gal-9 for 48 h, and untreated cells
were used as the controls. The cell cycle profiles were analyzed by
measuring the propidium iodide (PI)-labeled DNA content in
ethanol-fixed cells. Fixed cells were washed with
phosphate-buffered saline (PBS) and then stored at −20°C prior to
flow cytometric analysis. On the day of the analysis, the cells
were washed with cold PBS, suspended in 100 µl of PBS with 10 µl of
RNase A (250 µg/ml) and incubated for 30 min. Then, 110 µl of PI
stain (100 µg/ml) was added to each cell suspension, and the cells
were incubated at 4°C for at least 30 min prior to the analysis.
Apoptotic and necrotic cell death were analyzed by performing
double staining with FITC-conjugated Annexin V and PI; this
staining method is based on the binding of Annexin V to apoptotic
cells with exposed phosphatidylserine residues and the binding of
PI to late apoptotic/necrotic cells with membrane damage. Tumor
cells were treated with 0.3 µM Gal-9 for either 12 or 24 h and
untreated cells were used as the controls. Staining was performed
according to the manufacturers instructions. Flow cytometry was
performed using a Cytomics FC 500 flow cytometer (Beckman Coulter,
Indianapolis, IN, USA), and the proportions of stained cells were
analyzed using the Kaluza software (Beckman Coulter). All
experiments were performed in triplicate.
Gel electrophoresis and western
blotting
SK-GT4 cells were seeded at a density of
1.0×106 cells/100-mm dish and cultured for 24 h. Then,
0.3 µM Gal-9 was added, and the cells were cultured for an
additional 24–48 h. Next, the cells were lysed in a protease
inhibitor cocktail (Complete protease inhibitor mixture; iNtRON
Biotechnology, Sungnam, Korea) on ice for 20 min. Suspensions of
lysed cells were centrifuged at 13,000 × g at 4°C for 5 min, and
supernatants containing soluble cellular proteins were harvested
and stored at −80°C until use. The protein concentrations were
measured using a NanoDrop 2000 fluorospectrometer (Thermo Fisher
Scientific). Protein aliquots (1–10 µg) were resuspended in sample
buffer, separated on 10% Tris-glycine gradient gels by SDS-PAGE
(26), and then transferred to
nitrocellulose membranes. After blocking, the membranes were
incubated with primary antibodies, followed by incubation with
HRP-conjugated secondary antibodies (27). Immunoreactive proteins were
visualized with an enhanced chemiluminescence detection system
(Perkin-Elmer Co., Waltham, MA, USA) on X-ray film.
Angiogenic profile analysis
SK-GT4 cells were seeded (6.0×106
cells/100 mm-diameter dish) and treated with 0.3 µM Gal-9, while
control cells remained untreated; all cells were cultured with
RPMI-1640 medium supplemented with 10% FBS for 24 h and then lysed
in PRO-PREP. A RayBio Human Angiogenesis Antibody array
(RayBiotech, Inc., Norcross, GA, USA) was employed according to the
manufacturers protocol. This protocol includes a spot-based assay
that facilitates the detection and comparison of 20 angiogenic
cytokines. Each array membrane was exposed to X-ray film, and the
signals were detected using a chemiluminescence detection system
(Perkin-Elmer). The immunoreactive band densities obtained with
this array were analyzed by densitometric scanning (TIc scanner;
Shimizu, Co., Ltd., Kyoto, Japan).
Phosphorylated receptor tyrosine
kinase (p-RTK) antibody arrays
SK-GT4 cells were treated with 0.3 µM Gal-9 for 24 h
and then lysed in PRO-PREP. Human phospho-RTKs were assayed using a
Human Phospho-RTK Array kit (R&D Systems, Minneapolis, MN, USA)
according to the manufacturers instructions. Each array membrane
was exposed to X-ray film, and the signals were detected using a
chemiluminescence detection system (Perkin-Elmer).
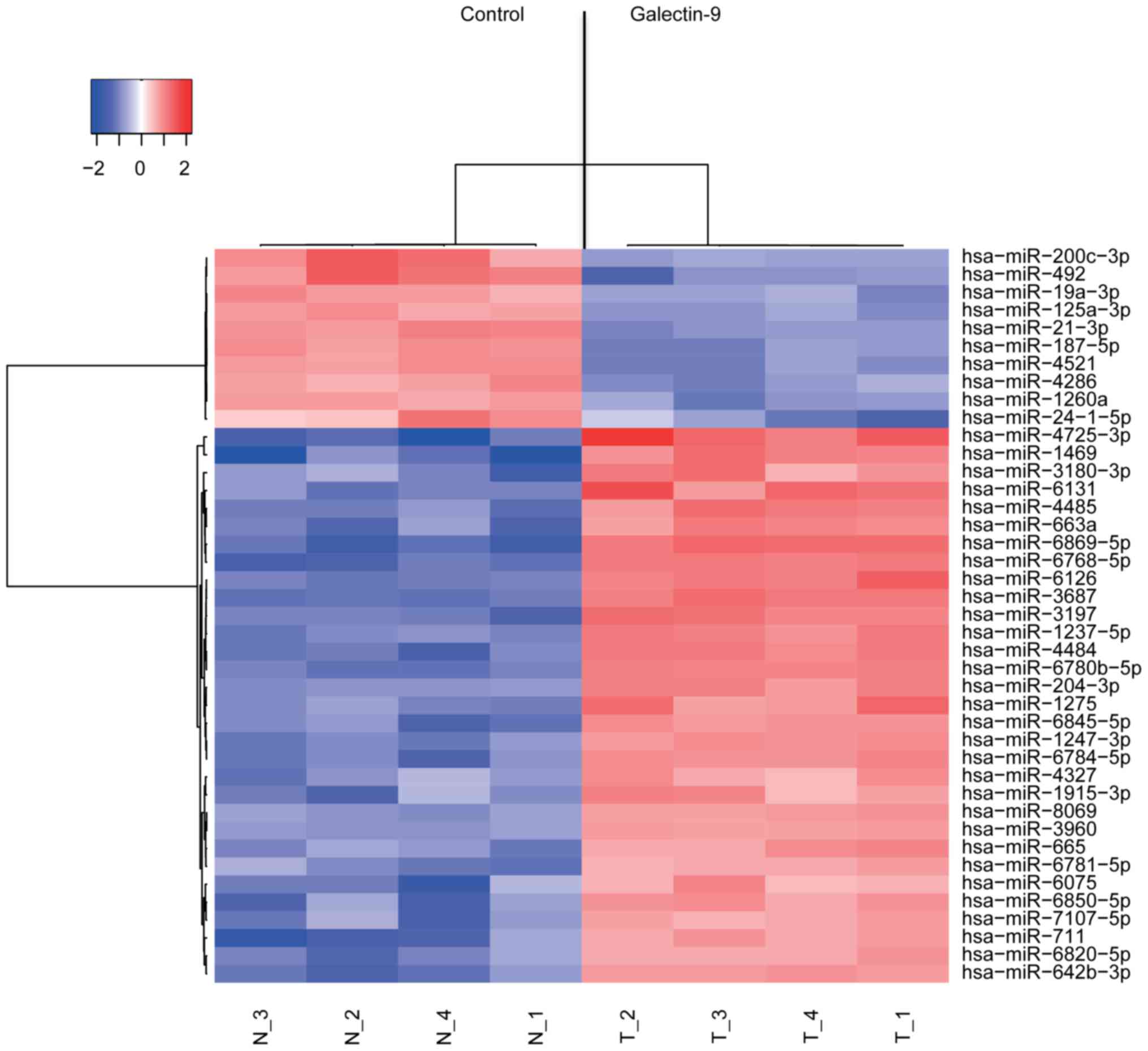
Analysis of miRNA arrays
SK-GT4 cells were treated with 0.3 µM Gal-9 for 24 h
and then stored in the RNAprotect reagent (Qiagen, Venlo, The
Netherlands). Total RNA was extracted from each cell line using a
miRNeasy Mini kit (Qiagen) according to the manufacturer's
instructions, and the RNA quantity and quality were measured using
an RNA 6000 Nano kit (Agilent Technologies, Santa Clara, CA, USA).
The samples were labeled using a miRCURY Hy3 Power Labeling kit
(Exiqon A/S, Vedbaek, Denmark) and hybridized to a human miRNA
oligo chip (v.21; Toray Industries, Tokyo, Japan). Scanning was
performed using a 3D-Gene Scanner 3000 (Toray Industries). The
3D-Gene extraction version 1.2 software (Toray Industries) was used
to read the raw intensities from the images. To detect differences
in miRNA expression between the Gal-9-treated and control samples,
the raw data were analyzed using the GeneSpring GX 10.0 software
(Agilent Technologies). Quantile normalization was performed for
raw data above background levels, and differentially expressed
miRNAs were identified using the Mann-Whitney U test. Hierarchical
clustering was performed using the farthest neighbor method with
the absolute uncentered Pearson's correlation coefficient as a
metric. A heat map was produced with the base-2 logarithms of the
intensities median-centered for each row to depict the relative
expression intensity of each miRNA.
Statistical analysis
All statistical analyses were performed using the
GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA,
USA). Comparisons between the treated and control groups were
performed using two-tailed paired or unpaired Students t-tests. A
P<0.05 was considered significant.
Results
Gal-9 suppresses human esophageal
adenocarcinoma cell proliferation
The effects of Gal-9 on the proliferation of four
EAC cell lines (OE19, OE33, SK-GT4 and OACM5.1c) were evaluated.
The cells were grown in 10% FBS and treated with 0, 0.1, 0.3 or 1.0
µmol/l Gal-9 for 48 h. Gal-9 inhibited the proliferation of all
four EAC cell lines in a dose-dependent manner (Fig. 1).
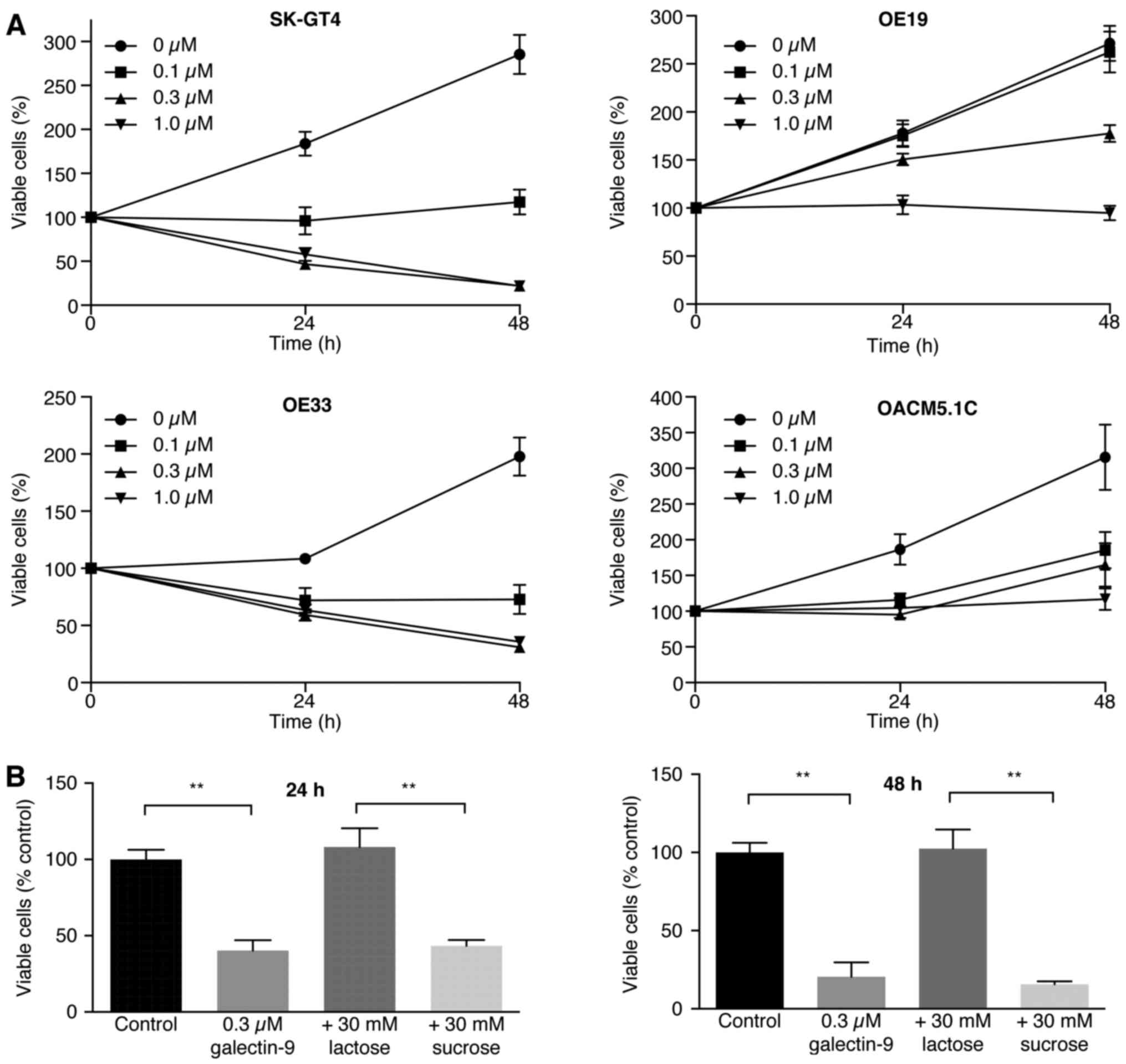 | Figure 1.Gal-9 suppresses the proliferation of
esophageal adenocarcinoma cells. (A) OE19, OE33, SK-GT4 and OACM
5.1C cells were seeded into 96-well plates. After 24 h, Gal-9 (0,
0.1, 0.3, or 1.0 µmol/l) or vehicle was added to the culture
medium; the cells were enumerated 24 h later by performing a CCK
assay. OE19, OE33, SK-GT4 and OACM 5.1C cells (5,000/well) were
seeded into 96-well plates, and Gal-9 was added as described above.
Cell viability was assayed daily from 0 to 48 h. The viability of
the Gal-9-treated cells differed significantly from the viability
of the control cells (P<0.05). (B) The antagonistic effect of
lactose against galectin-9 (Gal-9). SK-GT4 cells were incubated
with or without 30 mM lactose in addition to 0.3 µM Gal-9 for 48 h.
The effect of Gal-9 was antagonized by lactose, suggesting that the
β-galactosidase-binding nature of Gal-9 was essential for its
activity (**P<0.01). |
Gal-9 exhibits antitumor effects in
EAC cells by inducing apoptosis
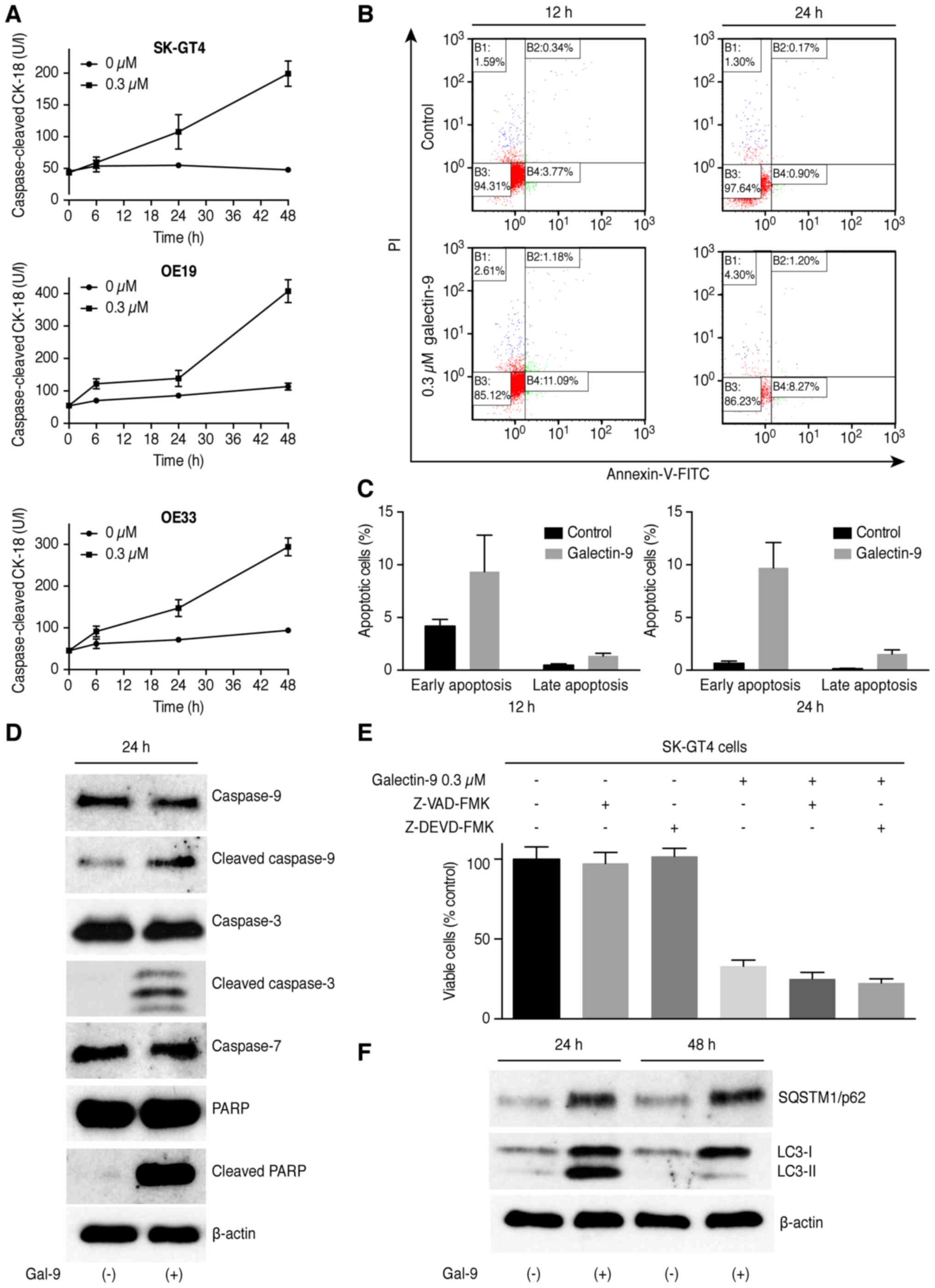
To determine whether Gal-9 induces apoptosis,
SK-GT4, OE19 and OE33 cells were treated with 0.3 µM Gal-9, and the
CCK18 levels were measured following treatment using an M30 ELISA
kit. The results of Annexin V-FITC/PI staining and the flow
cytometric analysis demonstrated that Gal-9 significantly increased
the CCK-18 levels in the three EAC cell lines (Fig. 2A) and also induced apoptosis of the
SK-GT4 cells in a dose- and time-dependent manner. The different
quadrants presented in Fig. 2B
represent living cells (lower left quadrant), early apoptotic cells
(lower right quadrant), and late apoptotic cells (upper right
quadrant). The increased numbers of early- and late-phase apoptotic
cells indicated that the antitumor effects of Gal-9 involved
induction of apoptosis (Fig. 2C).
This apoptotic induction was accompanied by increases in the
cleaved caspase-3, cleaved caspase-9 and cleaved PARP levels
(Fig. 2D). The blockade of caspase
activation by treatment with either the pan-caspase inhibitor
Z-VAD-FMK or caspase-3 inhibitor Z-DEVD-FMK did not protect the
cells from Gal-9-induced cell death (Fig. 2E). Thus, Gal-9 may suppress the
proliferation of EAC cells by inducing apoptosis via a
caspase-independent pathway. Because apoptosis often occurs
simultaneously with autophagy, this process is also involved in the
antitumor effect of Gal-9. Therefore, we evaluated the levels of
SQSTM1/p62 and LC3-II, which are key proteins involved in autophagy
regulation, after treatment with Gal-9 for 24 h. As shown in
Fig. 2F, the accumulation of LC3-II
and upregulation of SQSTM1/p62 were observed in the treated SK-GT4
cells, indicating that Gal-9 may inhibit the autophagic flux.
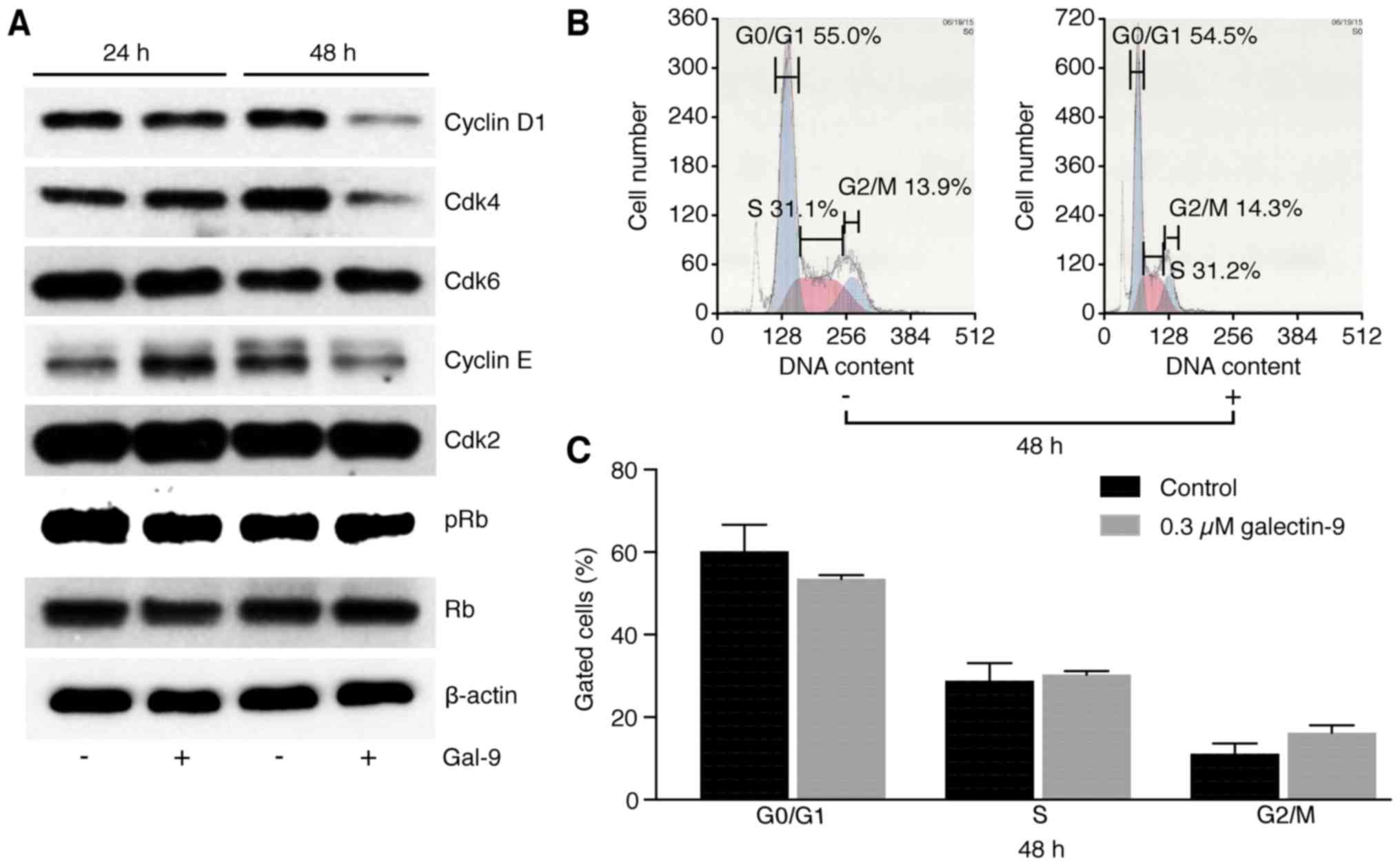
No specific effects of Gal-9 are
observed on cell cycle regulatory proteins in SK-GT4 cells
The effects of Gal-9 on the expression of various
cell cycle-related molecules in SK-GT4 cells were evaluated by
western blotting. SK-GT4 cells were treated with 0.3 µM Gal-9 for
48 h. Gal-9 treatment resulted in progressive decreases in the
cyclin D1, cyclin E and Cdk4 levels but had no effects on the
levels of other cell cycle regulatory proteins (Fig. 3A).
To elucidate the mechanism of action of Gal-9 in the
control of SK-GT4 cell proliferation, cell cycle progression was
examined by flow cytometry. No changes were observed in the cell
cycle profiles of SK-GT4 cells treated with 0.3 µM Gal-9 (Fig. 3B), suggesting that Gal-9 suppressed
EAC cell growth through tumor cell apoptosis but not through cell
cycle arrest.
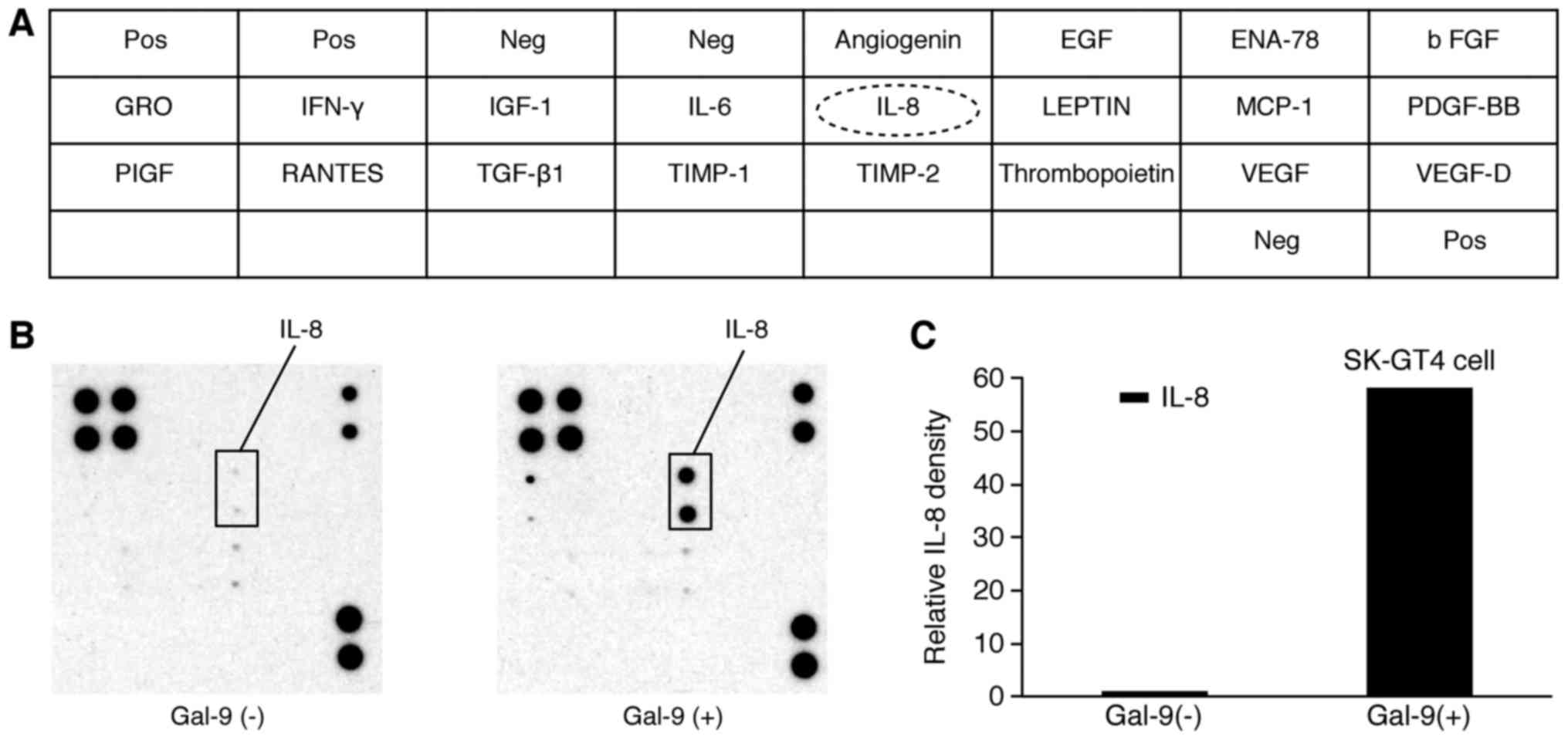
Gal-9 treatment affects the expression
of angiogenesis-related molecules
We used an angiogenesis array system (Fig. 4A) to identify the key
angiogenesis-related molecules associated with the antitumor
effects of Gal-9 in SK-GT4 cells. Of the 20 angiogenesis molecules
screened, only the interleukin (IL)-8 levels increased in
vitro following Gal-9 treatment (Fig. 4B). The densitometric ratio of IL-8
spots for Gal-9-treated vs. untreated cells was 58.2-fold (Fig. 4C).
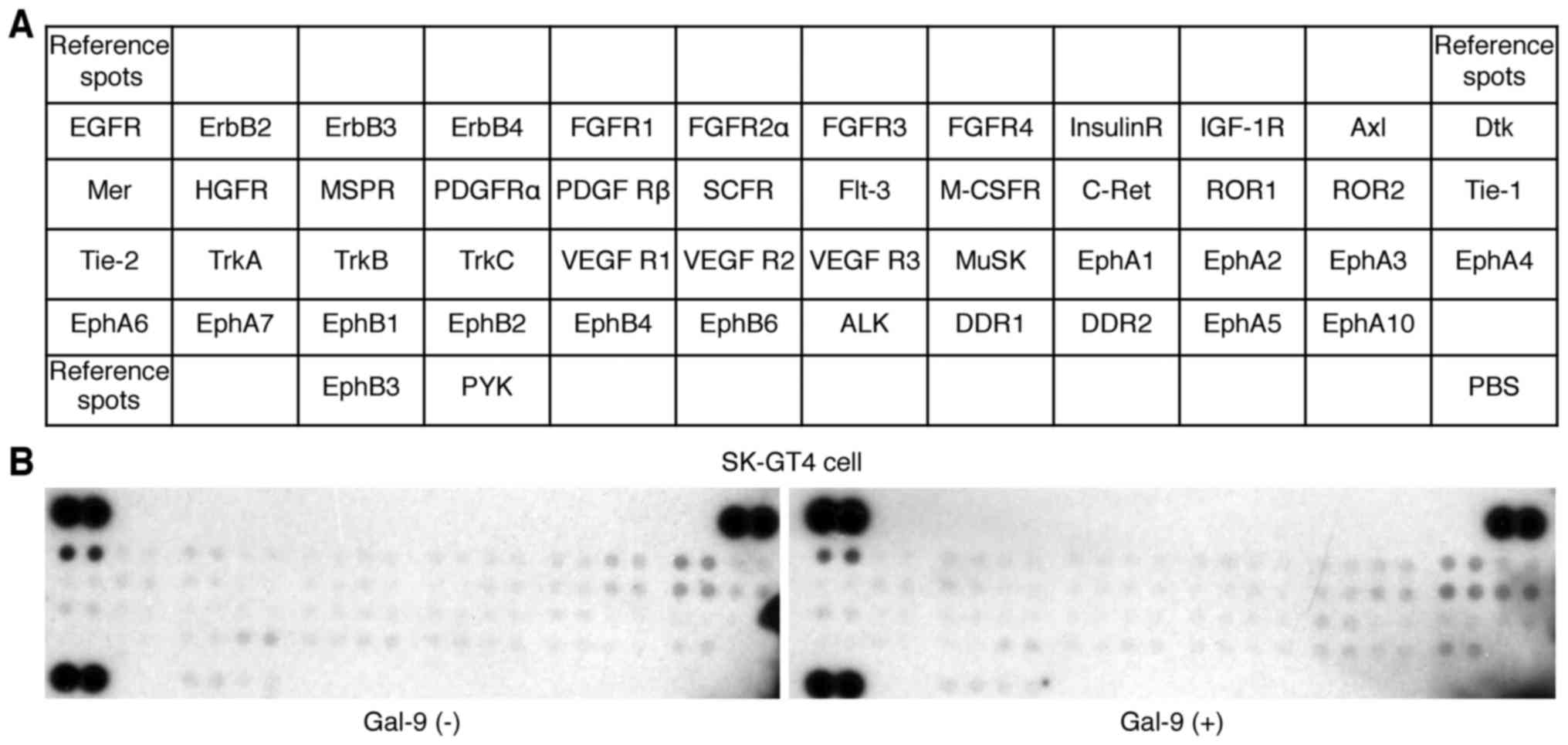
Effects of Gal-9 on p-RTKs in SK-GT4
cells
A p-RTK array system was used to identify the key
RTKs associated with the antitumor effects of Gal-9. The use of an
antibody array (Fig. 5A) enabled
the evaluation of the expression of 49 activated RTKs in SK-GT4
cells and tumors in the presence and absence of Gal-9. The
activated RTK levels did not change following Gal-9 treatment
(Fig. 5B).
Effects of Gal-9 on miRNA
expression
Using a custom microarray platform, we analyzed the
expression of 2,555 miRNA probes in cell lines cultured in the
presence and absence of Gal-9. Treatment of SK-GT4 cells with 0.3
µmol/l Gal-9 for 48 h resulted in the significant upregulation of
31 miRNAs and in the significant downregulation of 10 miRNAs
(Table I).
 | Table I.Relative expression levels and
chromosomal locations of miRNAs in SK-GT4 cells cultured with or
without Gal-9. |
Table I.
Relative expression levels and
chromosomal locations of miRNAs in SK-GT4 cells cultured with or
without Gal-9.
| miRNA | Fold change
(Treated/Chromosomal untreated) | P-value | Chromosomal
location |
|---|
| Upregulated |
|
hsa-miR-4725-3p | 7.72 | 0.0286 | 17 |
|
hsa-miR-6869-5p | 6.38 | 0.0294 | 20 |
|
hsa-miR-1469 | 5.95 | 0.0286 | 15q26.2 |
|
hsa-miR-6768-5p | 5.44 | 0.0294 | 16 |
|
hsa-miR-3687 | 5.13 | 0.0286 | 21 |
|
hsa-miR-6131 | 4.92 | 0.0286 | 5 |
|
hsa-miR-3197 | 4.76 | 0.0294 | 21 |
|
hsa-miR-4484 | 4.75 | 0.0286 | 10 |
|
hsa-miR-6126 | 4.69 | 0.0286 | 16 |
|
hsa-miR-4485 | 4.50 | 0.0286 | 11 |
|
hsa-miR-6780b-5p | 4.42 | 0.0294 | 6 |
|
hsa-miR-663a | 4.25 | 0.0286 | 20p11.1 |
|
hsa-miR-711 | 4.23 | 0.0286 | 3 |
|
hsa-miR-1237-5p | 4.23 | 0.0286 | 11 |
|
hsa-miR-1275 | 4.05 | 0.0286 | 6 |
|
hsa-miR-3180-3p | 3.96 | 0.0286 | 16 |
|
hsa-miR-6784-5p | 3.94 | 0.0286 | 17 |
|
hsa-miR-6845-5p | 3.91 | 0.0286 | 8 |
|
hsa-miR-642b-3p | 3.77 | 0.0294 | 19 |
|
hsa-miR-1247-3p | 3.70 | 0.0286 | 14q32.31 |
|
hsa-miR-6850-5p | 3.69 | 0.0286 | 8 |
|
hsa-miR-204-3p | 3.61 | 0.0294 | 9q21.12 |
|
hsa-miR-1915-3p | 3.50 | 0.0286 | 10p12.31 |
|
hsa-miR-6075 | 3.37 | 0.0286 | 5 |
|
hsa-miR-665 | 3.29 | 0.0286 | 14q32.2 |
|
hsa-miR-6820-5p | 3.27 | 0.0286 | 22 |
|
hsa-miR-7107-5p | 3.22 | 0.0286 | 12 |
|
hsa-miR-6781-5p | 3.14 | 0.0286 | 17 |
|
hsa-miR-4327 | 3.04 | 0.0286 | 21 |
|
hsa-miR-8069 | 3.04 | 0.0294 | 21 |
|
hsa-miR-3960 | 3.02 | 0.0275 | 9 |
| Downregulated |
|
hsa-miR-492 | 0.23 | 0.0286 | 12q22 |
|
hsa-miR-200c-3p | 0.27 | 0.0286 | 12p13.31 |
|
hsa-miR-21-3p | 0.28 | 0.0286 | 17q23.1 |
|
hsa-miR-4521 | 0.29 | 0.0286 | 17 |
|
hsa-miR-187-5p | 0.29 | 0.0294 | 18q12.2 |
|
hsa-miR-1260a | 0.32 | 0.0294 | 14 |
|
hsa-miR-24-1-5p | 0.32 | 0.0286 | 9q22.32 |
|
hsa-miR-4286 | 0.32 | 0.0294 | 8 |
|
hsa-miR-125a-3p | 0.32 | 0.0286 | 19q13.41 |
|
hsa-miR-19a-3p | 0.33 | 0.0286 | 13q31.3 |
An unsupervised hierarchical clustering analysis
performed by calculating Pearsons correlation coefficient revealed
clustering of the cell lines treated with Gal-9 in vitro;
the miRNA expression patterns of the treated cells were distinct
from the patterns of the untreated cell lines (Fig. 6).
Discussion
Based on the results of the present study, Gal-9
suppresses the cell proliferation and tumor growth of human EAC
cell lines in vitro. The antitumor effects of Gal-9 on T
cell homeostasis, cell aggregation and metastasis are well known
(13,14). Additionally, Gal-9 inhibits the
proliferation of hematologic malignancies, such as multiple myeloma
(17) and chronic myeloid leukemia
(18) and significantly retards the
growth of myeloma xenografts in mice (17). Furthermore, cell surface-associated
Gal-9 triggers the aggregation of melanoma cells, which is
indicative of the Gal-9-mediated activation of cellular adhesion
and inhibition of cell detachment (19,20).
Although Gal-9 may suppress the proliferation and tumor growth of
hematologic malignancies in vitro and in vivo, Gal-9
exerts different effects on solid malignancies. For example, breast
cancer cell lines with high endogenous Gal-9 levels exhibit a
strong tendency to aggregate, whereas cells with low Gal-9 levels
do not (28). Importantly, ectopic
expression of endogenous Gal-9 and treatment with recombinant Gal-9
trigger the formation of tight cellular clusters (19,28).
Therefore, Gal-9 directly suppresses cell proliferation and tumor
growth and has therapeutic potential for several solid tumors.
Recombinant Gal-9 induces apoptosis and cell death
through apoptotic signaling pathways (17,18).
In multiple myeloma cells, apoptotic signaling is induced via the
activation of the MAP kinases JNK and p38 (17). Additionally, Gal-9 induces the
pro-apoptotic Bcl-2 family member Noxa via activation of
transcription factor 3, leading to the death of chronic myeloma
cells (18). Moreover, various
hematologic malignancies are sensitive to apoptotic elimination by
recombinant Gal-9. Cleavage of cytokeratin 18 occurs as an early
event during apoptosis following the activation of apoptosis
executioners, particularly effector caspases, but remains intact
during other types of cell death, such as autophagy and necrosis
(29).
Gal-9 induces apoptosis through both
caspase-dependent and caspase-independent mechanisms (17,20).
In previous studies, Gal-9 increased the levels of cleaved
cytokeratin-18 in various cancer cell lines in a dose- and
time-dependent manner (21–23,31).
Based on our data, Gal-9 also increased CCK18 levels in the three
EAC cell lines. Additionally, Gal-9 increased the activated
caspase-3, caspase-9 and PARP levels. The death receptor and
mitochondrial pathways are the two major pathways that initiate
apoptotic responses, and caspase-3 is the key executioner caspase
in both pathways (32). The present
study revealed that the apoptosis of EAC cells was initiated
through caspase-independent pathways. Recently, Wiersma et
al (30) showed an association
of Gal-9 with impaired lysosomal function and fatal frustrated
autophagy. Gal-9 converts LC3-I to LC3-II, whereas SQSTM1/p62 is
increased after Gal-9 treatment for 24–48 h. Moreover, the
inhibition of autophagosome-lysosome fusion and LC3-II-SQSTM1/p62
accumulation by the lysosomal inhibitor chloroquine has previously
been reported. Combination therapies using autophagy inhibitors and
standard chemotherapies have been proposed for many cancer types,
and these findings indicate that Gal-9 may have a synergic effect
in such a combination therapy for acquired therapeutic
resistance.
The expression levels of cell cycle-related proteins
were unchanged or only slightly altered 48 h after the addition of
Gal-9. Additionally, the results of the flow cytometric analysis
revealed no effects of Gal-9 on the G0 to G1 transition in EAC
cells in vitro. Thus, the antitumor effects of Gal-9 may not
be related to a reduction in the levels of various cell
cycle-related proteins.
IL-8 in tumors and the tumor microenvironment
contributes to tumor progression by regulating angiogenesis and
cancer cell growth and survival (33). In patients with esophageal cancer,
the elevated expression of IL-8 and its receptor CXCR-2 has been
associated with a poor prognosis (34). Based on our data, Gal-9 increased
IL-8 expression in Gal-9-treated SK-GT4 cells. Acquired Gal-9
resistance in EAC cells may be attributable to Gal-9-induced IL-8
expression; thus, the applicability of Gal-9 for EAC treatment may
be limited to the tumor microenvironment and angiogenesis.
Conversely, the levels of 49 pRTKs did not change following
treatment of the human EAC cell lines with Gal-9.
The miRNAs associated with the antitumor effects of
Gal-9 were assessed using miRNA expression arrays. The cluster
analysis clearly showed the effects of Gal-9 treatment on the miRNA
expression levels in cancer cells. We identified 41 miRNAs that
were differentially expressed in the Gal-9-treated EAC cells. These
miRNAs are important candidates to gauge the effectiveness of Gal-9
treatment and provide insights into the molecular basis of
Gal-9-mediated antitumor effects, particularly those mediated by
miRNAs.
miR-200c expression was upregulated in
hepatocellular carcinoma (35) and
ovarian cancer (36) tissues
compared with their respective normal tissues. Additionally, the
overexpression of miR-200c in esophageal cancer was associated with
unfavorable responses to chemotherapy and poor prognoses (37) because this miRNA supported tumor
growth by directly suppressing PPP2R1 and promoting Akt activation
(38). The present study was not
able to determine whether miR-200c acted as an oncogenic or
tumor-suppressive molecule. However, Gal-9 treatment downregulated
miR-200c expression in EAC cells, which may be associated with the
antitumor effects of Gal-9.
In conclusion, Gal-9 suppresses human EAC cell
proliferation, possibly by inducing apoptosis in a miRNA-dependent
manner.
Acknowledgements
We thank Ms. Noriko Murao and Ms. Kana Ogawa for
providing technical assistance.
Glossary
Abbreviations
Abbreviations:
|
Gal-9
|
galectin-9
|
|
EAC
|
esophageal adenocarcinoma
|
|
CRDs
|
carbohydrate recognition domains
|
|
miRNAs
|
microRNAs
|
|
CCK-8
|
Cell Counting kit-8
|
|
IL-8
|
interleukin-8
|
|
phospho-RTKs
|
phosphorylated receptor tyrosine
kinases
|
|
CCK18
|
caspase-cleaved cytokeratin 18
|
References
|
1
|
Gaur P, Hunt CR and Pandita TK: Emerging
therapeutic targets in esophageal adenocarcinoma. Oncotarget.
7:48644–48655. 2016.PubMed/NCBI
|
|
2
|
Edgren G, Adami HO, Weiderpass E and Nyrén
O: A global assessment of the oesophageal adenocarcinoma epidemic.
Gut. 62:1406–1414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rubenstein JH and Shaheen NJ:
Epidemiology, diagnosis, and management of esophageal
adenocarcinoma. Gastroenterology. 149:302–17.e1. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pennathur A, Farkas A, Krasinskas AM,
Ferson PF, Gooding WE, Gibson MK, Schuchert MJ, Landreneau RJ and
Luketich JD: Esophagectomy for T1 esophageal cancer: Outcomes in
100 patients and implications for endoscopic therapy. Ann Thorac
Surg. 87:1048–1054, discussion 1054–1055. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matsumoto R, Matsumoto H, Seki M, Hata M,
Asano Y, Kanegasaki S, Stevens RL and Hirashima M: Human ecalectin,
a variant of human galectin-9, is a novel eosinophil
chemoattractant produced by T lymphocytes. J Biol Chem.
273:16976–16984. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsushita N, Nishi N, Seki M, Matsumoto
R, Kuwabara I, Liu FT, Hata Y, Nakamura T and Hirashima M:
Requirement of divalent galactoside-binding activity of
ecalectin/galectin-9 for eosinophil chemoattraction. J Biol Chem.
275:8355–8360. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matsumoto R, Hirashima M, Kita H and
Gleich GJ: Biological activities of ecalectin: A novel
eosinophil-activating factor. J Immunol. 168:1961–1967. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saita N, Goto E, Yamamoto T, Cho I,
Tsumori K, Kohrogi H, Maruo K, Ono T, Takeya M, Kashio Y, et al:
Association of galectin-9 with eosinophil apoptosis. Int Arch
Allergy Immunol. 128:42–50. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asakura H, Kashio Y, Nakamura K, Seki M,
Dai S, Shirato Y, Abedin MJ, Yoshida N, Nishi N, Imaizumi T, et al:
Selective eosinophil adhesion to fibroblast via IFN-gamma-induced
galectin-9. J Immunol. 169:5912–5918. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dai SY, Nakagawa R, Itoh A, Murakami H,
Kashio Y, Abe H, Katoh S, Kontani K, Kihara M, Zhang SL, et al:
Galectin-9 induces maturation of human monocyte-derived dendritic
cells. J Immunol. 175:2974–2981. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nobumoto A, Oomizu S, Arikawa T, Katoh S,
Nagahara K, Miyake M, Nishi N, Takeshita K, Niki T, Yamauchi A, et
al: Galectin-9 expands unique macrophages exhibiting plasmacytoid
dendritic cell-like phenotypes that activate NK cells in
tumor-bearing mice. Clin Immunol. 130:322–330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wiersma VR, De Bruyn M, Helfrich W and
Bremer E: Therapeutic potential of Galectin-9 in human disease. Med
Res Rev. 33 Suppl 1:E102–E126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fujihara S, Mori H, Kobara H, Rafiq K,
Niki T, Hirashima M and Masaki T: Galectin-9 in cancer therapy.
Recent Pat Endocr Metab Immune Drug Discov. 7:130–137. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kashio Y, Nakamura K, Abedin MJ, Seki M,
Nishi N, Yoshida N, Nakamura T and Hirashima M: Galectin-9 induces
apoptosis through the calcium-calpain-caspase-1 pathway. J Immunol.
170:3631–3636. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu LH, Nakagawa R, Kashio Y, Ito A, Shoji
H, Nishi N, Hirashima M, Yamauchi A and Nakamura T:
Characterization of galectin-9-induced death of Jurkat T cells. J
Biochem. 141:157–172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kobayashi T, Kuroda J, Ashihara E, Oomizu
S, Terui Y, Taniyama A, Adachi S, Takagi T, Yamamoto M, Sasaki N,
et al: Galectin-9 exhibits anti-myeloma activity through JNK and
p38 MAP kinase pathways. Leukemia. 24:843–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuroda J, Yamamoto M, Nagoshi H, Kobayashi
T, Sasaki N, Shimura Y, Horiike S, Kimura S, Yamauchi A, Hirashima
M, et al: Targeting activating transcription factor 3 by Galectin-9
induces apoptosis and overcomes various types of treatment
resistance in chronic myelogenous leukemia. Mol Cancer Res.
8:994–1001. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kageshita T, Kashio Y, Yamauchi A, Seki M,
Abedin MJ, Nishi N, Shoji H, Nakamura T, Ono T and Hirashima M:
Possible role of galectin-9 in cell aggregation and apoptosis of
human melanoma cell lines and its clinical significance. Int J
Cancer. 99:809–816. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wiersma VR, De Bruyn M, van Ginkel RJ,
Sigar E, Hirashima M, Niki T, Nishi N, Samplonius DF, Helfrich W
and Bremer E: The glycan-binding protein galectin-9 has direct
apoptotic activity toward melanoma cells. J Invest Dermatol.
132:2302–2305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fujita K, Iwama H, Sakamoto T, Okura R,
Kobayashi K, Takano J, Katsura A, Tatsuta M, Maeda E, Mimura S, et
al: Galectin-9 suppresses the growth of hepatocellular carcinoma
via apoptosis in vitroin vivo. Int J Oncol. 46:2419–2430.
2015.PubMed/NCBI
|
|
22
|
Kobayashi K, Morishita A, Iwama H, Fujita
K, Okura R, Fujihara S, Yamashita T, Fujimori T, Kato K, Kamada H,
et al: Galectin-9 suppresses cholangiocarcinoma cell proliferation
by inducing apoptosis but not cell cycle arrest. Oncol Rep.
34:1761–1770. 2015.PubMed/NCBI
|
|
23
|
Tadokoro T, Morishita A, Fujihara S, Iwama
H, Niki T, Fujita K, Akashi E, Mimura S, Oura K, Sakamoto T, et al:
Galectin-9: An anticancer molecule for gallbladder carcinoma. Int J
Oncol. 48:1165–1174. 2016.PubMed/NCBI
|
|
24
|
Nishi N, Itoh A, Fujiyama A, Yoshida N,
Araya S, Hirashima M, Shoji H and Nakamura T: Development of highly
stable galectins: Truncation of the linker peptide confers
protease-resistance on tandem-repeat type galectins. FEBS Lett.
579:2058–2064. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schutte B, Henfling M, Kölgen W, Bouman M,
Meex S, Leers MP, Nap M, Björklund V, Björklund P, Björklund B, et
al: Keratin 8/18 breakdown and reorganization during apoptosis. Exp
Cell Res. 297:11–26. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: Procedure and some applications. Proc Natl
Acad Sci USA. 76:4350–4354. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Irie A, Yamauchi A, Kontani K, Kihara M,
Liu D, Shirato Y, Seki M, Nishi N, Nakamura T, Yokomise H, et al:
Galectin-9 as a prognostic factor with antimetastatic potential in
breast cancer. Clin Cancer Res. 11:2962–2968. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kramer G, Erdal H, Mertens HJ, Nap M,
Mauermann J, Steiner G, Marberger M, Bivén K, Shoshan MC and Linder
S: Differentiation between cell death modes using measurements of
different soluble forms of extracellular cytokeratin 18. Cancer
Res. 64:1751–1756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wiersma VR, De Bruyn M, Wei Y, van Ginkel
RJ, Hirashima M, Niki T, Nishi N, Zhou J, Pouwels SD, Samplonius
DF, et al: The epithelial polarity regulator LGALS9/galectin-9
induces fatal frustrated autophagy in KRAS mutant colon carcinoma
that depends on elevated basal autophagic flux. Autophagy.
11:1373–1388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takano J, Morishita A, Fujihara S, Iwama
H, Kokado F, Fujikawa K, Fujita K, Chiyo T, Tadokoro T, Sakamoto T,
et al: Galectin-9 suppresses the proliferation of gastric cancer
cells in vitro. Oncol Rep. 35:851–860. 2016.PubMed/NCBI
|
|
32
|
Fulda S: Targeting apoptosis for
anticancer therapy. Semin Cancer Biol. 31:84–88. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Waugh DJ and Wilson C: The interleukin-8
pathway in cancer. Clin Cancer Res. 14:6735–6741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ogura M, Takeuchi H, Kawakubo H, Nishi T,
Fukuda K, Nakamura R, Takahashi T, Wada N, Saikawa Y, Omori T, et
al: Clinical significance of CXCL-8/CXCR-2 network in esophageal
squamous cell carcinoma. Surgery. 154:512–520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ladeiro Y, Couchy G, Balabaud C,
Bioulac-Sage P, Pelletier L, Rebouissou S and Zucman-Rossi J:
MicroRNA profiling in hepatocellular tumors is associated with
clinical features and oncogene/tumor suppressor gene mutations.
Hepatology. 47:1955–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iorio MV, Visone R, Di Leva G, Donati V,
Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, et
al: MicroRNA signatures in human ovarian cancer. Cancer Res.
67:8699–8707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tanaka K, Miyata H, Yamasaki M, Sugimura
K, Takahashi T, Kurokawa Y, Nakajima K, Takiguchi S, Mori M and
Doki Y: Circulating miR-200c levels significantly predict response
to chemotherapy and prognosis of patients undergoing neoadjuvant
chemotherapy for esophageal cancer. Ann Surg Oncol. 20 Suppl
3:S607–S615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hamano R, Miyata H, Yamasaki M, Kurokawa
Y, Hara J, Moon JH, Nakajima K, Takiguchi S, Fujiwara Y, Mori M, et
al: Overexpression of miR-200c induces chemoresistance in
esophageal cancers mediated through activation of the Akt signaling
pathway. Clin Cancer Res. 17:3029–3038. 2011. View Article : Google Scholar : PubMed/NCBI
|