Introduction
Cervical cancer is the second most common type of
cancer among women (1), with deaths
projected to rise by almost 25% over the next 10 years, according
to the World Health Organization (WHO) (2). One reason for the high death rate is
that cervical cancer has a high rate of invasion and metastasis
(3).
Aberrant activation of oncogenes contribute to
cancer progression (4). DEK is one
of the increasing number of oncogenes that have been reported
(5). DEK is primarily identified as
a fusion with the CAN nucleoporin in acute myeloid leukemia
(6,7). DEK protein overexpression has been
discovered in lung cancer, colorectal carcinoma, hepatocellular
carcinoma, breast cancer, bladder cancer and osteosarcoma (8–11). The
DEK protein promotes cancer cell proliferation,
epithelial-mesenchymal transition (EMT), metastasis, and DNA damage
(6,12,13).
DEK is an induced target of the human papillomavirus (HPV) E7
oncoprotein in head and neck cancer (10,14).
Moreover, DEK upregulate β-catenin in MCF-7 cells (15). On the contrary, DEK expression is
regulated by upstream regulators such as ERα, E2F, and NF-Y
(16,17). However, the role of DEK in cervical
cancer has been reported only rarely.
Glycogen synthase kinase-3 (GSK-3) is a
proline-directed serine/threonine kinase that was initially
identified as a phosphorylating and an inactivating agent of
glycogen synthase (18). Two
isoforms, α (GSK3α) and β (GSK3β), show a high degree of amino acid
homology (18,19). GSK3β is involved in energy
metabolism, neuronal cell development, and body pattern formation
(19,20). It can regulate nuclear transcription
factor-κB (NF-κB), p53 and β-catenin (21–24).
GSK-3β can be inactivated by phosphorylation at the N-terminal
Serine 9 (Ser9) residue which is the most frequently examined
mechanism that negatively regulates the activity of the kinase
(20,25). On the contrary, the phosphorylation
of Tyrosine 216 (Tyr216) residue positively regulates GSK-3β
activity (20,26). As a component of the protein complex
regulating the cellular level of β-catenin, GSK-3β increases
β-catenin degradation and maintains cell physiology (27,28).
When the Wnt/β-catenin pathway is aberrantly activated, cell
behavior changes and cancer cell malignant behavior increases
(29). The Wnt/β-catenin pathway
increases cancer cell invasion and migration (30,31).
Herein, we report on the function of DEK in
tumorigenesis and metastasis of cervical cancer cells. This study
provides insight into DEK-targeting treatment for cervical cancer
patients.
Materials and methods
Tissue specimens
Human cervical cancer samples (n=78) and non-cancer
samples (n=15) were collected at the Second Affiliated Hospital of
Chongqing Medical University, Chongqing, China. The experiments
were approved by the Research Ethics Committee of Chongqing Medical
University.
Cell lines
Human cervical cancer cell line SiHa was purchased
from the China Center for Type Culture Collection (Wuhan, China).
All cells were cultured in RPMI-1640 medium (Gibco, San Diego, CA,
USA), containing 10% fetal bovine serum (Gibco) and 1% penicillin
and streptomycin solution. All human cervical cancer cell lines
were maintained at 37°C in an atmosphere containing 5.0% carbon
dioxide.
Immunohistochemistry (IHC)
An IHC evaluation of DEK was performed according to
the streptavidin/peroxidase kit instructions (SPlink Detection
kits, ZSGB-Bio, Beijing, China). After being deparaffnized and
rehydrated, the sections were heated in citrate buffer-induced for
25 min for antigen retrieval. Endogenous peroxidase activity was
quenched with 3% H2O2 for 10 min. Thereafter,
the sections were blocked with goat serum for 10 min and incubated
with anti-DEK (1:100) overnight at 4°C. Then sections were
incubated with HRP-conjugated secondary antibodies for 10 min and
incubated in horseradish enzyme-labeled chain avidin solution for
10 min at 37°C. Visualization was performed with a DAB Horseradish
Peroxidase Color Development kit and the samples were
counterstained with hematoxylin. Staining intensity was graded on a
scale of 0–3, as follows: 0 (absence of staining), 1 (weakly
stained), 2 (moderately stained), and 3 (strongly stained). The
percentage of positive tumor cells was scored as follows: 0
(absence of tumor cells), 1 (<33% tumor cells), 2 (33–66% tumor
cells) and 3 (>66% tumor cells). Immunohistochemical score
(ranging from 0 to 9) was calculated by multiplying the intensity
score and the percentage score. Specimen was determined positive
when it was scored 4 or over. Specimen was determined negative when
it was scored 3 or under. The same qualified pathologist analyzed
all the IHC data to ensure scoring consistency.
Quantitative real-time polymerase
chain reaction (PCR)
Total RNA was extracted from the cancer cells using
an RNA Extraction kit (BioTeke, Beijing, China), according to the
manufacturer's protocol. Then RNA was quantified using a Nanodrop
spectrophotometer. cDNA was synthesized using a qPCR RT kit
(GeneCopoeia Inc., Guangzhou, China). The primers used for DEK and
GAPDH amplification were synthesized by GeneCopoeia Inc. The
real-time PCR kit was purchased from GeneCopoeia Inc. Each sample
was analyzed in triplicate. Quantification of gene transcription
was determined using the 2-∆∆Cq method (28). RT-PCR analyses were performed at
least three times.
Western blot analysis
Total protein extracted from each sample was
separated in 8% polyacrylamide gel, and electrotransferred to
polyvinylidene fluoride membranes (Millipore Corp., Billerica, MA,
USA). The bands were blocked in 5% powdered milk for 1 h at room
temperature. The membranes were incubated with primary antibodies
(1:1000-1:2000) against DEK, β-catenin, GSK-3β and p-GSK-3β
overnight at 4°C. After washing with tris-buffered saline
containing 0.1% Tween-20 (TBS-T), the membranes were incubated with
an anti-rabbit IgG antibody conjugated with horseradish peroxidase
(Bioss, China) for 1 h. After washing with TBS-T, the membranes
were visualized with an ECL detection system (KeyGen Biotech Inc.,
Nanjing, China). All of the western blot analyses were repeated at
least three times.
Transfections
The shRNA lentiviral vector targeting DEK (LV3-DEK,
sense CGAACCAAAUGUCCUGAAA dTdT; antisense UUUCAGGACAUUUGGUUCG dTdT)
and Negative control (LV3-NC, 5′-UUCUUCGAACGUGUCAGUTT-3′) were
provided by Genepharma Co., Ltd. (Shanghai, China). SiHa cells were
cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum
(Gibco), and were transduced with the lentivirus (multiplicity of
infection = 20), containing 5 µg/ml polybrene. Medium was refreshed
48 h post-transduction and replaced by RPMI-1640 medium, containing
10% fetal bovine serum and 1% penicillin and streptomycin solution.
Following transduction for 48 h, stably infection colonies of cells
were selected using 10 µg/ml puromycin.
Colony forming assay
Cells were cultured by seeding 1000 cells per well
in 6-well plates. All cells were incubated at 37°C in an atmosphere
containing 5.0% carbon dioxide for 14 days, then the number of
colonies formed was counted. All experiments were performed in
triplicate.
Proliferation assay
Cells were seeded into 96-well plates at a density
of 1000 cells per well. Cell proliferation was tested using a CCK-8
Kit (DNDOJAN, Japan) every 24 h after transfection for 6 days (the
reactions were incubated for 1 h at 37°C and 5% CO2;
detection: 450 nm, reference: 630 nm). The experiments were
repeated in triplicate.
Wound healing assay
Cell migratory capacity was analyzed using the
wound-healing assay in vitro. Cells were cultured in 6-well
plates and cultivated until 95% confluent. Wounds were incised in
the cell monolayer using a sterile pipette tip. At 0 and 72 h after
the wounding, cells were observed under the light microscope. The
distance between the two wounds were measured and the ability to
close the wound channel was expressed as the average percent of
wound closure at 72 h, compared to that at 0 h. The experiment was
repeated in triplicate.
Transwell membrane based invasion
assay
The ability of cells to migrate was analyzed using
the Matrigel Transwell assay. For Matrigel migration assays, the
upper side of an 8 µm pore, 6.5-mm polycarbonate Transwell filter
chamber (Corning Inc., New York, NY, USA) was uniformly coated with
Matrigel basement membrane matrix (BD Biosciences, Bedford, MA,
USA) for 2 h at 37°C before the cells were added. The cells were
seeded (2×105 cells/well) in the upper chambers in 200
µl serum-free media, and the lower chambers were filled with 750 µl
complete media containing fetal bovine serum, which can induce cell
migration. After 24 h, cells that invaded to the lower surface of
the filter were fixed with 4% paraformaldehyde, stained with 0.5%
crystal violet, and counted using a microscope. Each experiment was
performed in triplicate and repeated thrice.
Dual-luciferase reporter gene
assay
Luciferase reporter gene assay was performed using
Dual-Luciferase Reporter assay System (Promega, Madison, WI, USA)
according to the manufacturer's protocol. TOPflash reporter plasmid
was provided by Shanghai Qcbio Science & Technologies Co., Ltd.
(Shanghai, China). Reporter assay was performed 48 h
post-transfection using the Dual Luciferase Assay System (Promega).
Firefly luciferase activity was normalized for transfection
efficiency using the corresponding Renilla luciferase
activity. The experiment was repeated in triplicate.
In vivo tumor xenograft study
Six-week-old female BALB/c nude mice were purchased
from the Experimental Animal Center of Chongqing Medical
University. The research protocol used in this study was approved
by, and the mice were maintained, in accordance with the
institutional guidelines set forth by the Committee on the Use and
Care on Animals (Chongqing Medical University). Cells were infected
with the indicated lentiviral vectors and injected
(5×106 cells per mouse in 200 µl) subcutaneously into
the left armpit of 6-week-old BALB/c nude mice. Twenty-one days
later, the animals were sacrificed to confirm the the weight of the
established tumors.
Statistical analysis
All statistical analyses were performed using SPSS
software v17.0 (Chicago, IL, USA). Comparisons between groups were
analyzed using a Student's t-test or a Mann-Whitney U test. The
Chi-squared test was used to compare the associations between DEK
overexpression and clinicopathological variables of cervical cancer
and non-tumor samples. All experiments were performed in
triplicate. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
DEK is overexpressed in cervical
cancer tissues
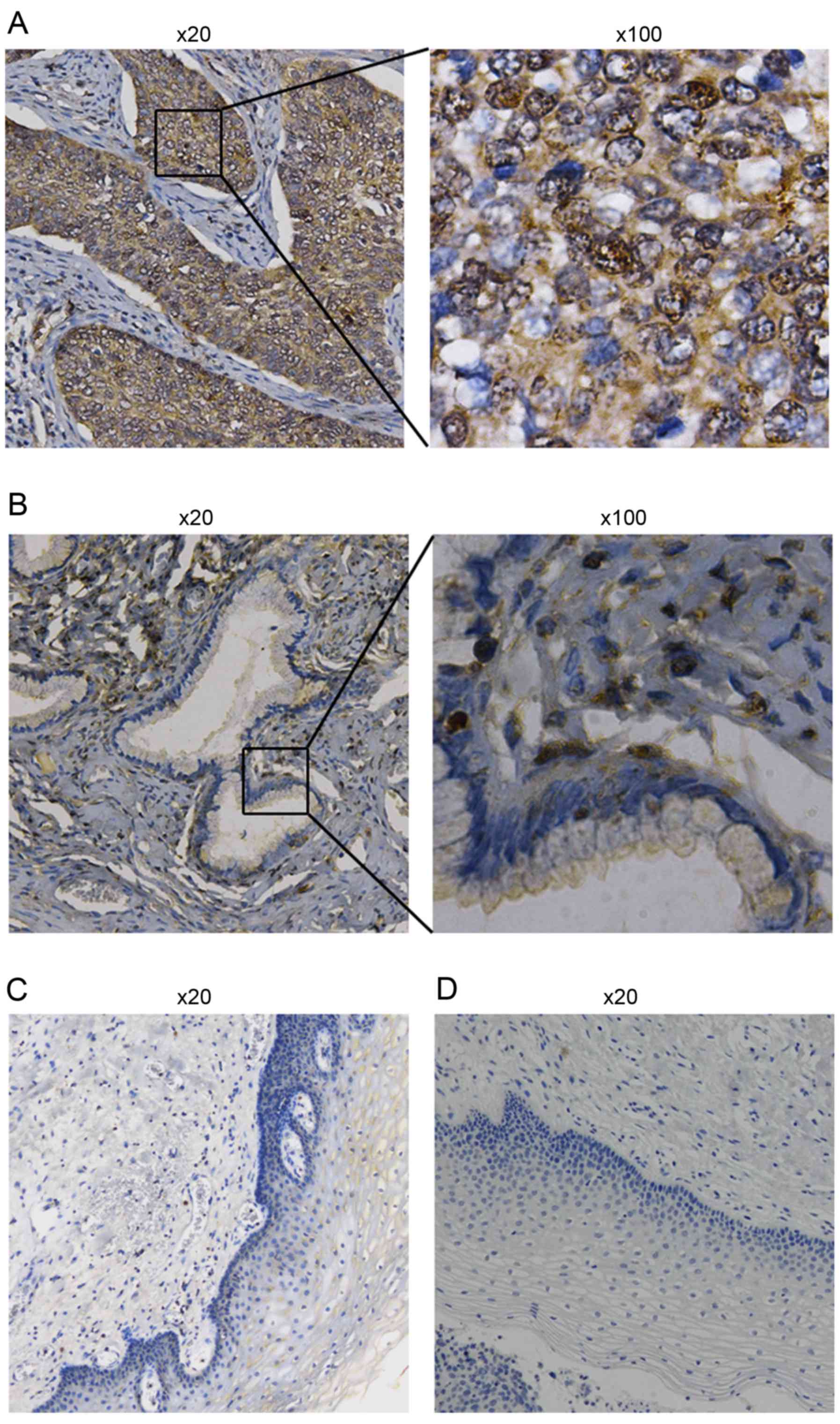
To evaluate the expression of DEK, IHC analyses were
performed in 78 cervical cancer and 15 non-cancer cervical tissue
samples (the clinicopathological parameters of the tumor specimens
examined in the study are summarized in Table I). DEK was shown to be expressed at
significantly higher levels in cervical cancer tissues than that in
normal cervical tissues (P<0.05, Fig. 1, Table
I). According to the FIGO staging system, the IHC results
indicated that high DEK expression was positively correlated with
FIGO stage classification (P<0.05). Patients having higher FIGO
stages tended to overexpress DEK. Additionally, the positive levels
of DEK expression was higher in squamous cell carcinoma than in
adenocarcinoma (P<0.05). However, there was no obvious
correlation between DEK expression and patient age.
 | Table I.Association of DEK expression with
clinicopathological characteristics in 78 patients with EOC. |
Table I.
Association of DEK expression with
clinicopathological characteristics in 78 patients with EOC.
|
|
| DEK expression |
|
|---|
|
|
|
|
|
|---|
|
Characteristics | No. of pts.
(n=78) | Low no. (%) | High no. (%) | P-value |
|---|
| Age (years) |
|
|
| 0.452 |
|
<51 | 48 | 23 (47.92) | 25 (52.08) |
|
|
≥51 | 30 | 17 (56.67) | 13 (43.33) |
|
| FIGO stage |
|
|
| 0.043 |
|
I–II | 52 | 22 (42.31) | 30 (57.69) |
|
|
III–IV | 26 | 5
(19.23) | 21 (80.77) |
|
| Grade |
|
|
| 0.157 |
|
1-2 | 51 | 18 (35.29) | 33 (65.71) |
|
| 3 | 27 | 14 (51.85) | 13 (48.15) |
|
| Tumor type |
|
|
| 0.038 |
|
Squamous carcinoma | 63 | 10 (15.87) | 53 (84.13) |
|
|
Adenocarcinoma | 15 | 6
(40.00) | 9
(60.00) |
|
Silencing DEK inhibits SiHa cell
proliferation
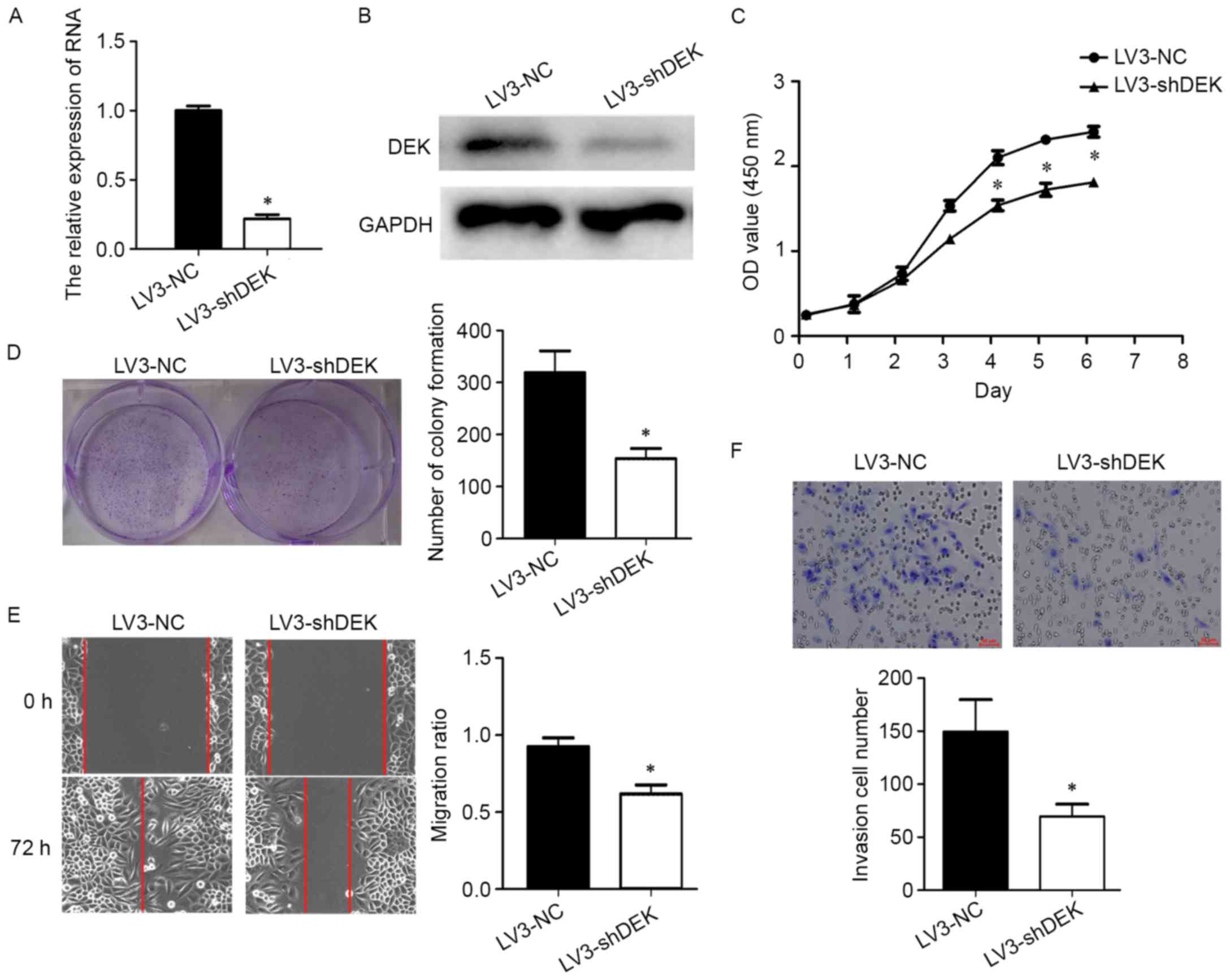
In this study, DEK was silenced using the shRNA
lentiviral vector (LV3-DEK). The efficiency was tested by qPCR and
western blotting. In the silenced group, DEK expression at mRNA
level was 78% lower compared to the negative control group (LV3-NC)
(Fig. 2A and B).
To understand better the role of DEK in cervical
cancer cells, cell proliferation was analyzed by CCK-8 and
colony-forming assays. The results showed that cell proliferation
was inhibited in the LV3-DEK group (Fig. 2C). The OD value of LV3-shDEK group
was significantly lower than that of LV-NC group from day 4.
Consistent with the CCK-8 assay, the colony-forming ability was
reduced in the LV3-DEK group (Fig.
2D).
Silencing DEK impaired SiHa cell
migration and invasion
Cell migration capability was determined with a
wound healing assay. After 72 h, the wound was filled in the LV3-NC
group, while there was still a gap in the LV3-DEK group (Fig. 2E). Moreover, cell invasion
capability was observed using a Matrigel-Transwell assay (Fig. 2F). It was found that silencing DEK
impaired the migration and invasion capacity of cervical cancer
cells.
Silencing DEK downregulates the Wnt
pathway
The Wnt pathway is a classic signaling pathway which
regulates cell proliferation, migration and invasion. Previous
studies have reported that DEK regulates the Wnt signaling pathway
in acute leukemia cells (5).
However, whether DEK has an effect on cervical cancer cells is not
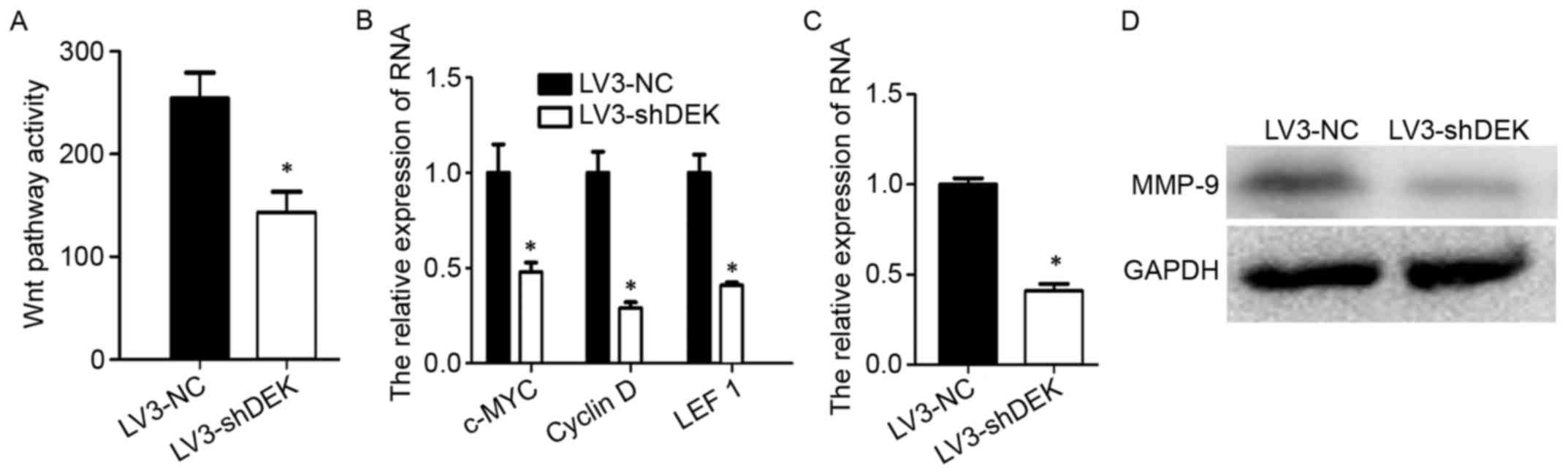
clear. Thus, we investigated Wnt pathway activity by luciferase
reporter assay. The Wnt pathway was found to be inhibited when DEK
was silenced (Fig. 3A). At the same
time, silencing DEK reduced the expression of c-MYC, cyclin D and
LEF1 (Fig. 3B), the downstream
targets of Wnt signaling. MMP-9 has been reported as a Wnt
targeting gene, and is closely related to tumor metastasis
(32). We examined MMP-9 in
cervical cancer cells. Silencing DEK resulted in reduced expression
of MMP-9 at the mRNA level (Fig.
3C) and the protein level (Fig.
3D). These data indicated that DEK promoted cervical cancer
cell metastasis via upregulating the Wnt pathway and MMP-9
expression.
Silencing DEK downregulates the
Wnt/β-catenin pathway by mediating p-GSK-3β
In order to identify which proteins interacted with
DEK, we utilized String10.0 (http://string.embl.de/). We found that GSK-3β was
predicted to interact with DEK, although this had not yet been
proven. Therefore, we investigated the relationship between DEK and
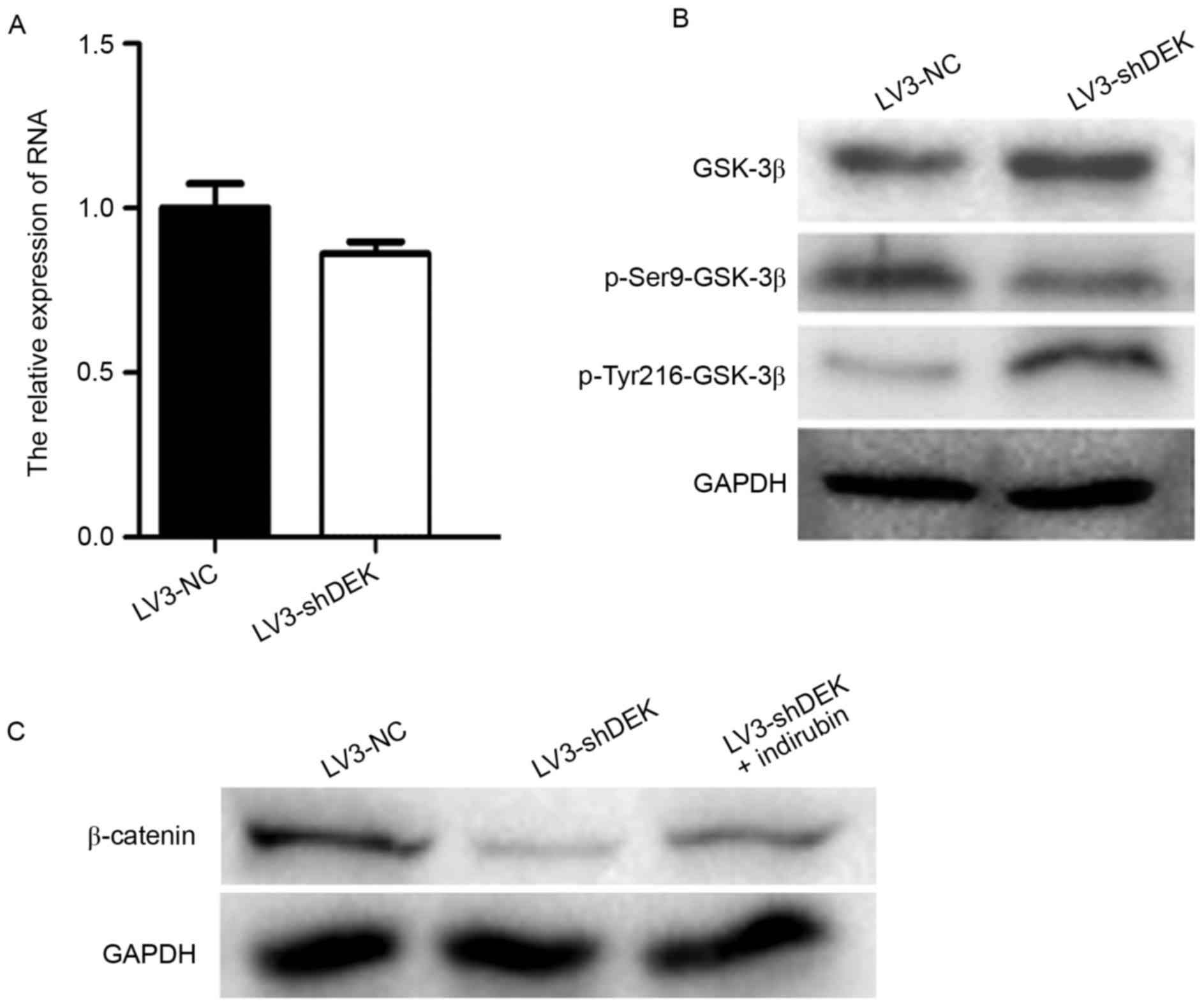
GSK-3β. First we found that the expression of GSK-3β at the mRNA
and protein levels displayed no significant difference between the
LV3-DEK cells and LV3-NC cells (Fig. 4A
and B). It has been reported that, in the normal state, GSK-3β,
Axin, CK-I and β-catenin form a compound and promote β-catenin
degradation. When Wnt signaling terminates, the compound
disintegrates and β-catenin accumulates and becomes active. Thus,
we hypothesized that DEK regulated the Wnt/GSK-3β/β-catenin pathway
via regulation of GSK-3β phosphorylation. Levels of p-Ser9-GSK-3β
and p-Tyr216-GSK-3β were measured in the LV3-DEK and control groups
to confirm this hypothesis. The results showed that p-Ser9-GSK-3β
expression was lower in the LV3-DEK group than that in LV3-NC group
(Fig. 4B) and that p-Tyr216-GSK-3β
expression was higher in DEK silenced cells (Fig. 4B). This indicated that DEK regulated
the Wnt/β-catenin by mediating GSK-3β phosphorylation rather than
GSK-3β translation. To further prove this idea, indirubin, a
powerful inhibitor of GSK-3β (33),
was added. The result showed that DEK-induced downregulation of
β-catenin could be partially reversed (Fig. 4C).
DEK impaired in vivo
tumorigenesis
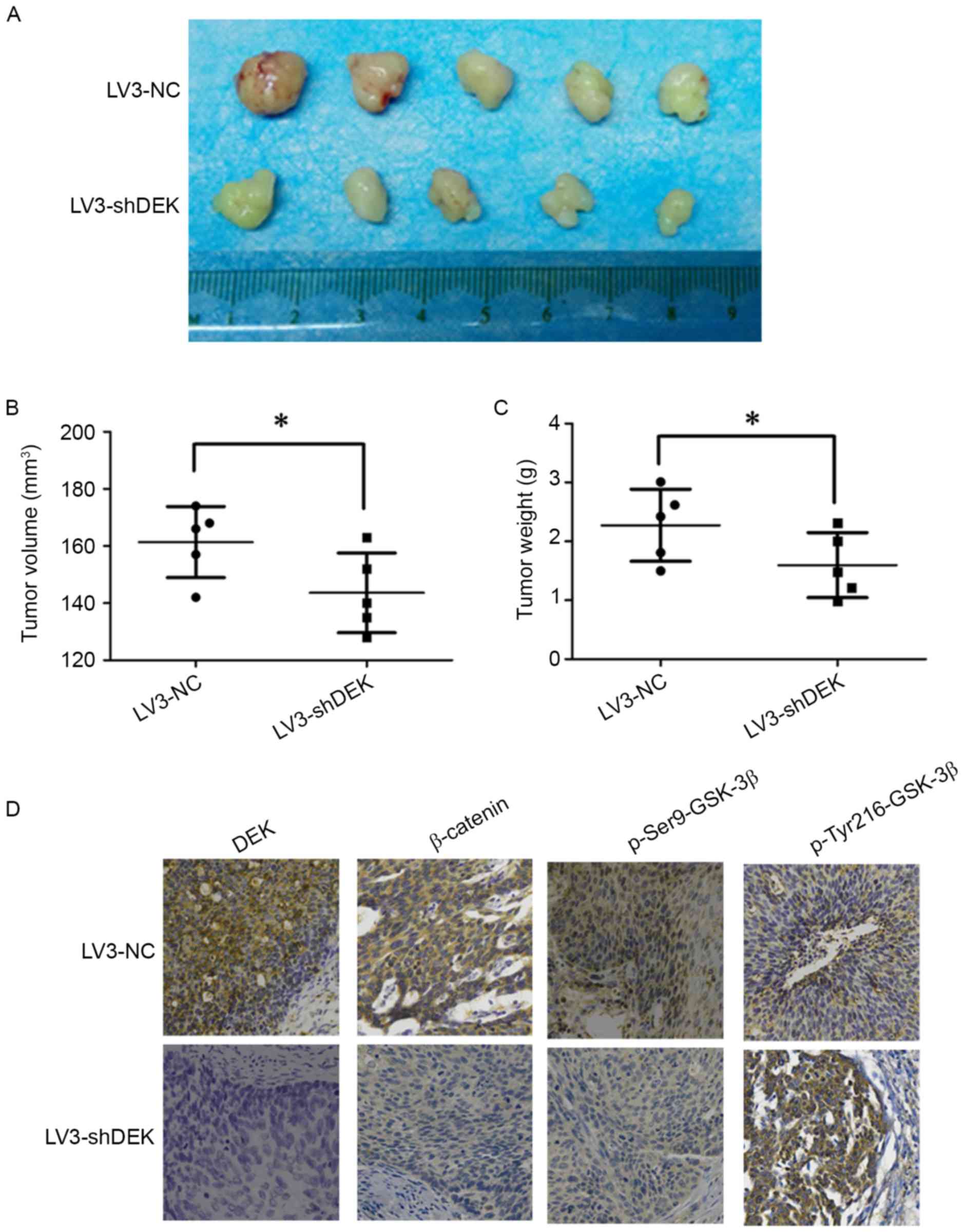
Xenograft tumorigenesis in nude mice was used to
explore the effect of DEK on tumor formation in cervical cancer.
The LV3-DEK cells and LV3-NC cells were implanted subcutaneously
into the left armpit of nude female mice. Twenty-one days after
transplantation, the tumors were harvested from mice (Fig. 5A). The average volume and weight of
tumors in the LV3-DEK group were significantly smaller and lighter
than those of the LV3-NC group (Fig. 5B
and C). The expression of DEK, p-Ser9-GSK-3β and β-catenin was
reduced in the LV3-DEK, compared to the LV3-NC group (Fig. 5D). Conversely, p-Tyr216-GSK-3β
expression was increased in the LV3-DEK group (Fig. 5D). These data showed that silencing
DEK blocked in vivo tumor formation and inhibited the in
vivo expression of p-GSK-3β.
Discussion
Cervical cancer is the most common gynecological
malignancy and is intimately linked with HPV infection (34). DEK has been reported as an oncogene
in acute leukemia, lung cancer, hepatocellular carcinoma, breast
cancer, and other forms of cancer (35,36).
The research has focused on its role in apoptosis, metastasis, and
DNA damage. However, the role of DEK in cervical cancer has not
been well established. In the present study, it was found that DEK
is significantly overexpressed in cervical cancer tissues, compared
to non-cancerous cervical tissue. Furthermore, high DEK expression
in samples had a positive correlation with FIGO staging and tumor
type. A higher proportion of DEK positive staining was found in
squamous carcinoma, compared to adenocarcinoma. These results
demonstrate that DEK is an oncogene in cervical cancer and is
related to squamous carcinoma, which is highly linked to HPV
infection. Determine whether or not DEK expression is correlated to
HPV infection, however, requires further research.
DEK promotes proliferation, epithelial-mesenchymal
transition (EMT), and metastasis in various cancer cells. In order
to investigate the function of DEK in cervical cancer, the present
study employed a lentiviral vector to inhibit DEK expression in
functional assays. The results showed that silencing DEK inhibited
cervical cancer cell proliferation, migration and invasion. The
results indicate that DEK acts as a promoter in cervical cancer
cell metastasis, a result in line with a previous study of DEK in
acute leukemia cells, hepatoma cells, and colorectal cancer cells.
These results partially explain the relationship between
overexpression of DEK and FIGO staging in cervical cancer
tissues.
The Wnt pathway is a classic signaling pathway which
regulates cell proliferation, migration and invasion. Thus we
investigated the effect of DEK on Wnt/β-catenin pathway. Our
results showed that silencing DEK inhibited not only Wnt pathway
activity but also MMP-9 expression. MMP-9 is a downstream target
for Wnt signaling, which is also closely related to tumor
metastasis (32,37). Taken together, these data could be
the explanation to the decrease of invasion and migration after DEK
silencing in SiHa cells.
Glycogen synthase kinase-3β (GSK-3β) is a
serine/threonine protein kinase (25) that plays an important role in early
embryo development, neurodegenerative disease, diabetes,
inflammatory conditions and oncogenesis (38). It has also been reported to regulate
transcription factors, such as nuclear transcription factor-κB
(NF-κB), p53 and β-catenin (19,22,23).
While GSK-3β can be inactivated by phosphorylation at the
N-terminal Serine 9 residue, phosphorylation at the Tyrosine 216
residue activates GSK-3β (20). In
the present study, we found that DEK regulated GSK-3β at
post-translational protein modification level rather than at the
translation level. DEK inhibited GSK-3β activity such that the
downstream target was activated. Under normal conditions, GSK-3β,
Axin, CK-I and β-catenin form a compound and promote β-catenin
degradation (27). In the absence
of Wnt signaling, the compound disintegrates and β-catenin
accumulates and becomes active. Therefore, abnormally high DEK
expression can phosphorylate GSK-3β at Ser9 and inhibit Tyr216
phosphorylation, thereby inactivating GSK-3β. Consequently,
β-catenin accumulated and aberrantly increased downstream target
genes, which facilitated tumor metastasis, proliferation and other
malignant behavior. These data explained the results of high
positive rate in IHC staining in cervical cancer samples and the
phenotype of decreased proliferation, migration and invasion in DEK
silenced cervical cancer cells.
In summary, the results reported herein highlight
the determination that DEK is an oncogene in cervical cancer and
may serve as a novel target for cervical cancer research and
treatment.
Acknowledgements
This work was supported by the National Natural
Science Foundation of China (grant no. 81372800).
References
|
1
|
Parekh N, Donohue JM, Men A, Corbelli J
and Jarlenski M: Cervical Cancer Screening Guideline Adherence
Before and After Guideline Changes in Pennsylvania Medicaid. Obstet
Gynecol. 129:66–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Machalek DA, Wark JD, Tabrizi SN, Hopper
JL, Bui M, Dite GS, Cornall AM, Pitts M, Gertig D, Erbas B, et al:
Genetic and environmental factors in invasive cervical cancer:
Design and methods of a classical twin study. Twin Res Hum Genet.
20:10–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sawaya GF and Smith-McCune K: Cervical
cancer screening. Obstet Gynecol. 127:459–467. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Knudson AG Jr: Overview: Genes that
predispose to cancer. Mutat Res. 247:185–190. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sandén C and Gullberg U: The DEK
oncoprotein and its emerging roles in gene regulation. Leukemia.
29:1632–1636. 2015.PubMed/NCBI
|
|
6
|
Deutzmann A, Ganz M, Schönenberger F,
Vervoorts J, Kappes F and Ferrando-May E: The human oncoprotein and
chromatin architectural factor DEK counteracts DNA replication
stress. Oncogene. 34:4270–4277. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sandén C, Nilsson HJ and Gullberg U: The
DEK oncoprotein is upregulated by multiple leukemia-associated
fusion genes. Blood Cells Mol Dis. 54:284–285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu X, Qi D, Qi J, Mao Z, Li X, Zhang J,
Li J and Gao W: Significance of DEK overexpression for the
prognostic evaluation of non-small cell lung carcinoma. Oncol Rep.
35:155–162. 2016.PubMed/NCBI
|
|
9
|
Yi HC, Liu YL, You P, Pan JS, Zhou JY, Liu
ZJ and Zhang ZY: Overexpression of DEK gene is correlated with poor
prognosis in hepatocellular carcinoma. Mol Med Rep. 11:1318–1323.
2015.PubMed/NCBI
|
|
10
|
Wise-Draper TM, Morreale RJ, Morris TA,
Mintz-Cole RA, Hoskins EE, Balsitis SJ, Husseinzadeh N, Witte DP,
Wikenheiser-Brokamp KA, Lambert PF, et al: DEK proto-oncogene
expression interferes with the normal epithelial differentiation
program. Am J Pathol. 174:71–81. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinez-Useros J, Rodriguez-Remirez M,
Borrero-Palacios A, Moreno I, Cebrian A, del Gomez Pulgar T, del
Puerto-Nevado L, Vega-Bravo R, Puime-Otin A, Perez N, et al: DEK is
a potential marker for aggressive phenotype and irinotecan-based
therapy response in metastatic colorectal cancer. BMC Cancer.
14:9652014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu HG, Scholten I, Gruss C and Knippers R:
The distribution of the DEK protein in mammalian chromatin. Biochem
Biophys Res Commun. 358:1008–1014. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sawatsubashi S, Murata T, Lim J, Fujiki R,
Ito S, Suzuki E, Tanabe M, Zhao Y, Kimura S, Fujiyama S, et al: A
histone chaperone, DEK, transcriptionally coactivates a nuclear
receptor. Genes Dev. 24:159–170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu K, Feng T, Liu J, Zhong M and Zhang S:
Silencing of the DEK gene induces apoptosis and senescence in CaSki
cervical carcinoma cells via the up-regulation of NF-κB p65. Biosci
Rep. 32:323–332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vinnedge Privette LM, McClaine R, Wagh PK,
Wikenheiser-Brokamp KA, Waltz SE and Wells SI: The human DEK
oncogene stimulates β-catenin signaling, invasion and mammosphere
formation in breast cancer. Oncogene. 30:2741–2752. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vinnedge Privette LM, Ho SM,
Wikenheiser-Brokamp KA and Wells SI: The DEK oncogene is a target
of steroid hormone receptor signaling in breast cancer. PLoS One.
7:e469852012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carro MS, Spiga FM, Quarto M, Di Ninni V,
Volorio S, Alcalay M and Müller H: DEK Expression is controlled by
E2F and deregulated in diverse tumor types. Cell Cycle.
5:1202–1207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Woodgett JR: Molecular cloning and
expression of glycogen synthase kinase-3/factor A. EMBO J.
9:2431–2438. 1990.PubMed/NCBI
|
|
19
|
Eldar-Finkelman H: Glycogen synthase
kinase 3: An emerging therapeutic target. Trends Mol Med.
8:126–132. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park CH, Lee BH, Ahn SG, Yoon JH and Oh
SH: Serine 9 and tyrosine 216 phosphorylation of GSK-3β
differentially regulates autophagy in acquired cadmium resistance.
Toxicol Sci. 135:380–389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu Y, Gu X, Li R, Luo Q and Xu Y: Glycogen
synthase kinase-3β inhibition induces nuclear factor-κB-mediated
apoptosis in pediatric acute lymphocyte leukemia cells. J Exp Clin
Cancer Res. 29:1542010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He F, Chen H, Yang P, Wu Q, Zhang T, Wang
C, Wei J, Chen Z, Hu H, Li W, et al: Gankyrin sustains
PI3K/GSK-3β/β-catenin signal activation and promotes colorectal
cancer aggressiveness and progression. Oncotarget. 7:81156–81171.
2016.PubMed/NCBI
|
|
23
|
Hartigan JA, Xiong WC and Johnson GV:
Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2.
Biochem Biophys Res Commun. 284:485–489. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miyashita K, Nakada M, Shakoori A,
Ishigaki Y, Shimasaki T, Motoo Y, Kawakami K and Minamoto T: An
emerging strategy for cancer treatment targeting aberrant glycogen
synthase kinase 3 beta. Anticancer Agents Med Chem. 9:1114–1122.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cole A, Frame S and Cohen P: Further
evidence that the tyrosine phosphorylation of glycogen synthase
kinase-3 (GSK3) in mammalian cells is an autophosphorylation event.
Biochem J. 377:249–255. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bhat RV, Shanley J, Correll MP, Fieles WE,
Keith RA, Scott CW and Lee CM: Regulation and localization of
tyrosine216 phosphorylation of glycogen synthase kinase-3beta in
cellular and animal models of neuronal degeneration. Proc Natl Acad
Sci USA. 97:11074–11079. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hsieh CH, Hsu HH, Shibu MA, Day CH, Bau
DT, Ho CC, Lin YM, Chen MC, Wang SH and Huang CY: Down-regulation
of β-catenin and the associated migration ability by Taiwanin C in
arecoline and 4-NQO-induced oral cancer cells via GSK-3β
activation. Mol Carcinog. 56:1055–1067. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu C, Dong T, Li R, Lu J, Wei X and Liu P:
Emodin inhibits epithelial to mesenchymal transition in epithelial
ovarian cancer cells by regulation of GSK-3β/β-catenin/ZEB1
signaling pathway. Oncol Rep. 35:2027–2034. 2016.PubMed/NCBI
|
|
29
|
Fu Y, Zheng S, An N, Athanasopoulos T,
Popplewell L, Liang A, Li K, Hu C and Zhu Y: β-catenin as a
potential key target for tumor suppression. Int J Cancer.
129:1541–1551. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gupta A, Verma A, Mishra AK, Wadhwa G,
Sharma SK and Jain CK: The Wnt pathway: Emerging anticancer
strategies. Recent Pat Endocr Metab Immune Drug Discov. 7:138–147.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nair RR, Solway J and Boyd DD: Expression
cloning identifies transgelin (SM22) as a novel repressor of 92-kDa
type IV collagenase (MMP-9) expression. J Biol Chem.
281:26424–26436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Varela AT, Simões AM, Teodoro JS, Duarte
FV, Gomes AP, Palmeira CM and Rolo AP: Indirubin-3′-oxime prevents
hepatic I/R damage by inhibiting GSK-3beta and mitochondrial
permeability transition. Mitochondrion. 10:456–463. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wilson KL, Cowart CJ, Rosen BL, Pulczinski
JC, Solari KD, Ory MG and Smith ML: Characteristics associated with
HPV diagnosis and perceived risk for cervical cancer among
unmarried, sexually active college women. J Cancer Educ. Nov
28–2016.(Epub ahead of print). View Article : Google Scholar
|
|
35
|
Liu S, Wang X, Sun F, Kong J, Li Z and Lin
Z: DEK overexpression is correlated with the clinical features of
breast cancer. Pathol Int. 62:176–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Datta A, Adelson ME, Mogilevkin Y,
Mordechai E, Sidi AA and Trama JP: Oncoprotein DEK as a tissue and
urinary biomarker for bladder cancer. BMC Cancer. 11:2342011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Deryugina EI and Quigley JP: Matrix
metalloproteinases and tumor metastasis. Cancer Metastasis Rev.
25:9–34. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Domoto T, Pyko IV, Furuta T, Miyashita K,
Uehara M, Shimasaki T, Nakada M and Minamoto T: Glycogen synthase
kinase-3β is a pivotal mediator of cancer invasion and resistance
to therapy. Cancer Sci. 107:1363–1372. 2016. View Article : Google Scholar : PubMed/NCBI
|