Introduction
Angiogenesis refers to the formation of new blood
vessels from existing capillaries or via intravenous development.
The complete process of angiogenesis involves a variety of cells
and molecules that play roles in vascular basement membrane
degradation as well as in the subsequent vascular endothelial cell
activation, proliferation, migration and reconstruction (1). In the process of tumour development,
angiogenesis is a necessary prerequisite and is regulated by a
balance between pro-angiogenic factors, such as vascular
endothelial cell growth factor (VEGF), basic fibroblast growth
factor (bFGF) and angiopoietin, and anti-angiogenic factors, such
as tumstatin, endostatin and thrombospondin-1 (2–4).
Hepatocellular carcinoma (HCC) is one of the most common types of
carcinoma and is characterized by an enriched blood supply. The
progression of angiogenesis is vital for tumour occurrence and
development and is closely associated with HCC metastasis, invasion
and resistance to therapy (5,6). Thus,
several anti-angiogenic drugs (such as sorafenib) have been
recommended for clinical applications as treatment options for
patients with advanced stage HCC (7,8).
Pigment epithelial-derived factor (PEDF) is a
multifunctional glycoprotein that belongs to a family of
non-inhibitory serpins. Among naturally occurring anti-angiogenic
factors, PEDF is considered an effective angiogenesis inhibitor
(9,10). However, PEDF comprises multiple
functional fragments that are responsible for several functions,
such as inhibition of angiogenesis and promotion of survival and
neuro-differentiation. Previous studies have demonstrated that the
anti-angiogenic functional fragment of PEDF is located at the
NH2-terminal surface epitope (the 34-mer amino acid residues 24–57,
PEDF-34mer), while the pro-survival and neuro-differentiating
functional fragments are located at the adjacent epitope (44-mer
residues 58–101, PEDF 44-mer) (11–13).
Studies have revealed that PEDF inhibits the development of several
malignant tumours, such as lung carcinoma, osteosarcoma and
pancreatic carcinoma (14–16). However, although the in vivo
overexpression of full-length PEDF is beneficial for inhibiting
tumour growth, its application in clinical treatments is limited
due to its low stability and immune antigenicity. Therefore, short
but stable peptides derived from PEDF that are as effective at
inhibiting angiogenesis as the full-length PEDF fragments could
have more practical value in clinical practice. The P18 peptide is
an angioinhibitory epitope of PEDF 34-mer (18-mer residues 40–57)
that has been proposed to be a bioactive anti-angiogenic fragment.
A previous study demonstrated that the P18 peptide exerted activity
analogous to that of full-length PEDF in prostate and renal
carcinoma (17). However, the
effects of the P18 peptide on angiogenesis in HCC and its
applicability in tumour therapy remain unclear.
VEGF is a potent pro-angiogenic cytokine that is
also known to promote proliferation and survival of endothelial
cells (ECs) as well as vascular permeability (18,19).
VEGF is expressed in vascularized tissues and is critical for
normal and pathological angiogenesis. Substantial evidence has
implicated VEGF in the induction of tumour proliferation,
metastasis and angiogenesis (3,20,21).
VEGF165, the predominant isoform of VEGF in humans, signals through
three receptors: fms-like tyrosine kinase (flt-1, also VEGFR1), KDR
gene product (KDR, also VEGFR2) and the flt4 gene product (flt-4,
also VEGFR3). Among these three receptors, VEGFR2 is a major
receptor for VEGF-induced signalling in endothelial cells (3,22–24).
Previous studies have detected VEGFR2 not only in vascular
endothelial cells but also its overexpression in many types of
malignant solid tumours (25). Upon
binding with VEGF, VEGFR2 undergoes autophosphorylation and becomes
activated. Phosphorylation of Tyr1175 allows binding with the p85
subunit of the PI-3 kinase (PI3K), which leads to activation of the
PI3K/Akt signalling pathway (26).
This VEGFR2 signalling is necessary for the execution of
VEGF-stimulated proliferation, chemotaxis and sprouting as well as
for the survival of cultured endothelial cells in vitro and
angiogenesis in vivo (20,24).
Existing evidence has indicated that VEGFR2 is a
target of PEDF (9,27). However, whether the P18 peptide can
block VEGF/VEGFR2 signal transduction and ultimately lead to the
inhibition of angiogenesis in HCC remains unclear. Thus, we
designed the present study to identify the mechanism by which the
P18 peptide inhibits angiogenesis in HCC. We observed that the P18
peptide counteracted the bioactivity of VEGF and suppressed cell
activity by reducing the secretion of cadherins (VE-cadherin and
E-cadherin) and matrix metalloproteinases (matrix
metalloproteinase-2, MMP-2 and MMP-9) in both human umbilical vein
and microvascular endothelial cells (HUVECs) and HepG2 hepatoma
cells in vitro, which resulted in the suppression of
invasiveness and pro-angiogenesis of endothelial cells. Moreover,
our xenograft tumour model also provided evidence that the P18
peptide downregulates the phosphorylation of VEGFR2 and inhibits
angiogenesis of HCC in vivo. We demonstrated that the P18
peptide may act on ECs by modulating the VEGF/VEGFR2 signalling
pathway and it induced PI3K/Akt cascades, which would lead to
apoptosis of ECs and reduced neovascularization.
Materials and methods
Cell lines and culture
Human umbilical vein and microvascular endothelial
cells (HUVECs) and human HCC cell line HepG2 cells were purchased
from the Typical Culture Reserve Center of China (Shanghai, China)
and cultured under 5% CO2 in ECM-2 medium (ScienCell
Research Laboratories, Carlsbad, CA, USA) supplemented with 5%
foetal bovine serum (FBS, Gibco, Grand Island, NY, USA), 100 U/ml
penicillin, 100 µg/ml streptomycin and 1% endothelial cell growth
supplement (ECGS, ScienCell Research Laboratories). The human HCC
cell line HepG2 was obtained from the American Type Culture
Collection (Rockville, MD, USA) and cultured in Dulbecco's modified
Eagle's medium (DMEM, Hyclone, Thermo Fisher Scientific Inc.,
Logan, UT, USA) supplemented with 10% FBS (Gibco), 100 U/ml
penicillin and 100 µg/ml streptomycin. A hypoxia incubator was used
to simulate hypoxic conditions (1% O2, 5% CO2
and 94% N2).
Antibodies and reagents
Antibodies against VEGFR2, phosphorylated (p)-VEGFR2
(Tyr1175), PI3K, p-PI3K p85 (Tyr458)/p55 (Tyr199), Akt, p-Akt
(Ser473), Bax, Bcl-2, caspase-3, cleaved caspase-3, MMP-2, MMP-9
and CD31 were purchased from Cell Signalling Technology (Danvers,
MA, USA). Antibodies against VEGF, VE-cadherin, E-cadherin, Ki67,
GAPDH and β-actin were purchased from Abcam (Cambridge, UK).
Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and
HRP-conjugated mouse IgG were purchased from Beyotime Institute of
Biotechnology (Jiangsu, China). Recombinant P18 peptide (>95%
purity) was purchased from GL Biochem Ltd. (Shanghai, China) and
characterized by mass spectrometry. The peptides were acetylated at
their NH2 termini and amidated at their COOH termini for stability.
SU1498, a selective VEGFR2 inhibitor, was acquired from Abcam.
Recombinant human VEGF (VEGF165) and recombinant human IGF-1 were
obtained from PeproTech (Rocky Hill, NJ, USA). Matrigel was
purchased from BD Biosciences (San Jose, CA, USA).
Cell viability assay
HUVECs (5×103/well) or HepG2 cells
(5×103/well) were seeded in three 96-well plates. After
cell attachment, the supernatants in the plates was replaced with
ECM containing gradient concentrations of the P18 peptide (10, 20,
40, 80, 160, 320, 640 and 1280 nM). The plates were numbered as
plate 1, plate 2 and plate 3 and incubated at 37°C under 5%
CO2. Viable cells were quantified by Cell Counting Kit-8
(CCK-8, Dojindo Molecular Technologies, Kumamoto, Japan) assay at
various time-points (plate 1:24 h, plate 24:8 h and plate 3:72 h).
The optical density (OD) at 450 nm was measured using a Spectra Max
190 (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
The cells or frozen tumour samples were lysed in
cold RIPA lysis buffer (Beyotime, Beijing, China) with 1 nM
phenylmethylsufonyl fluoride. A BCA Protein Assay kit (Beyotime)
was used to measure the concentration of the protein samples. Total
protein (20–25 µg) was separated via SDS-PAGE gels and transferred
to PVDF membranes (Millipore, Billerica, MA, USA). The membranes
were blocked with 5% skimmed milk for 2 h at room temperature and
then incubated with primary antibodies at 4°C overnight according
to the manufacturer's instructions. The membranes were then
incubated with HRP-conjugated anti-mouse or rabbit IgG secondary
antibody (Beyotime) for 2 h at room temperature, followed by
detection using enhanced chemiluminescence (ECL) immunoblotting
detection reagents (Millipore). Protein band intensities were
quantified via densitometric analysis using ImageJ software
(National Institutes of Health, Bethesda, MD, USA).
Xenograft tumour growth assay
HepG2 cells (5×106/0.1 ml) in PBS were
inoculated into the dorsal area near front leg of 4-week-old BALB/c
nude mice (Shanghai Laboratory Animal Company, Shanghai, China).
The mice were observed until tumours reached a volume of 100
mm3. Then, the mice were randomized into three groups (5
in each group) and marked. The two experimental groups were
administered an intraperitoneal injection of P18 peptide (dissolved
with 0.9% normal saline) at doses of 0.1 mg/kg (dose/mouse body
weight) or 0.5 mg/kg. The control group was treated with the same
volume of 0.9% normal saline (0.9% NS). Tumour growth was monitored
by the length and width of the tumour acquired from external
measurements every 2 days. Tumour volume was determined according
to the following equation: volume (mm3) = (length ×
width2)/2. Fourteen days after the first injection, the
mice were sacrificed, and the tumours were excised and weighed. All
experiments were performed following approval by the ethics
committee of Qianfoshan Hospital.
Immunofluorescence (IF) assay
The xenogeneic tumours were frozen in liquid
nitrogen immediately after the mice were sacrificed and then frozen
in 5-µm sections. The sections were fixed in 4% paraformaldehyde
for 20 min. After blocking in 5% BSA for 1 h, the sections were
incubated at 4°C overnight with goat polyclonal anti-VEGF (Novus,
San Diego, USA) and rabbit monoclonal anti-phosphorylated VEGFR2.
Cells or mouse sections were then incubated with donkey anti-goat
FITC-labelled (Abcam) and donkey anti-rabbit TRITC-labelled (Abcam)
secondary antibodies for 2 h and were stained with DAPI (Abcam).
Fluorescent images from tissues were photographed using a light
microscope, and double immunofluorescent staining were merged using
Image-Pro Plus software.
Immunohistochemistry (IHC) assay
Tumour simples were fixed in 10% formalin, embedded
in paraffin and then processed for immunohistochemistry. The
sections were deparaffinized in graded xylene and rehydrated in
graded ethanol, and then washed with PBS 3 times. After
heat-induced antigen retrieval in citrate buffer, endogenous
peroxidase was inhibited by treatment with 3% hydrogen peroxide at
room temperature for 10 min, followed by washing 3 times with PBS.
The sections were then incubated with primary anti-VE cadherin,
anti-CD31 and anti-Ki67 antibodies overnight. After washing with
PBS, the sections were treated with horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG for 1 h at 37°C. Negative
control sections were incubated with PBS instead of the primary
antibody.
Wound-healing assay
HUVECs were seeded in 6-well plates. When the cells
were 90% confluent, a wound line was made using a 10-µl plastic
pipette tip. The cells were then incubated in serum-free ECM-2
medium with VEGF (8 ng/ml) with or without P18 peptide (0.2 µM).
The wound-healing processes were photographed at time points 0, 12
and 24 h, and the cell migration distance was quantified by
analysing the images.
Migration and invasion assay
A Transwell cell migration system (8-µm, Corning
Inc., Corning, MA, USA) coated with Matrigel was used to perform
the cell migration and invasion assays. HUVECs
(1×105/well) or HepG2 cells (5×104/well) were
added to the upper chamber of a Transwell plate, and 500 µl of
serum-free medium with or without VEGF (8 ng/ml) and the P18
peptide (0.2 µM) was added to the lower chamber. After 48 h of
incubation, the migrated cells were fixed with 95% methanol and
stained with 0.1% crystal violet for 30 min followed by washing 5
times with PBS.
Tube formation assay
Each well of 96-well plates was coated with 50 µl of
Matrigel. HUVECs (1×104/well) were seeded in the wells
following solidification of the Matrigel. The cells in the
experimental groups were treated with P18 peptide (0.2 µM), and the
control groups were treated with an equal volume of PBS. The cells
were incubated at 37°C with 5% CO2 for 6 h and then
photographed using a light microscope (Olympus, Tokyo, Japan).
Cell apoptosis assay
HUVECs (4×105/well) or HepG2 cells
(4×105/well) were treated with or without the P18
peptide and incubated for 24 h. Then, the cultured cells were
suspended in PBS, counted and resuspended with binding buffer.
Annexin V-FITC and propidium iodide (PI; NeoBioscience Ltd.,
Shenzhen, China) were used to detect the apoptosis rate of the
cells via a flow cytometry assay.
Statistical analysis
The data were analysed with SPSS software (version
17.0, SPSS China, Shanghai, China) and expressed as the mean ±
standard deviation (SD). Student's t-tests were used for
comparisons between 2 groups, and one-way analysis of variance was
used for comparisons between multiple groups. P-values <0.05
were considered to indicate statistically significant results.
Results
P18 peptide inhibits ECs proliferation
in vitro
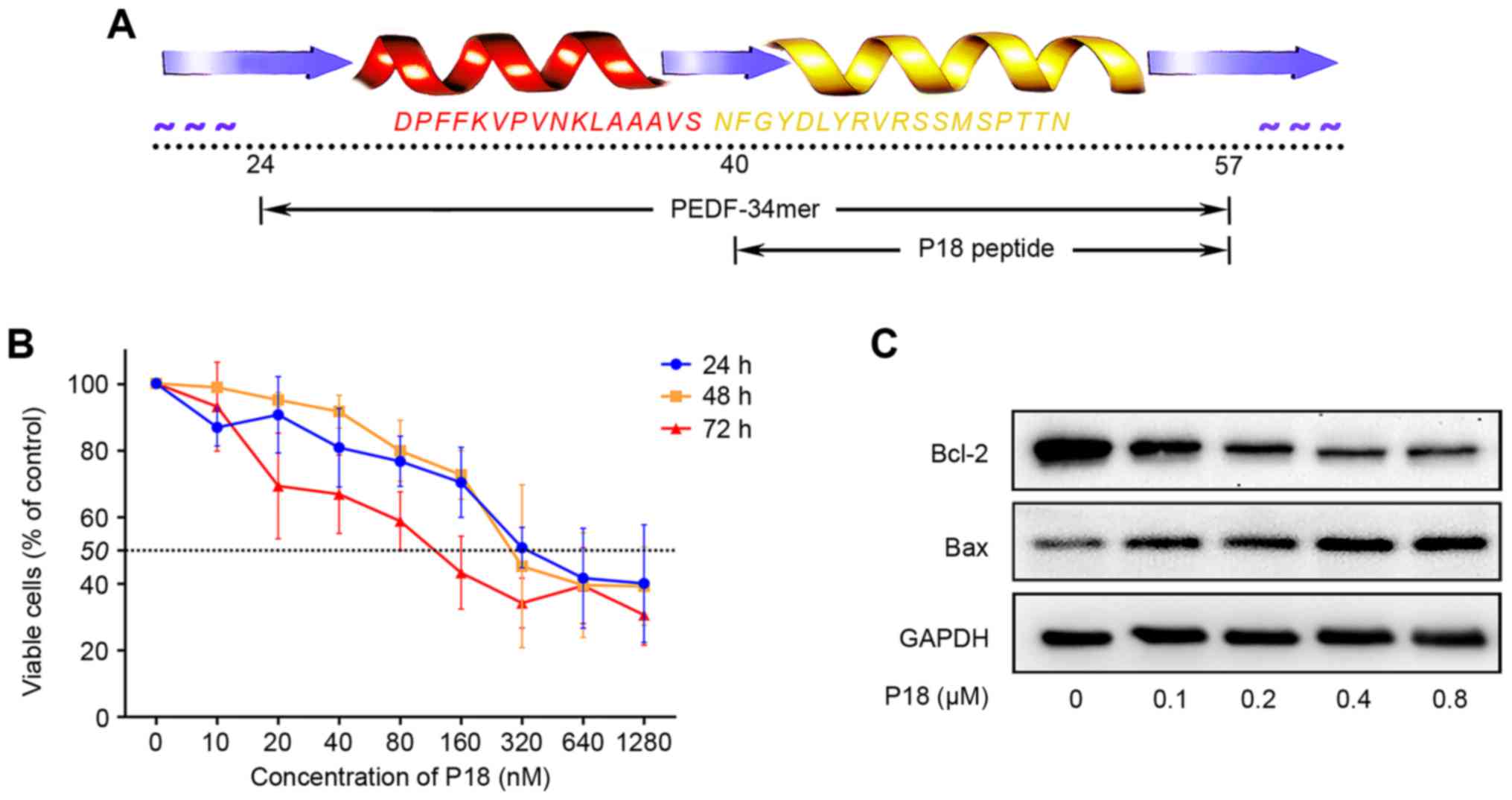
The P18 peptide was synthesized according to the
amino acid sequence shown in Fig.
1A. Dose-response analysis achieved by CCK-8 assay confirmed an
IC50 of ~320 nmol/l for the P18 peptide in vitro
(Fig. 1B). As a biomarker for the
level of apoptosis, Bcl-2 and Bax were detected by western
blotting. We treated HUVECs with different concentrations of P18
peptide and observed a dose-dependent downregulation of Bcl-2 but
upregulation of Bax (Fig. 1C).
P18 peptide suppresses tumour growth
and angiogenesis of HCC in vivo
To evaluate the effect of the P18 peptide on tumour
growth in vivo, we designed a xenograft tumour growth assay
with HepG2 cells in nude mice. Two experimental groups received the
P18 peptide at a dose of 0.1 mg/kg or 0.5 mg/kg, while the control
group received the same volume of 0.9% normal saline (0.9% NS). The
average tumour volume was 47.40% in the P18-treated (0.1 mg/kg)
group and 20.64% in the P18-treated (0.5 mg/kg) group normalized to
the control group (Fig. 2A and B).
As hallmarks of angiogenesis, expression levels of CD31 and
VE-cadherin in tumour tissues were detected by IHC analysis, which
revealed a gradual decrease in CD31 and VE-cadherin with increasing
dosage of P18 peptide (Fig. 2C-E).
In addition, western blot assays showed a significant
downregulation of expression levels of CD31 and VE-cadherin
(Fig. 2F and G).
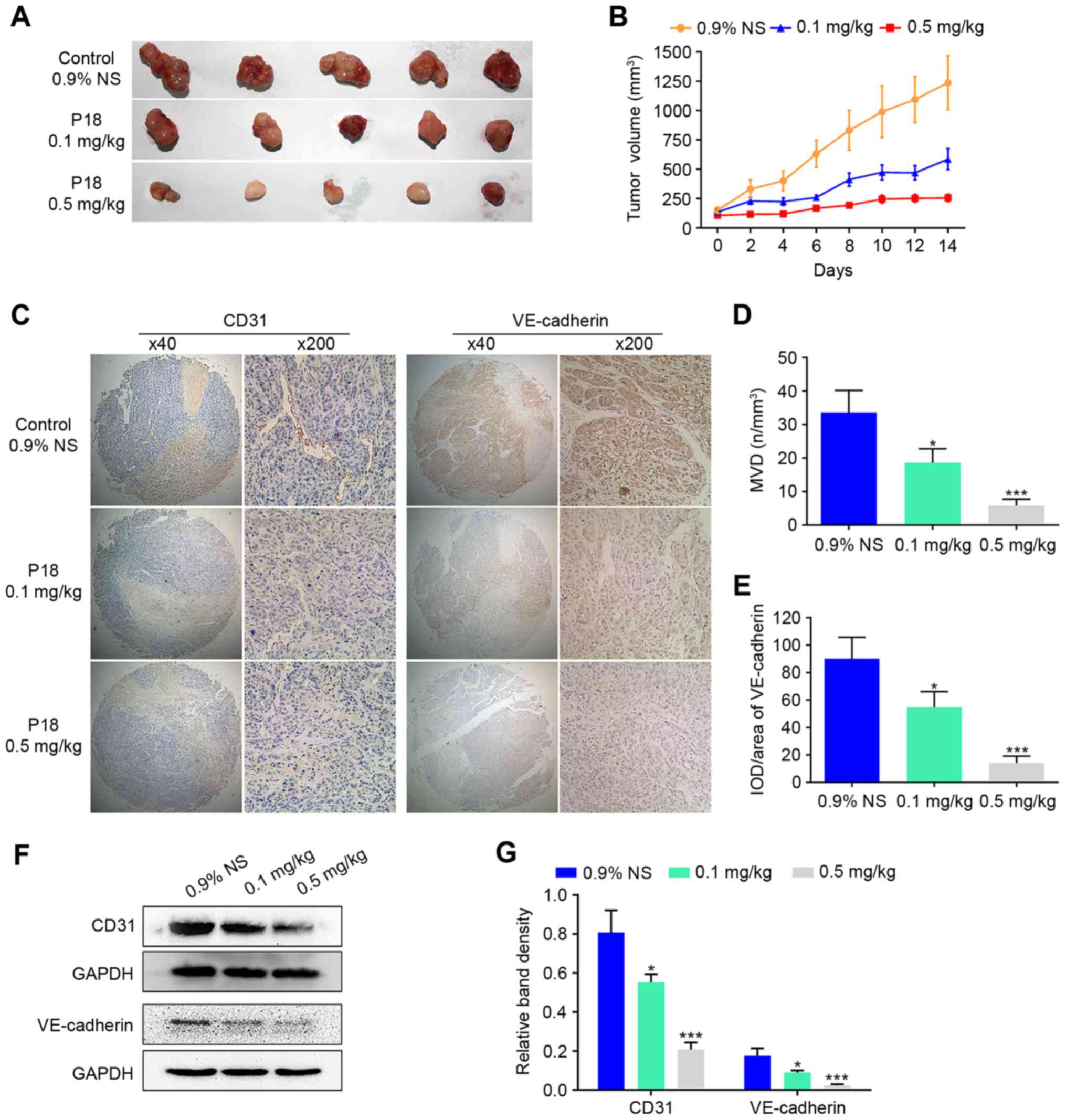 | Figure 2.The P18 peptide suppresses tumour
growth and angiogenesis of HCC in vivo. (A) Tumour tissue on
day 14 after injection with 0.9% NS or different doses of P18. (B)
Curve of tumour growth suppression is shown as tumour volume after
treatment with 0.9% NS or different doses of P18 (mean ± SD). (C)
Results of IHC staining for CD31 and VE-cadherin in tumour tissues
(magnification, ×200). (D) Data represent MVD in tumour tissues
(mean ± SD, *P<0.05, ***P<0.01). MVD was determined via IHC
staining with an endothelial-specific antibody against CD31
(magnification, ×200). (E) The P18 peptide inhibited expression of
VE-cadherin, a molecular marker of angiogenesis, in HCC tumour
tissue (magnification, ×200). The data represent IOD/Area (mean ±
SD, *P<0.05, ***P<0.01). (F) Expression of CD31 and
VE-cadherin were detected by western blot assays. (G) The density
of each band in western blot assay was quantified and normalized to
that of GAPDH (mean ± SD, *P<0.05, ***P<0.01). |
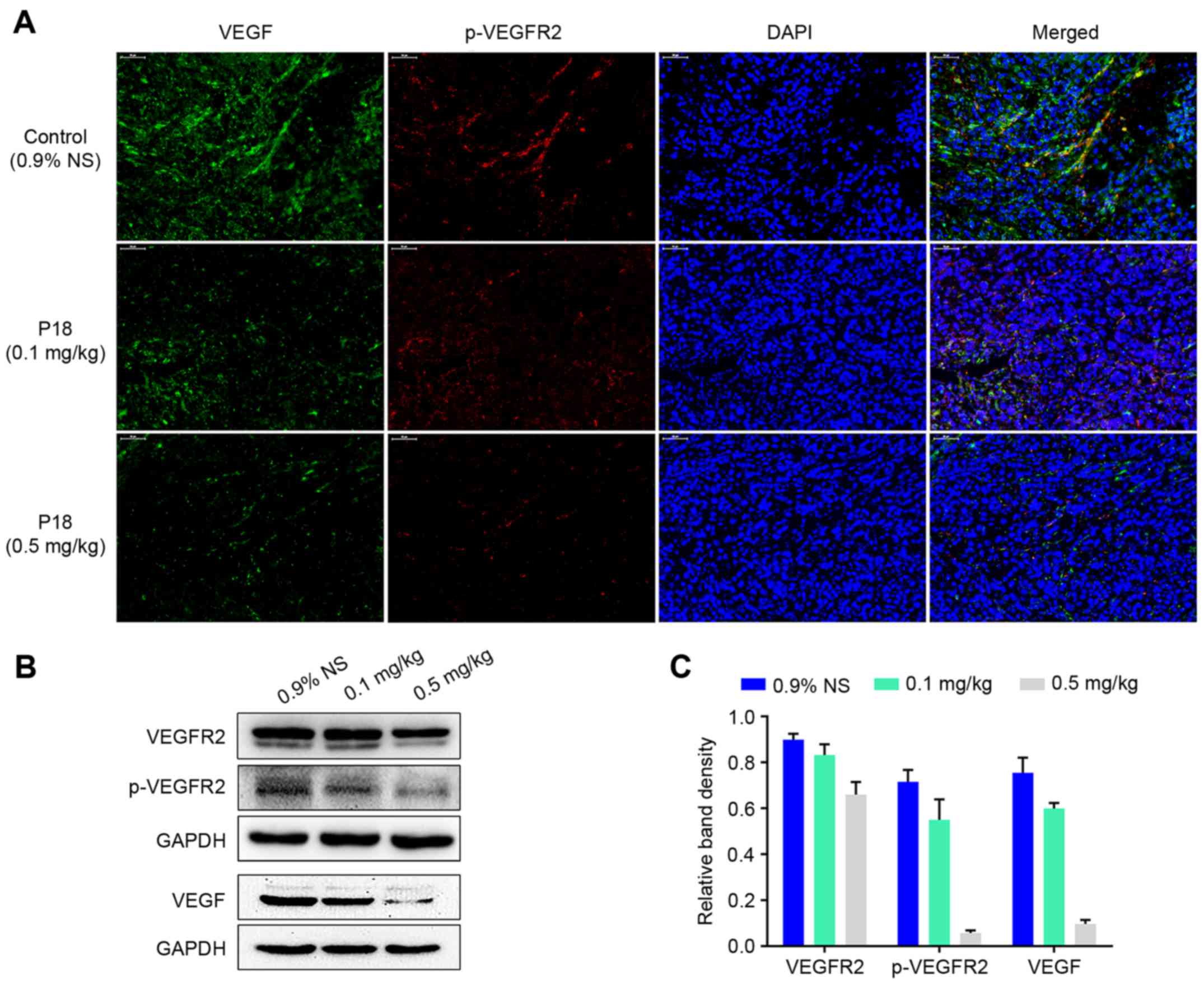
P18 peptide inhibits phosphorylation
of VEGFR2 in vivo
Expression levels of VEGF and p-VEGFR2 in tumour
tissues were detected by IF analysis. The results indicated that
there was a dose-dependent suppression in expression of VEGF
followed by a decreased phosphorylation level of VEGFR2 in
P18-treated groups (Fig. 3A).
Western blot assays also demonstrated that the P18 peptide
downregulated the expression of VEGF and suppressed the
phosphorylation of VEGFR2 in HCC tumour tissues (Fig. 3B and C).
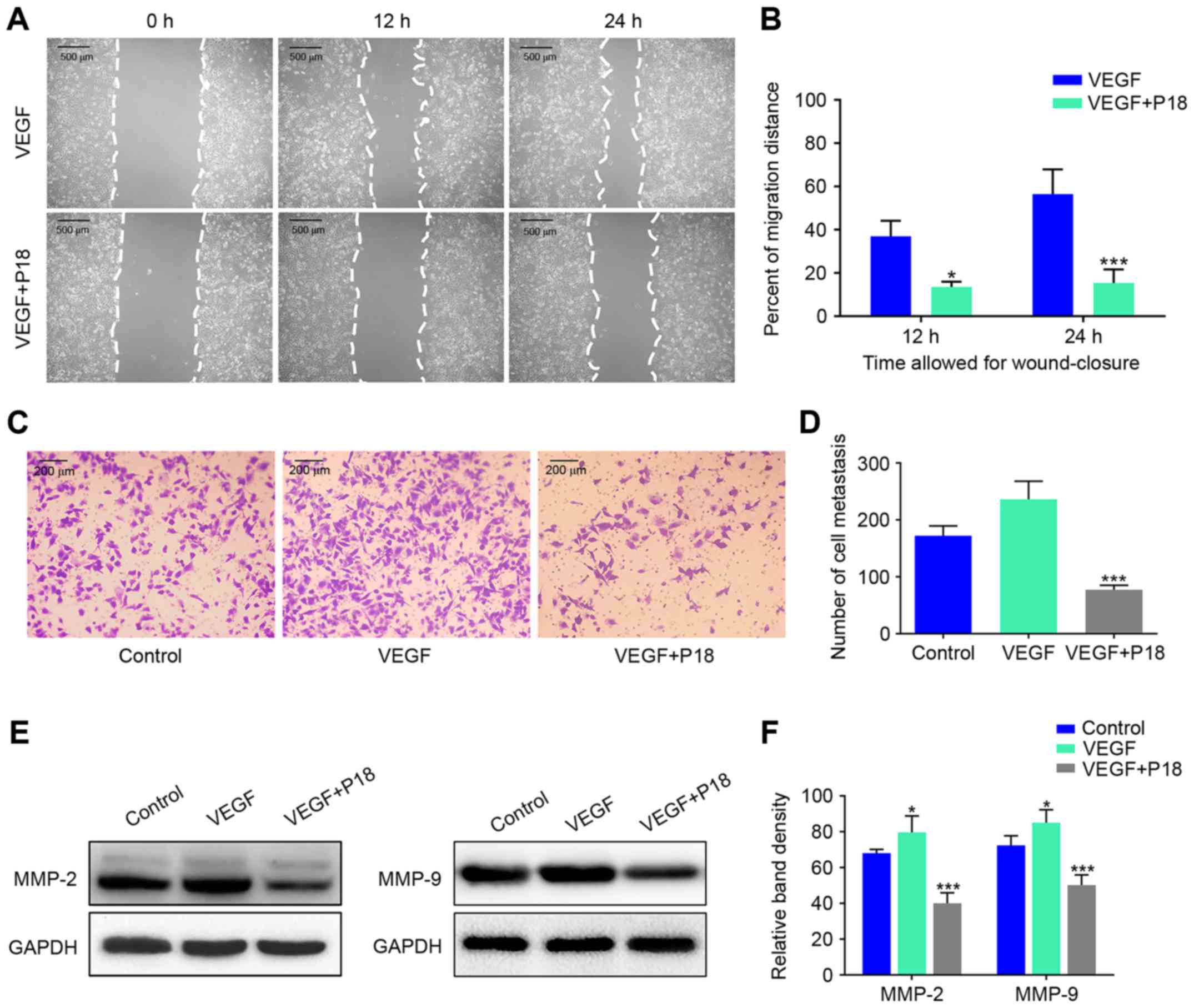
P18 peptide suppresses EC viability by
counteracting the bioactivity of VEGF in vitro
Wound healing assays and Transwell assays were
performed to investigate the effects of the P18 peptide on the
migration and invasive ability of ECs in the presence of VEGF, and
the results suggested that the P18 peptide significantly inhibited
HUVEC migration and invasion at a concentration of 0.2 µM (Fig. 4A-D). Results of western blot assays
suggested that the P18 peptide achieved this bioactivity primarily
through downregulating expression levels of MMP-2 and MMP-9 in
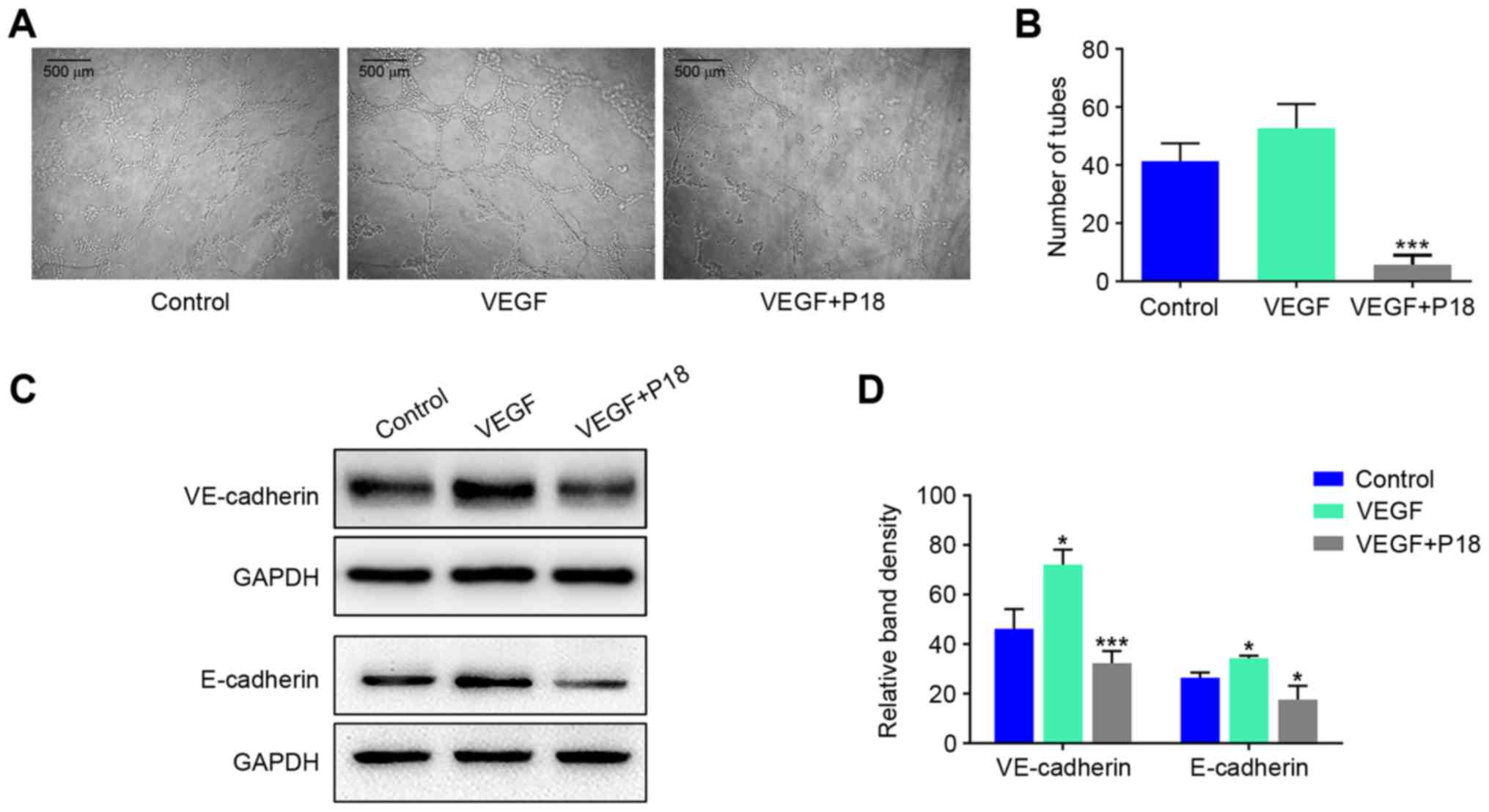
HUVECs (Fig. 4E and F). To further
explore the anti-angiogenic potency of the P18 peptide in
vitro, we seeded HUVECs on Matrigel-coated 96-well plates for 8
h using additional VEGF (8 ng/ml) as a positive control. The
results indicated that VEGF could enhance angiogenesis of HUVECs
but the P18 peptide reversed this effect and inhibited angiogenesis
in vitro (Fig. 5A and B). At
the same time, western blot assays confirmed that additional VEGF
could upregulate expression levels of VE-cadherin and E-cadherin in
HUVECs, while the P18 peptide counteracted this bioactivity
(Fig. 5C and D).
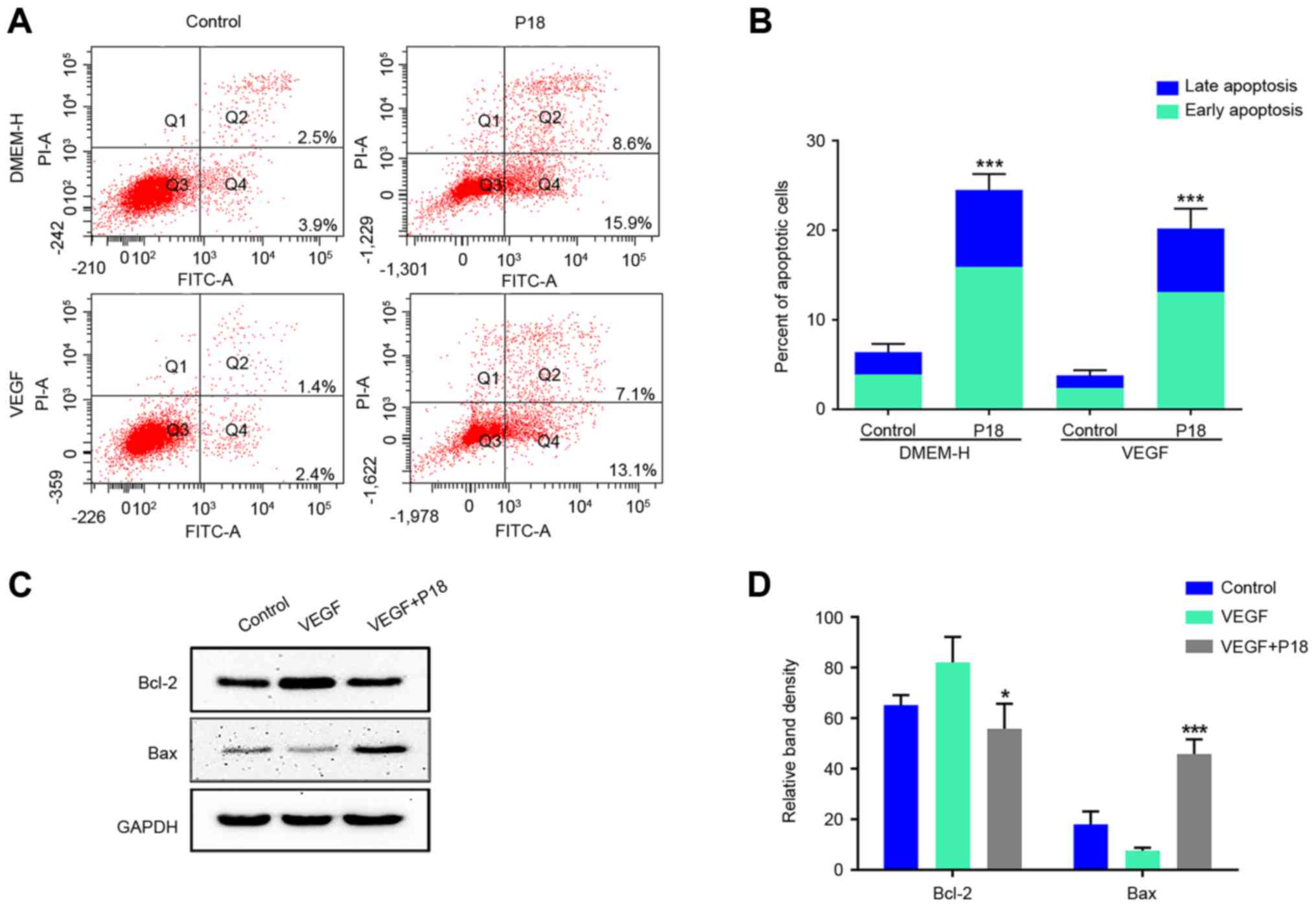
P18 peptide induces apoptosis of ECs
in vitro
Since the current study preliminarily determined an
IC50 of ~320 nmol/l for the P18 peptide in vitro,
we further cultured HUVECs in DMEM medium with or without VEGF.
After 12 h, PBS- dissolved P18 peptide (0.32 µM) was added to the
experimental group, and the cells were cultured for another 24 h.
Annexin V-FITC/PI flow cytometry and western blot assays were used
to determine the apoptosis levels of HUVECs. The results
consistently indicated that additional VEGF may suppress the
apoptosis of HUVECs and this process could be reversed by the P18
peptide (Fig. 6A-D).
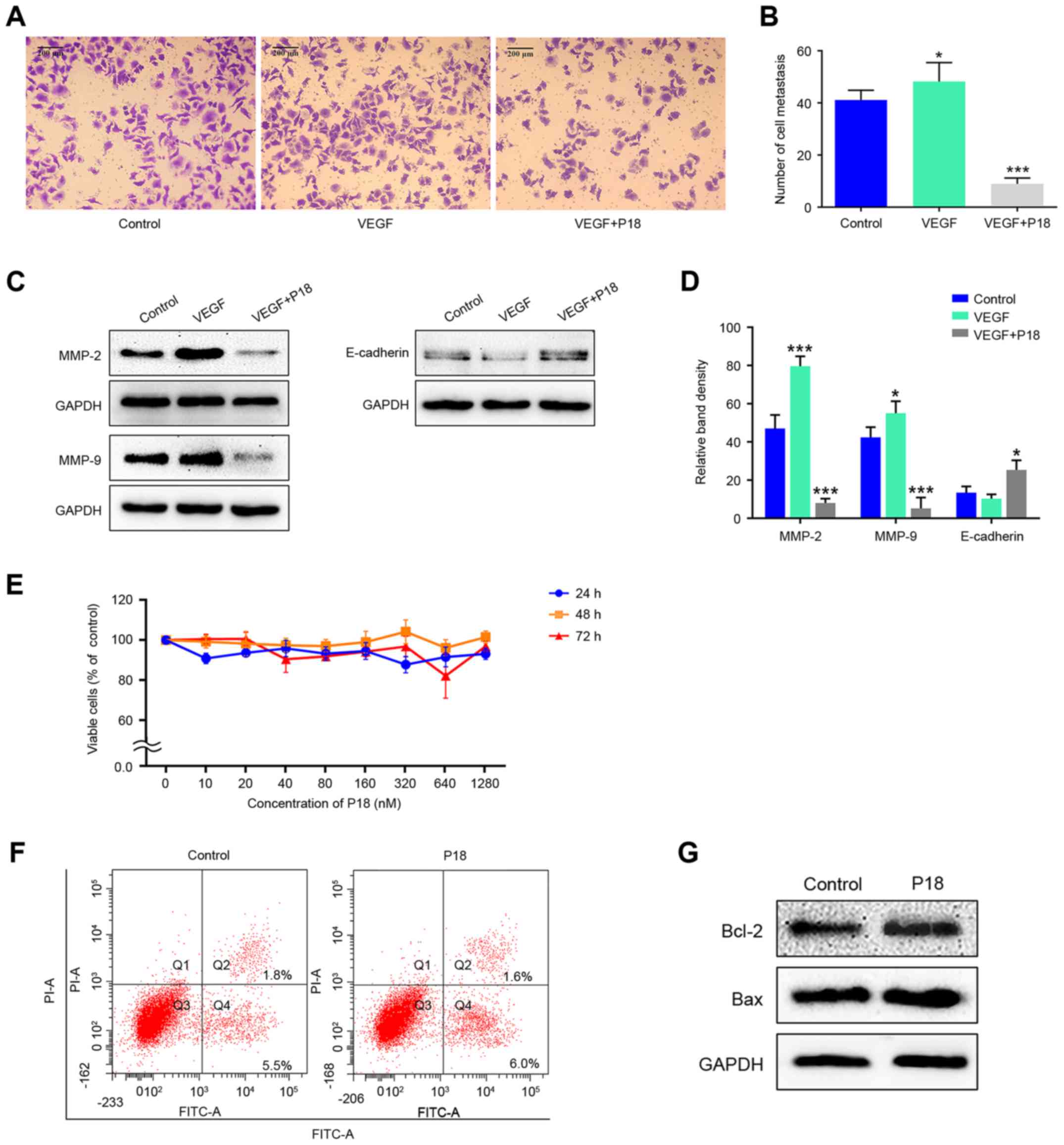
P18 peptide suppresses HepG2 cell
viability by suppressing cell migration rather than inhibiting cell
proliferation inducing apoptosis
Transwell assays were performed to investigate the
effects of the P18 peptide on the migration ability of HepG2 cells
in the presence of VEGF. Results indicated that the P18 peptide
inhibited the migration of tumour cells and counteracted the
bioactivity of VEGF in vitro (Fig. 7A and B). Expression levels of MMP-2,
MMP-9 and E-cadherin in HepG2 cells treated with VEGF (8 ng/ml) and
the P18 peptide (0.32 µM) was determined by western blot assays.
The high expression levels of MMP-2 and MMP-9 stimulated by VEGF in
HepG2 cells were downregulated by the P18 peptide while the
expression of E-cadherin was slightly upregulated in P18
peptide-treated groups, which contribute to the suppression of cell
migration (Fig. 7C and D). However,
after treatment with different concentrations of the P18 peptide
for different times, no significant differences were observed in
the cell proliferation of HepG2 cells in CCK-8 assays (Fig. 7E). In addition, the results of
Annexin V-FITC/PI flow cytometry and western blot assays showed no
significant difference in HepG2 cell apoptosis between the control
and P18 peptide-treated groups (Fig. 7F
and G). Of note, the experiments in Fig. 7E-G were performed in the absence of
VEGF but HepG2 cells were cultured in DMEM medium with 10% FBS.
Since HepG2 cells can maintain a high level of proliferation under
this condition without the need for additional VEGF, we consider
that cells in normal serum medium is sufficient as a control group
(28,29). The above results suggest that the
P18 peptide suppresses HepG2 cell viability by suppressing cell
migration rather than by inhibiting cell proliferation and inducing
apoptosis.
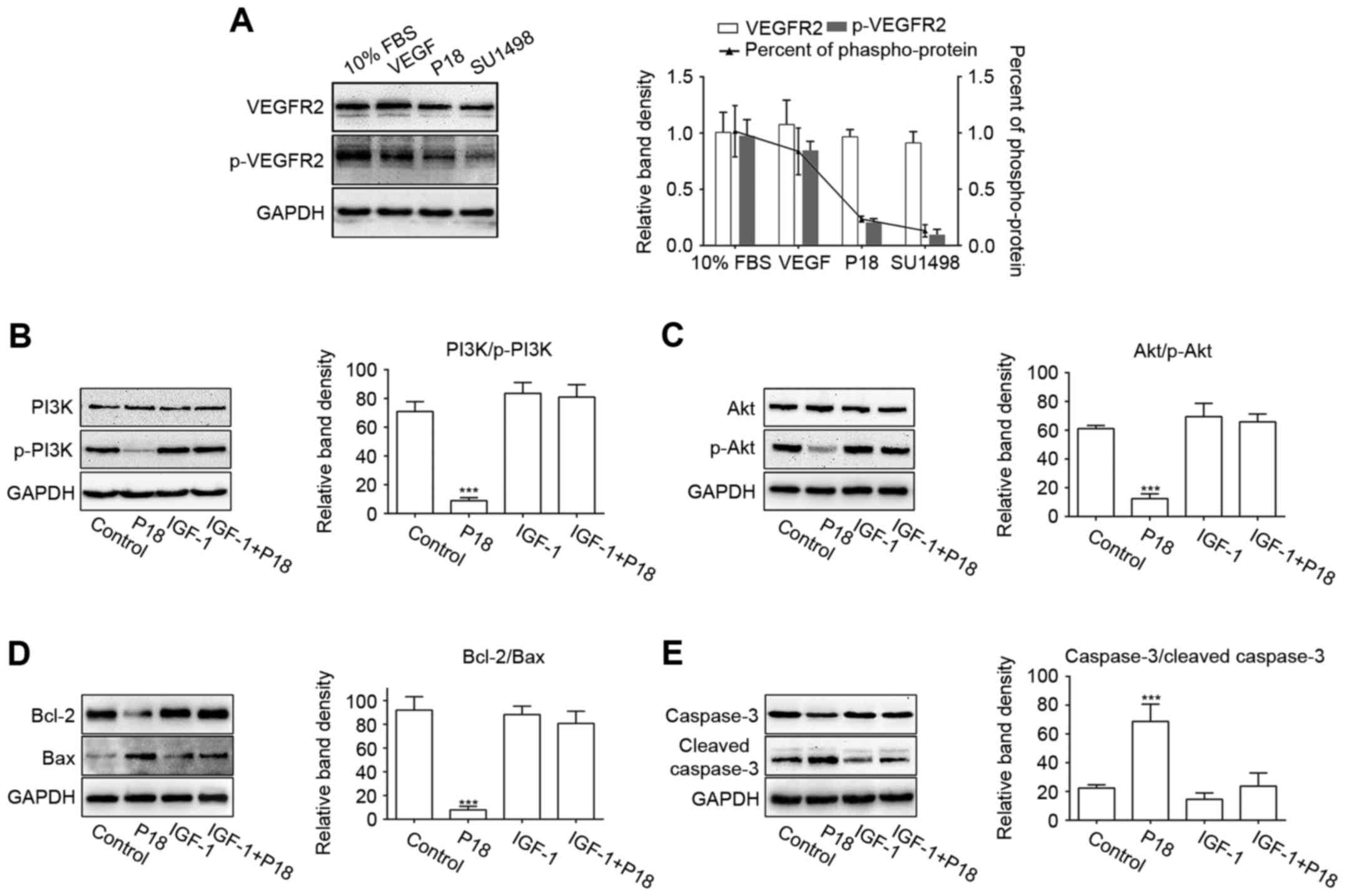
P18 peptide induces EC apoptosis via
blocking VEGF/VEGFR2 and induces the PI3K/Akt signalling
pathway
SU1498 is a selective inhibitor of VEGFR2, which is
the major receptor for VEGF-induced bioactivity in endothelial
cells (23,30). In this study, we cultured HUVECs in
serum-free medium with additional VEGF and used SU1498 as a
positive control for the P18 peptide. HUVECs cultured in medium
with 10% FBS or VEGF served as a negative control. The western blot
results showed that the P18 peptide inhibited VEGF-induced
phosphorylation of VEGFR2 in a manner similar to that of SU1498
(Fig. 8A). To further demonstrate
the mechanism by which P18 inhibits angiogenesis, we treated HUVECs
in serum-free medium for 24 h and then replaced the medium with
additional VEGF with or without the P18 peptide (0.2 µM). IGF-1, an
activator of PI3K/Akt pathway, was used as a negative control
(31). As downstream signalling
axis of VEGFR2, the activation of PI3K and Akt through
phosphorylation in the HUVECs were significantly suppressed after
treatment with the P18 peptide for 90 min, which could be reversed
with presence of IGF-1 (0.2 ng/ml) (Fig. 8B and C). After culturing with the
P18 peptide or IGF-1 for 4 h, Bcl-2/Bax, as biomarkers for the
extent of apoptosis, and their downstream target molecules, cleaved
caspase-3/caspase-3, were also detected by western blotting. The
results indicated that the P18 peptide suppressed the expression of
Bcl-2, induced the expression of Bax and enhanced the proteolytic
processing of inactive caspase-3 into cleaved caspase-3. However,
these effects were counteracted by IGF-1 (Fig. 8D and E). These results indicated
that deactivation of the VEGF/VEGFR2 signalling pathway and it
induced PI3K/Akt cascades may be a protective mechanism involved in
the P18 peptide-induced apoptosis of HUVECs and its anti-angiogenic
activity.
Discussion
As a type of richly vascularized solid tumour, the
occurrence and development of HCC is highly dependent on
angiogenesis (5). Therefore,
anti-angiogenic treatments have become clinically significant
therapeutic options for the treatment of HCC patients (7). Angiogenesis is primarily subject to
pro-angiogenic and anti-angiogenic factors. PEDF, a type of
endogenous angiogenesis inhibitor, has been confirmed as a
multifunctional antitumour factor. It has been reported that PEDF
inhibits angiogenesis by inducing apoptosis of ECs and several
types of tumour cells (10,16). Full-length PEDF has several
functional sections, and the anti-angiogenic functional fragment is
located from residues 24 to 57 (known as PEDF-34mer) (13,32).
In the current study, our data indicated that the P18 peptide, a
functional fragment mapping to residues 40 to 57 of full-length
PEDF, exerts anti-angiogenic activity both in vitro and
in vivo. Compared with full-length PEDF, the P18 peptide is
more stable and biocompatible but exhibits low antigenicity, which
indicates the potential of the P18 peptide for application in
anti-angiogenic therapy in HCC patients.
In the current study, the P18 peptide was confirmed
to have an anti-angiogenic effect on HCC in vivo. CD31 is
primarily concentrated at the borders between endothelial cells and
can be considered as a biomarker of angiogenesis. Studies have
demonstrated that in endothelial cells, VE-cadherin signalling,
expression, and localization correlate with vascular permeability
and tumour angiogenesis (33). IHC
staining of CD31 and VE-cadherin in tumour tissues suggested that
the P18 peptide functions as a potent inhibitor of angiogenesis
in vivo. Moreover, IF staining images indicated that
suppression of the phosphorylation of VEGFR2 may be the mechanism
by which the P18 peptide achieves its functions.
Angiogenesis relies on EC destabilization,
dissociation and migration. With the rapid growth of solid tumours,
the hypoxic environment inside tumour tissues stimulates the
expression of matrix metalloproteinases and pro-angiogenic
cytokines, such as MMP-2, MMP-9, VE-cadherin and E-cadherin
(34). MMP-2 and MMP-9 are
available from HUVECs and is responsible for tissue remodelling
during carcinogenesis, tumour metastasis and angiogenesis (35,36).
Our study showed that additional VEGF may upregulate secretions of
MMP-2 and MMP-9 in HUVECs, which corresponds to the results of
HUVEC migration and invasion assays. However, HUVECs treated with
P18 peptide show no response to additional VEGF. Moreover, we
observed similar results in expression levels of MMP-2 and MMP-9 in
HepG2 cells treated with P18 peptide. The results above certainly
suggest that the P18 peptide may perform its anti-angiogenic
function by inhibiting biological activity of VEGF. However,
opposite results were shown in the effects of P18 peptide on
E-cadherin expression in HUVECs and HepG2 cells. The upregulation
of E-cadherin in HepG2 cells is responsible for the suppression of
cell migration (37,38). The expression of E-cadherin in
HUVECs showed a decreasing trend. We consider that the decline in
cell activity after treatment with the P18 peptide may be connected
to this phenomenon. As an adhesion protein between cells, the
reduced expression of E-cadherin may lead to instability and
disintegration of vascular structures. However, the exact mechanism
by which the P18 peptide downregulated expression of E-cadherin in
HUVECs needs further study, which will be the next step of our
research.
Similar to parental PEDF and PEDF-34mer, the P18
peptide inhibits proliferation and induces apoptosis of HUVECs
in vitro and has an IC50 of 320 nM. The Bcl-2
family consists of many evolutionarily conserved proteins that can
regulate cell apoptosis through the classical mitochondrial
apoptosis pathway (39). Bcl-2 is
an anti-apoptotic protein in this family, whereas Bax is a
pro-apoptotic protein. Interactions between death-promoting and
death-suppressing factors regulate a dynamic equilibrium in which
the ratio between anti-apoptotic and pro-apoptotic proteins
controls cell apoptosis (40).
According to our results, HUVECs maintain a low level of apoptosis
under a certain concentration of VEGF (8 ng/ml). The P18 peptide
significantly increases the rate of apoptosis accompanied by
downregulation of both the expression of Bcl-2 and the ratio of
Bcl-2/Bax. However, according to our results, treatment with the
P18 peptide does not change the apoptosis rate of HepG2 cells.
Based on these results, we conjecture that the P18
peptide inhibits angiogenesis by blocking VEGF/VEGFR2 axis and
inducing apoptosis of endothelial cells. VEGF, which is produced by
a number of cells including endothelial cells, macrophages and
different types of tumour cells, is involved in angiogenesis,
vascular endothelial cell survival, proliferation and vascular
permeability (3,20). The results of the migration and
tube-formation assays showed that the P18 peptide could inhibit ECs
migration and angiogenic capacity induced by VEGF treatment. To
further account for these phenomena, the phosphorylation of VEGFR2
in HUVECs was detected. As a major receptor of VEGF, VEGFR2-induced
signalling is necessary for the execution of VEGF-stimulated
survival, the migration of ECs and angiogenesis in the development
of tumours (18,22). Phosphorylation of VEGFR2 leads to
activation of downstream signalling pathways, including the
MAPK/Erk and PI3K/Akt pathways (22,26).
According to our data, lower phosphorylation levels
of VEGFR2 in HUVECs were observed after culturing in serum-free
medium for 24 h, and those changes were rapidly reversed in the
presence of serum or additional VEGF. However, this reversal was
abrogated when the P18 peptide was simultaneously added to the
medium. The same result was also observed when SU1498 was added
instead of the P18 peptide. We noted that treatment with the P18
peptide also downregulated the phosphorylation levels of PI3K and
Akt induced by VEGF, whereas the ratio of p-PI3K/PI3K and p-Akt/Akt
can be upregulated upon treatment with IGF-1, an activator of the
PI3K/Akt pathway. In addition, we investigated variations in the
expression of Bcl-2/Bax among HUVECs treated with the P18 peptide,
the control group and the IGF-1-treated group. Since previous
studies have demonstrated that a reduction in Bcl-2/Bax leads to
the cleavage of caspase members and initiates a caspase cascade,
which results in activation and amplification of cell apoptotic
responses (41–43), we detected cleaved caspase-3 and
total caspase-3 as indexes for evaluating apoptosis. According to
the western blot results, we confirmed that the P18 peptide
downregulated the expression of Bcl-2 and the ratio of Bcl-2/Bax,
accompanied by enhanced cleavage of caspase-3 in synchronism with
an inhibition of PI3K/Akt pathway. Consequently, treatment of the
P18 peptide was associated with enhanced mitochondrial-mediated
apoptosis.
In summary, the P18 peptide exerts its
anti-angiogenic bioactivity by inhibiting endothelial cell
viability and inducing apoptosis. Simultaneously, the P18 peptide
suppresses tumour cell viability by inhibiting cell migration
rather than inducing apoptosis in HCC. VEGFR2 is the primary target
of the P18 peptide acting in ECs. Via modulating VEGF/VEGFR2 and it
induced PI3K/Akt signalling pathway, the P18 peptide enhances
mitochondrial-mediated apoptosis in ECs and suppresses vessel
formation in tumour tissues. This anti-angiogenic activity of the
P18 peptide suggests that it may be a potential agent for the
treatment of HCC.
Acknowledgements
This study was supported by the National Major
Research and Development Program of China (2016YFC0106004),
Shandong Provincial Science and Technology Development Planning,
China (2015GGB14168) and Shandong Provincial Natural Science
Foundation, China (ZR2015HL080).
References
|
1
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Folkman J: Patterns and
emerging mechanisms of the angiogenic switch during tumorigenesis.
Cell. 86:353–364. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schlieve CR, Mojica SG, Holoyda KA, Hou X,
Fowler KL and Grikscheit TC: Vascular endothelial growth factor
(VEGF) bioavailability regulates angiogenesis and intestinal stem
and progenitor cell proliferation during postnatal small intestinal
development. PLoS One. 11:e01513962016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li Y, Turpin CP and Wang S: Role of
thrombospondin 1 in liver diseases. Hepatol Res. Aug 4–2016.(Epub
ahead of print).
|
|
5
|
Zhu AX, Duda DG, Sahani DV and Jain RK:
HCC and angiogenesis: Possible targets and future directions. Nat
Rev Clin Oncol. 8:292–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Atta MM, Atta HM, Gad MA, Rashed LA, Said
EM, Hassanien Sel-S and Kaseb AO: Clinical significance of vascular
endothelial growth factor in hepatitis C related hepatocellular
carcinoma in Egyptian patients. J Hepatocell Carcinoma. 3:19–24.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Welker MW and Trojan J: Anti-angiogenesis
in hepatocellular carcinoma treatment: Current evidence and future
perspectives. World J Gastroenterol. 17:3075–3081. 2011.PubMed/NCBI
|
|
8
|
Edeline J, Boucher E, Rolland Y, Vauléon
E, Pracht M, Perrin C, Le Roux C and Raoul JL: Comparison of tumor
response by Response Evaluation Criteria in Solid Tumors (RECIST)
and modified RECIST in patients treated with sorafenib for
hepatocellular carcinoma. Cancer. 118:147–156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dawson DW, Volpert OV, Gillis P, Crawford
SE, Xu H, Benedict W and Bouck NP: Pigment epithelium-derived
factor: A potent inhibitor of angiogenesis. Science. 285:245–248.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
He X, Cheng R, Benyajati S and Ma JX: PEDF
and its roles in physiological and pathological conditions:
Implication in diabetic and hypoxia-induced angiogenic diseases.
Clin Sci (Lond). 128:805–823. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Belkacemi L and Zhang SX: Anti-tumor
effects of pigment epithelium-derived factor (PEDF): Implication
for cancer therapy. A mini-review. J Exp Clin Cancer Res. 35:42016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smith ND, Schulze-Hoepfner FT, Veliceasa
D, Filleur S, Shareef S, Huang L, Huang XM and Volpert OV: Pigment
epithelium-derived factor and interleukin-6 control prostate
neuroendocrine differentiation via feed-forward mechanism. J Urol.
179:2427–2434. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Filleur S, Volz K, Nelius T, Mirochnik Y,
Huang H, Zaichuk TA, Aymerich MS, Becerra SP, Yap R, Veliceasa D,
et al: Two functional epitopes of pigment epithelial-derived factor
block angiogenesis and induce differentiation in prostate cancer.
Cancer Res. 65:5144–5152. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He SS, Shi HS, Yin T, Li YX, Luo ST, Wu
QJ, Lu L, Wei YQ and Yang L: AAV-mediated gene transfer of human
pigment epithelium-derived factor inhibits Lewis lung carcinoma
growth in mice. Oncol Rep. 27:1142–1148. 2012.PubMed/NCBI
|
|
15
|
Broadhead ML, Dass CR and Choong PF:
Systemically administered PEDF against primary and secondary
tumours in a clinically relevant osteosarcoma model. Br J Cancer.
105:1503–1511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hase R, Miyamoto M, Uehara H, Kadoya M,
Ebihara Y, Murakami Y, Takahashi R, Mega S, Li L, Shichinohe T, et
al: Pigment epithelium-derived factor gene therapy inhibits human
pancreatic cancer in mice. Clin Cancer Res. 11:8737–8744. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mirochnik Y, Aurora A, Schulze-Hoepfner
FT, Deabes A, Shifrin V, Beckmann R, Polsky C and Volpert OV: Short
pigment epithelial-derived factor-derived peptide inhibits
angiogenesis and tumor growth. Clin Cancer Res. 15:1655–1663. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zsebik B, Symonowicz K, Saleh Y,
Ziolkowski P, Bronowicz A and Vereb G: Photodynamic therapy
combined with a cysteine proteinase inhibitor synergistically
decrease VEGF production and promote tumour necrosis in a rat
mammary carcinoma. Cell Prolif. 40:38–49. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kazemi M, Carrer A, Moimas S, Zandonà L,
Bussani R, Casagranda B, Palmisano S, Prelazzi P, Giacca M,
Zentilin L, et al: VEGF121 and VEGF165 differentially promote
vessel maturation and tumor growth in mice and humans. Cancer Gene
Ther. 23:125–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frezzetti D, Gallo M, Roma C, D'Alessio A,
Maiello MR, Bevilacqua S, Normanno N and De Luca A: Vascular
endothelial growth factor a regulates the secretion of different
angiogenic factors in lung cancer cells. J Cell Physiol.
231:1514–1521. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robinson CJ and Stringer SE: The splice
variants of vascular endothelial growth factor (VEGF) and their
receptors. J Cell Sci. 114:853–865. 2001.PubMed/NCBI
|
|
24
|
Domigan CK, Ziyad S and Iruela-Arispe ML:
Canonical and noncanonical vascular endothelial growth factor
pathways: New developments in biology and signal transduction.
Arterioscler Thromb Vasc Biol. 35:30–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smith NR, Baker D, James NH, Ratcliffe K,
Jenkins M, Ashton SE, Sproat G, Swann R, Gray N, Ryan A, et al:
Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3
are localized primarily to the vasculature in human primary solid
cancers. Clin Cancer Res. 16:3548–3561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Holmqvist K, Cross MJ, Rolny C, Hägerkvist
R, Rahimi N, Matsumoto T, Claesson-Welsh L and Welsh M: The adaptor
protein shb binds to tyrosine 1175 in vascular endothelial growth
factor (VEGF) receptor-2 and regulates VEGF-dependent cellular
migration. J Biol Chem. 279:22267–22275. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Johnston EK, Francis MK and Knepper JE:
Recombinant pigment epithelium-derived factor PEDF binds vascular
endothelial growth factor receptors 1 and 2. In Vitro Cell Dev Biol
Anim. 51:730–738. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu LF, Ye YQ, Huang GY, Li HB, Li GP, Pu
ZJ, Wei BL and Feng JL: Involvement of endoplasmic reticulum stress
in adenosine-induced human hepatoma HepG2 cell apoptosis. Oncol
Rep. 26:73–79. 2011.PubMed/NCBI
|
|
29
|
Donato MTTL, Tolosa L and Gómez-Lechón MJ:
Culture and functional characterization of human hepatoma HepG2
cells. Methods Mol Biol. 1250:77–93. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kisielewska J, Ligeza J and Klein A: The
effect of tyrosine kinase inhibitors, tyrphostins: AG1024 and
SU1498, on autocrine growth of prostate cancer cells (DU145). Folia
Histochem Cytobiol. 46:185–191. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Laurino L, Wang XX, de la Houssaye BA,
Sosa L, Dupraz S, Cáceres A, Pfenninger KH and Quiroga S: PI3K
activation by IGF-1 is essential for the regulation of membrane
expansion at the nerve growth cone. J Cell Sci. 118:3653–3662.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gong Q, Qiu S, Li S, Ma Y, Chen M, Yao Y,
Che D, Feng J, Cai W, Ma J, et al: Proapoptotic PEDF functional
peptides inhibit prostate tumor growth - a mechanistic study.
Biochem Pharmacol. 92:425–437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Coon BG, Baeyens N, Han J, Budatha M, Ross
TD, Fang JS, Yun S, Thomas JL and Schwartz MA: Intramembrane
binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the
endothelial mechanosensory complex. J Cell Biol. 208:975–986. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liao D and Johnson RS: Hypoxia: A key
regulator of angiogenesis in cancer. Cancer Metastasis Rev.
26:281–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sternlicht MD and Werb Z: How matrix
metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol.
17:463–516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li H, Daculsi R, Bareille R, Bourget C and
Amedee J: uPA and MMP-2 were involved in self-assembled network
formation in a two dimensional co-culture model of bone marrow
stromal cells and endothelial cells. J Cell Biochem. 114:650–657.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao Y, Peng S, Jia C, Xu F, Xu Y and Dai
C: Armc8 regulates the invasive ability of hepatocellular carcinoma
through E-cadherin/catenin complex. Tumour Biol. 37:11219–11224.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu AQ, Ding Y, Li CL, Yang Y, Yan SR and
Li DS: TALEN-induced disruption of Nanog expression results in
reduced proliferation, invasiveness and migration, increased
chemosensitivity and reversal of EMT in HepG2 cells. Oncol Rep.
35:1657–1663. 2016.PubMed/NCBI
|
|
39
|
Cory S, Huang DC and Adams JM: The Bcl-2
family: Roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li LY, Luo X and Wang X: Endonuclease G is
an apoptotic DNase when released from mitochondria. Nature.
412:95–99. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tian X, Shi Y, Liu N, Yan Y, Li T, Hua P
and Liu B: Upregulation of DAPK contributes to homocysteine-induced
endothelial apoptosis via the modulation of Bcl2/Bax and activation
of caspase 3. Mol Med Rep. 14:4173–4179. 2016.PubMed/NCBI
|
|
43
|
Cao Y, Jiang Z, Zeng Z, Liu Y, Gu Y, Ji Y,
Zhao Y and Li Y: Bcl-2 silencing attenuates hypoxia-induced
apoptosis resistance in pulmonary microvascular endothelial cells.
Apoptosis. 21:69–84. 2016. View Article : Google Scholar : PubMed/NCBI
|