Introduction
Imatinib is a small molecule kinase inhibitor with
potent activity toward tumors whose growth is driven by mutations
in KIT, including gastrointestinal stromal tumor (GIST) (1) and certain subsets of mastocytosis
(2) and melanoma (3). Despite their excellent initial
response to imatinib, these tumors are generally not cured by this
drug due to the acquisition of resistance. The mechanism of the
resistance of KIT mutation-driven tumors to imatinib has been
extensively studied in GIST; however, it is still not well
understood. Although acquisition of secondary mutations in KIT is a
major mechanism of the acquired resistance to imatinib, there are
patients whose tumor does not have a secondary mutation in KIT
(4,5). The mechanisms of the acquired
resistance in these patients are heterogeneous and overexpression
of KIT is one of the factors that contributes to imatinib
resistance in such patients (6).
KIT overexpression could be caused by mechanisms such as genomic
amplification (7,8) and upregulation of KIT
transcription by overexpression of p55PIK, an isoform of
phosphoinositide 3-kinase (9);
however, the underlying mechanism of KIT overexpression is still
unclear.
Similar to their development in humans, KIT
mutation-driven tumors spontaneously develop in dogs (10–12).
The neoplastic proliferation of mast cells in dogs, referred to as
a mast cell tumor (MCT), is one of the most common tumors in this
species (13), and ~30% of all MCT
cases and 70% of aggressive types of MCT possess an activating
mutation in KIT (14). Canine MCT
with KIT mutations respond well to kinase inhibitors including
imatinib; however, they are usually not cured and eventually
acquire resistance to the kinase inhibitors (14,15).
One of the mechanisms by which canine MCTs acquire resistance to
imatinib could be by the generation of secondary mutations in KIT
(16); however, similar to the case
in human GIST, there are imatinib-resistant cases without such
mutations in KIT.
Due to its potential ability to acquire resistance
to imatinib, canine MCT is considered a valuable spontaneous model
for translational research in investigation of the mechanisms, and
in overcoming strategies of imatinib resistance in KIT
mutation-driven human tumors. A canine MCT cell line, CoMS, has
been established from a dog with oral mucosa MCT (17). CoMS cells have an imatinib-sensitive
activating mutation in the juxtamembrane domain of KIT (KIT
exon 11, c.1720_1772+1dup). In the present study, we generated a
KIT-overexpressing imatinib-resistant subline from CoMS cells and
investigated the mechanisms underlying KIT overexpression that
caused imatinib resistance.
Materials and methods
Cell line
The canine MCT cell line CoMS was kindly provided by
Dr Takagi (University of Hokkaido, Hokkaido, Japan). Madin-Darby
canine kidney (MDCK) cells were purchased from the American Type
Culture Collection (Manassas, VA, USA). CoMS cells were cultured in
RPMI-1640 medium (Life Technologies, Carlsbad, CA, USA)
supplemented with 10% fetal calf serum (Nippon Bio-Supply, Tokyo,
Japan), 50 U/ml penicillin (Life Technologies), and 50 µg/ml
streptomycin (Life Technologies) (cRPMI) in a humidified incubator
at 37°C under 5% CO2. MDCK cells were cultured in
Dulbecco's modified Eagle's medium (Life Technologies) supplemented
with the same additives, under the same conditions.
Generation of an imatinib-resistant
CoMS subline
CoMS cells were exposed to increasing concentrations
of imatinib (LC Laboratories, Woburn, MA, USA), starting with a
concentration of 0.02 µM and then gradually increasing up to a
concentration of 1 µM over two months. The cells that grew
logarithmically in the presence of 1 µM of imatinib were named
rCoMS1 cells and were maintained in cRPMI supplemented with 1 µM of
imatinib.
Cell growth inhibition assay
CoMS and rCoMS1 cells suspended in cRPMI were seeded
in 96-well plates (2×104 cells/well) and treated with
various concentrations of imatinib (0–100 µM) for 24 h. Cell
viability was then measured using a WST-1 cell proliferation assay
kit (Takara, Otsu, Japan). The half maximal inhibitory
concentration (IC50) of imatinib was calculated using
the GraphPad Prism software program (GraphPad Software, San Diego,
CA, USA).
Flow cytometric analysis of KIT
Cellular expression of KIT was analyzed using flow
cytometry. For detection of cell surface KIT, CoMS and rCoMS1 cells
were fixed with 4% paraformaldehyde for 20 min and washed twice
with PBS-2% BSA. As a negative control, MDCK cells were also
subjected to the same assay. The cells were stained with
phycoerythrin (PE)-conjugated monoclonal rat anti-mouse KIT (ACK45)
or PE-conjugated isotype-matched control IgG (rat IgG2b, κ) (both
from BD Biosciences, San Diego, CA, USA) for 1 h in PBS-2% BSA. The
cells were then analyzed by flow cytometry using a FACSCalibur (BD
Biosciences). For detection of total (cell surface and
intracellular) KIT, PBS-2% BSA was replaced with permeabilization
buffer (eBioscience, San Diego, CA, USA) and the cellular
expression of KIT was analyzed using flow cytometry in the same
manner.
Detection of KIT and phosphorylated
KIT by western blot analysis
CoMS and rCoMS1 cells suspended in cRPMI were seeded
in 6-well plates (5×105 cells/well) and treated with
different concentrations of imatinib (0–10 µM) for 4 h. The cells
were then lysed, and aliquots were subjected to western blot
analysis using the following antibodies: polyclonal goat anti-human
KIT (C-14) (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
monoclonal rabbit anti-human phospho-KIT (Tyr703, D12E12) (Cell
Signaling, Danvers, MA, USA), or polyclonal goat anti-human GAPDH
(Santa Cruz Biotechnology), followed by biotin-conjugated rabbit
anti-goat or goat anti-rabbit IgG (Life Technologies). After
incubation of the membranes with peroxidase-conjugated
streptavidin, immunoreactive bands were visualised using an
enhanced chemiluminescence system (GE Healthcare, Chalfont, UK) and
the LAS-4000 (Fujifilm, Tokyo, Japan).
Semi-quantitative RT-PCR analysis of
KIT expression
Total RNA extracted from CoMS and rCoMS1 cells was
reverse-transcribed into cDNA and subjected to PCR amplification
using Platinum Taq DNA Polymerase (Life Technologies) and a primer
set for canine KIT: 5′ primer, 5′-GCTGGTCCGCTGCCCTCTGA-3′; 3′
primer, 5′-GGCGTAACACATGAACACTCCA-3′. After PCR amplification for
22 cycles, the products (10 µl aliquot) were size-fractionated on a
1.2% agarose gel and visualized with ethidium bromide staining and
the LAS-500 (Fujifilm). Band intensities were semi-quantified using
ImageQuant TL software (Fujifilm).
Cycloheximide (CHX) chase assay
CoMS and rCoMS1 cells seeded in a 6-well plate
(5×105 cells/well) were incubated with 50 µg/ml of CHX
(Life Technologies) for 0, 30, 60 and 120 min. After culture, these
cells were washed twice with PBS and lysed. The cell lysates were
then subjected to western blot analysis for detection of KIT and
GAPDH.
Analysis of the ubiquitination status
of KIT
CoMS and rCoMS1 cells were lysed and the KIT protein
was immunoprecipitated using Protein G-agarose beads (Life
Technologies) bound with polyclonal rabbit anti-human KIT (Dako
Corp., Carpinteria, CA, USA). The immunoprecipitated proteins were
subjected to western blot analysis using polyclonal rabbit
anti-ubiquitin antibody (Abcam, Cambridge, MA, USA) or polyclonal
goat anti-human KIT (C-14) (Santa Cruz Biotechnology).
Analysis of KIT status in rCoMS1 cells
following imatinib withdrawal and subsequent imatinib
re-treatment
rCoMS1 cells that were maintained in cRPMI
supplemented with 1 µM of imatinib were transferred into
imatinib-free cRPMI and culture was continued for a further 0, 7,
14 and 21 days. The rCoMS1 cells that were cultured in
imatinib-free cRPMI for 14 days (rCoMS1-IM14d cells) were further
cultured in cRPMI supplemented with 1 µM of imatinib for 0, 6, 24,
48 and 72 h. The expression levels of KIT in rCoMS1 cells under
these conditions were examined by western blot analysis. The
rCoMS1-IM14d cells and the cells that had been re-treated with
imatinib for 48 h (rCoMS1-IM14d+IM48h cells) were subjected to a
CHX chase assay, ubiquitination status analysis, KIT and
phosphorylated KIT analysis, and cell growth inhibition assay using
the methods described above. The rCoMS1-IM14d cells were also
cultured in cRPMI with imatinib (0 or 1 µM) and the pan
deubiquitinating enzyme inhibitor PR619 (Sigma-Aldrich, St. Louis,
MO, USA) (0 or 20 µM) for 48 h, following which the expression
level of KIT was examined using western blot analysis.
Statistical analysis
Statistical analysis was performed using an unpaired
two-tailed Student's t-test. Significance was accepted at
P<0.05.
Results
Overexpression of activated KIT in the
rCoMS1 imatinib-resistant cell line
A canine MCT cell line, CoMS, has been used in this
study because canine MCT can be a spontaneous model for
translational research of imatinib resistance in KIT
mutation-driven human tumors and CoMS cells have an
imatinib-sensitive activating mutation in KIT. An
imatinib-resistant CoMS cell line, rCoMS1, was generated by the
continuous exposure of CoMS cells to increasing concentrations of
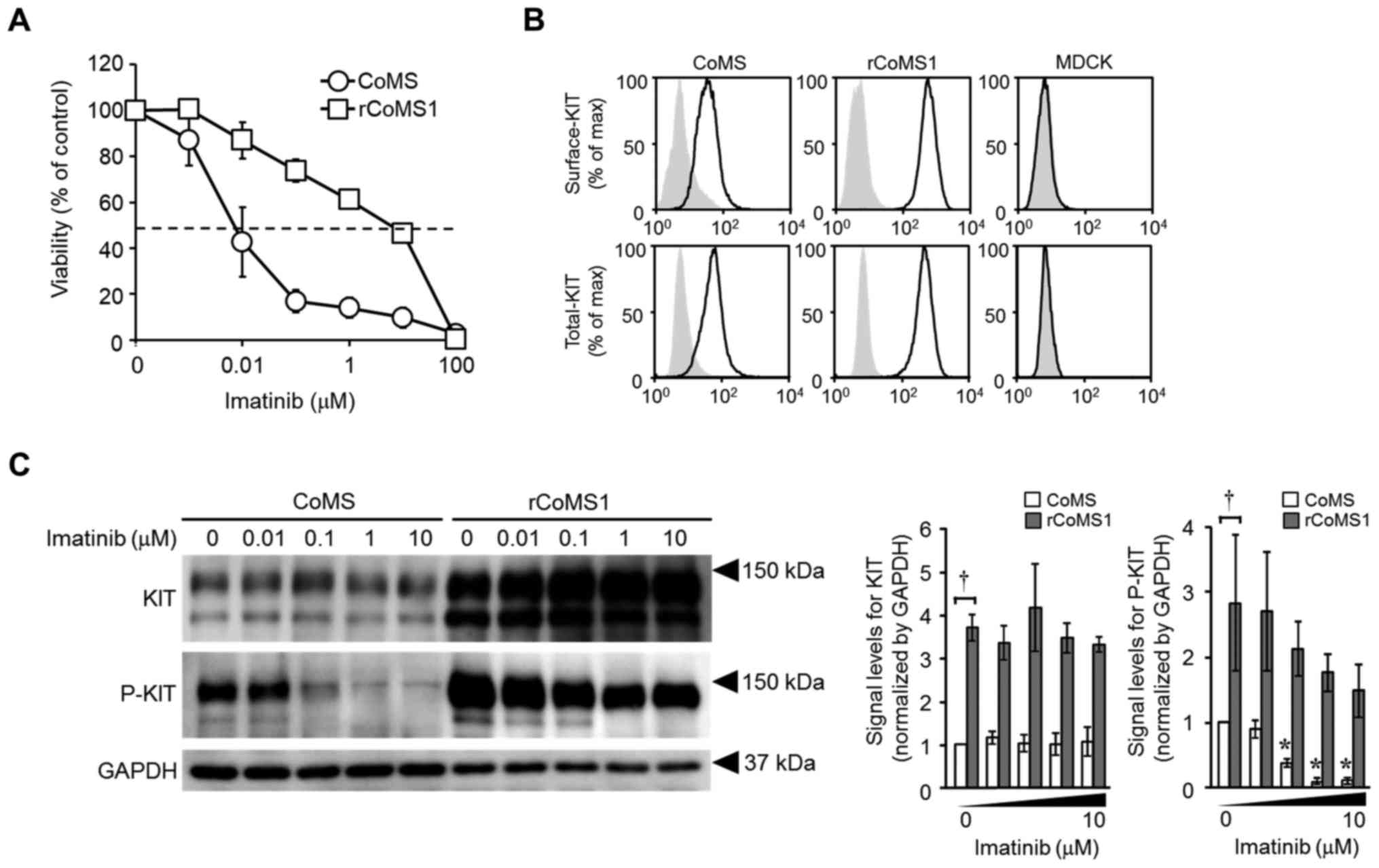
imatinib (Fig. 1A). The calculated
IC50 value of imatinib for rCoMS1 cells was >200-fold
higher than that for CoMS cells (IC50, CoMS, 0.038±0.022
µM; rCoMS1, 9.0±2.7 µM; P<0.01). KIT was mostly expressed on the
cell surface of both CoMS and rCoMS1 cells but the expression level
of KIT was much higher on rCoMS1 cells compared with CoMS cells
(Fig. 1B). The entire nucleotide
sequence of KIT was examined in CoMS and rCoMS1 cells but no
mutation other than the original mutation of c.1720_1772+1dup was
found in both cell lines (data not shown).
In western blot analysis of KIT, the signals of both
KIT and phosphorylated KIT were much higher in rCoMS1 cells
compared with those in CoMS cells under the non-treated condition
(Fig. 1C, imatinib 0 µM). The
phosphorylation of KIT was concentration dependently suppressed by
imatinib in CoMS cells and strong suppression of phosphorylation
was detected following treatment with 0.1 µM or more of imatinib
(Fig. 1C). In contrast, there was
only a small imatinib-induced decrease in KIT phosphorylation in
rCoMS1 cells and a high level of KIT phosphorylation was maintained
under treatment with the maximum concentration of imatinib (10 µM)
(Fig. 1C).
Prolonged life-span of KIT caused by a
decrease in its ubiquitination in rCoMS1 cells
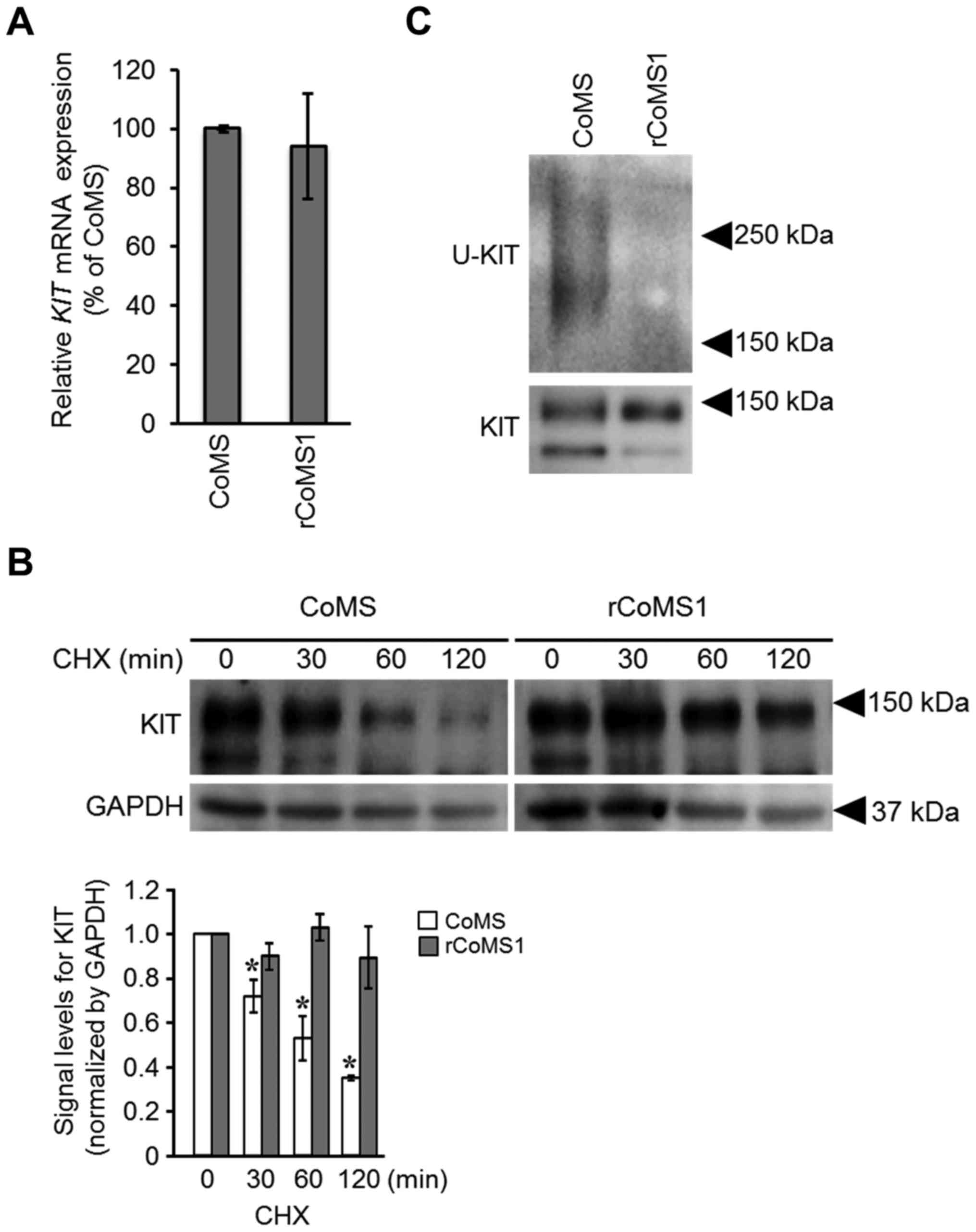
To examine if overexpression of KIT in rCoMS1 cells
is due to an increase in the level of the KIT transcripts,
the expression level of KIT mRNA was evaluated. No
difference in the expression level of KIT mRNA was found
between CoMS and rCoMS1 cells (Fig.
2A). We then examined the difference in KIT protein life-span
between CoMS and rCoMS1 cells. For this purpose, a CHX chase assay,
which monitors the time course of protein degradation after
translation is arrested by CHX treatment, was employed (Fig. 2B). In CoMS cells, the amount of KIT
was time-dependently decreased after treatment of the cells with
CHX and KIT was only weakly detected after treatment of 60 min. In
contrast, there was only a small decrease in the level of KIT after
treatment of rCoMS1 cells with CHX, and a high level of KIT
remained at 120 min of CHX treatment, indicating a prolonged
life-span of KIT in rCoMS1 cells. Since this prolonged life-span
could be caused by retardation of KIT degradation, the
ubiquitination status of KIT in CoMS and rCoMS1 cells was examined
using western blot analysis (Fig.
2C). Ubiquitinated KIT was clearly detected in CoMS cells,
while it was barely detected in rCoMS1 cells (Fig. 2C).
Imatinib-induced KIT
deubiquitination-mediated reversible overexpression of KIT in
rCoMS1 cells
To clarify if the prolonged life-span of KIT in
rCoMS1 cells was caused in response to imatinib, we examined the
changes in the KIT expression level in rCoMS1 cells under
conditions of imatinib-withdrawal and subsequent imatinib
re-treatment (Fig. 3). The
expression level of KIT in rCoMS1 cells gradually decreased over
time after removal of imatinib from the culture medium and a strong
decrease was noted on day 14 and thereafter (Fig. 3A, upper panel). The rCoMS1 cells
that had been cultured for 14 days without imatinib (rCoMS1-IM14d
cells) were then re-treated with imatinib (Fig. 3A, lower panel). KIT expression
increased again at 24 h, reaching a maximum at 48 h after
re-treatment of the rCoMS1-IM14d cells with imatinib. In contrast,
no difference in the expression level of KIT mRNA was found
between rCoMS1 cells, rCoMS1-IM14d cells and rCoMS1-IM14d cells
that had been re-treated with imatinib for 48 h (rCoMS1-IM14d+IM48h
cells) (Fig. 3B). To examine the
difference in the life-span of KIT between rCoMS1-IM14d cells and
rCoMS1-IM14d+IM48h cells, a CHX chase assay was performed (Fig. 3C). After protein synthesis was
inhibited by treatment with CHX, the KIT in the rCoMS1-IM14d cells
was degraded in a time-dependent manner, while little KIT
degradation was observed over time in the rCoMS1-IM14d+IM48h cells
(Fig. 3C). Fig. 3D shows the ubiquitination status of
KIT in rCoMS1-IM14d and rCoMS1-IM14d+IM48h cells. Ubiquitinated KIT
was clearly detected in the rCoMS1-IM14d cells, while it was barely
detected in the rCoMS1-IM14d+IM48h cells. Since dysregulation of
deubiquitinating enzyme(s) is one potential mechanism that might
impair ubiquitination-mediated downregulation of receptor tyrosine
kinases (18,19), the effects of the deubiquitinating
enzyme inhibitor PR619 on the induction of KIT overexpression by
imatinib in rCoMS1-IM14d cells was examined. As shown in Fig. 3E, PR619 strongly suppressed
imatinib-induced KIT overexpression in rCoMS1-IM14d cells.
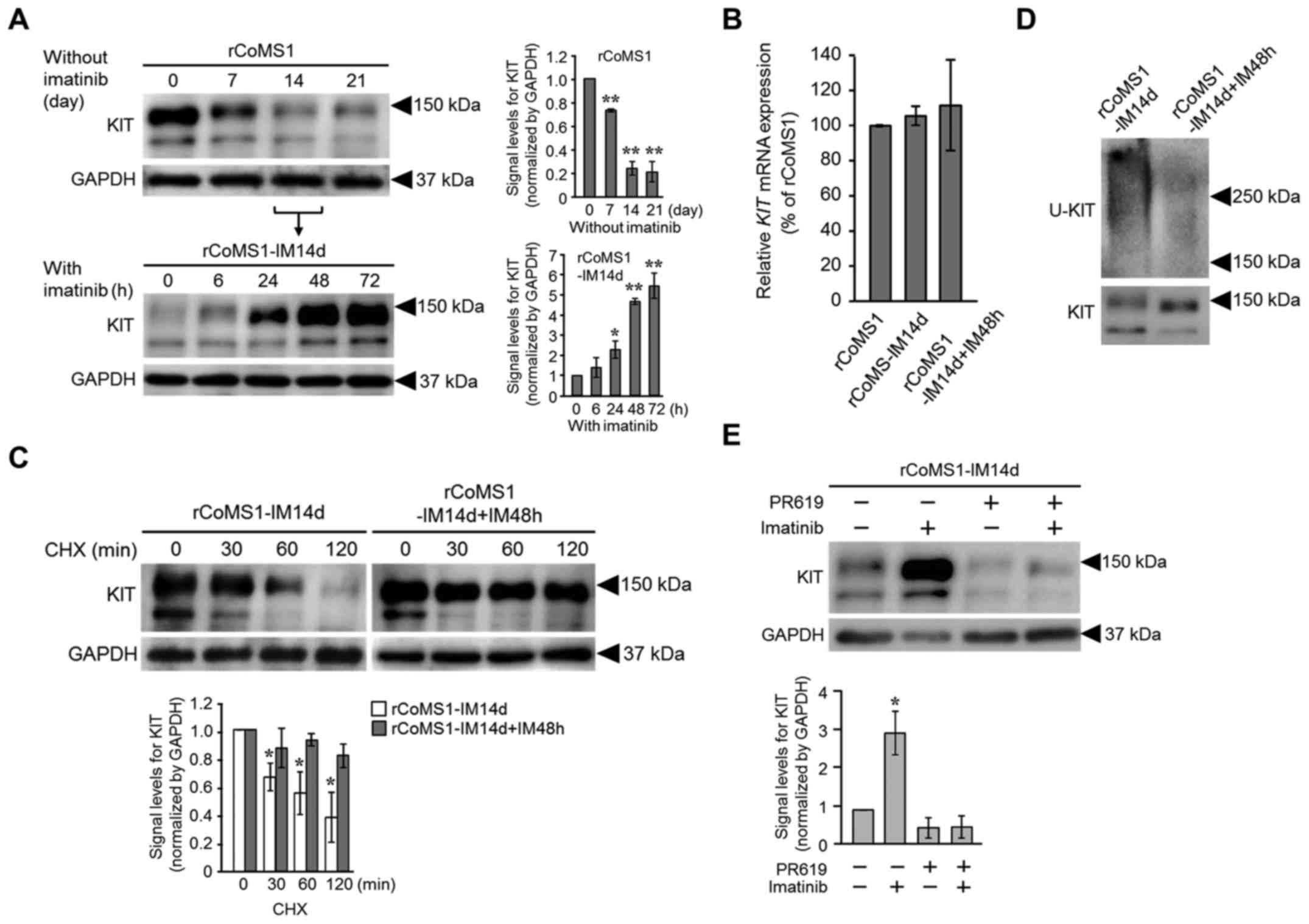 | Figure 3.Effects of imatinib-withdrawal and
subsequent imatinib re-treatment on KIT expression and
ubiquitination status in rCoMS1 cells. (A) Changes in the
expression level of KIT in rCoMS1 cells under imatinib-withdrawal
and subsequent imatinib re-treatment. rCoMS1 cells were cultured
for 0–21 days without imatinib (upper panel). rCoMS1 cells cultured
for 14 days in the absence of imatinib (rCoMS1-IM14d) were then
re-treated with 1 µM imatinib for 0–72 h (lower panel). Expression
of KIT in rCoMS1 cells under these conditions was examined by
western blot analysis. GAPDH was blotted as an internal control.
Left panel, representative western blot image of three independent
experiments. Right panel, semi-quantification of the signal levels
of KIT normalized to GAPDH is shown. The normalized signal level of
KIT on day 0 (rCoMS1) and at 0 h (rCoMS1-IM14d) was set at 1.0.
Data are expressed as means ± SD (n=3). *P<0.05, **P<0.01.
Significant difference vs. day 0 (rCoMS1) or 0 h (rCoMS1-IM14d).
(B) Semi-quantitative RT-PCR analysis of KIT mRNA
expression. The expression levels of KIT mRNA in
rCoMS1-IM14d cells and rCoMS1-IM14d+IM48h cells were expressed as a
percentage of that in rCoMS1 cells. Data are expressed as means ±
SD (n=4). No statistically significant difference in KIT
expression was found between these cell lines. (C) Cycloheximide
(CHX) chase assay for KIT expression in rCoMS1-IM14d cells and in
the same cells that were further cultured for 48 h in the presence
of 1 µM imatinib (rCoMS1-IM14d+IM48h). After treatment of the cells
with CHX (50 µg/ml) for the indicated time (0–120 min), the cells
were subjected to western blot analysis for detection of KIT and
control GAPDH. Upper panel, representative western blot image of
three independent experiments. Lower panel, semi-quantification of
the signal levels of KIT normalized to GAPDH is shown (white bars,
rCoMS1-IM14d; grey bars, rCoMS1-IM14d+IM48h). The normalized signal
level of KIT in CHX 0 min cells was set at 1.0. Data are expressed
as means ± SD (n=3). *Significant difference vs. 0 min (P<0.01).
(D) Western blot analysis of the ubiquitination status of KIT
(U-KIT) in rCoMS1-IM14d and rCoMS1-IM14d+IM48h cells. An
ubiquitin-positive smear band was detected in the lane of
rCoMS1-IM14d cells but was barely detected in that of
rCoMS1-IM14d+IM48h cells. The data are representative of three
independent experiments. (E) Effect of the pan deubiquitinating
enzyme inhibitor PR619 (20 µM) on the expression status of KIT in
rCoMS1-IM14d cells cultured in the presence or absence of imatinib
(1 µM) for 48 h. KIT and control GAPDH were detected by western
blot analysis. Upper panel, representative western blot image of
three independent experiments. Lower panel, semi-quantification of
the signal levels of KIT normalized to GAPDH is shown. The
normalized signal level of KIT in the condition of imatinib
(−)/PR619 (−) was set at 1.0. Data are expressed as means ± SD
(n=3). *Significant difference vs. imatinib (−)/PR619 (−)
(P<0.01). |
Linkage of recovery of imatinib
sensitivity and re-acquirement of resistance to imatinib of rCoMS1
cells to KIT expression status
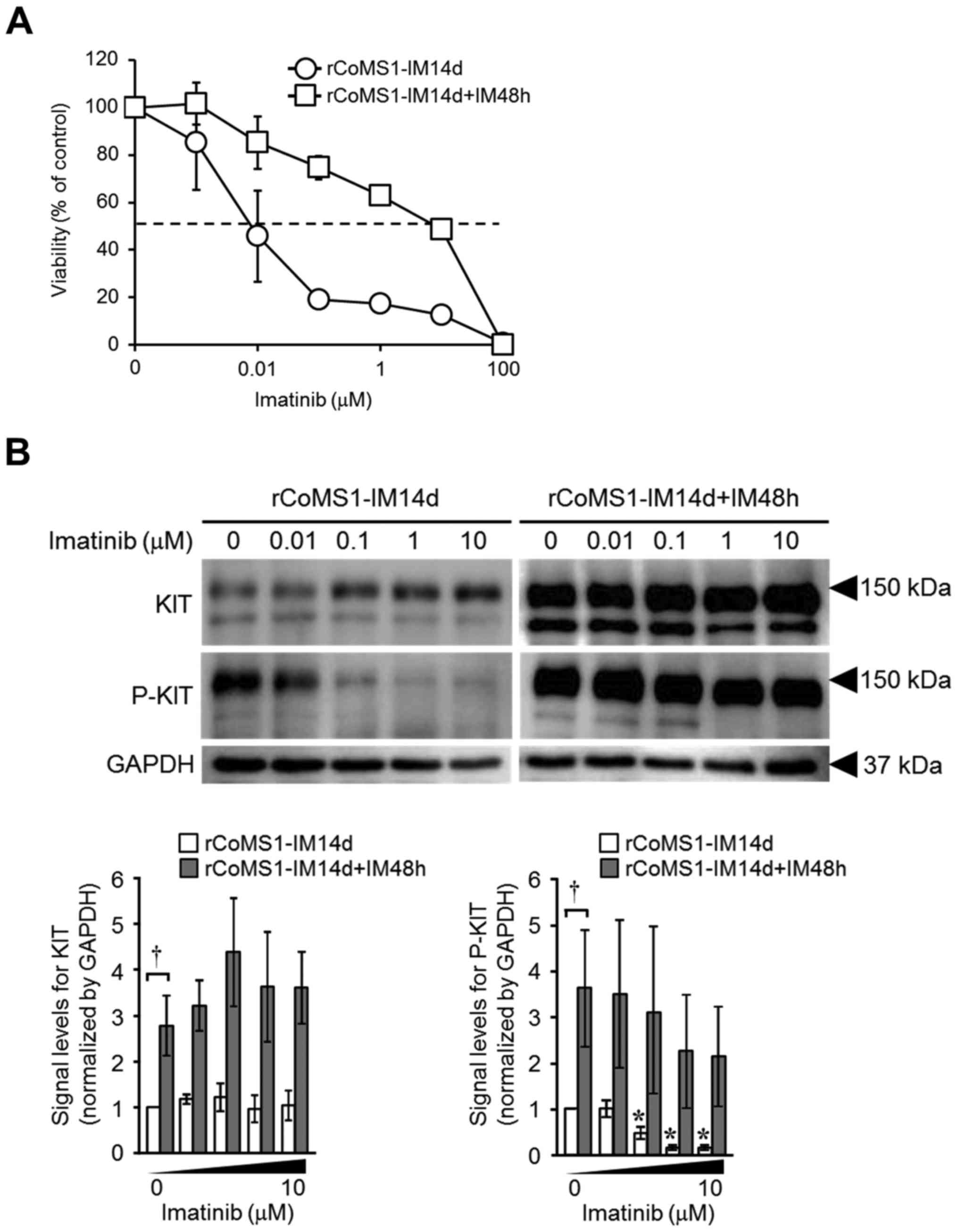
To confirm that the sensitivity of rCoMS1 cells to
imatinib was linked to the expression status of KIT, we examined
the effects of imatinib on the growth of rCoMS1-IM14d and
rCoMS1-IM14d+IM48h cells. As shown in Fig. 4A, growth of the rCoMS1-IM14d cells
was efficiently inhibited by imatinib, while rCoMS1-IM14d+IM48h
cells had re-acquired resistance to imatinib (IC50:
rCoMS1-IM14d, 0.043±0.023 µM; rCoMS1-IM14d+IM48h, 9.45±2.85 µM;
P<0.01). Consistent with these findings, imatinib efficiently
suppressed the phosphorylation of KIT in rCoMS1-IM14d cells, while
it failed to suppress the phosphorylation of the re-overexpressed
KIT in rCoMS1-IM14d+IM48h cells (Fig.
4B).
Discussion
This is the first report to suggest that an
imatinib-induced decrease in ubiquitination and a subsequent
prolonged life-span of KIT underlies the overexpression of KIT that
contributes to imatinib resistance of KIT mutation-driven
tumors.
The role of KIT overexpression in the acquisition of
resistance to imatinib in CoMS cells was supported by the findings
that: i) a high level of KIT phosphorylation was maintained in the
presence of imatinib; ii) the resistance of rCoMS1 cells to
imatinib was clearly linked to changes in the expression status of
KIT; and iii) no secondary mutation in KIT was identified in
rCoMS1 cells. Analyses of the expression of KIT mRNA and the
life-span of KIT, suggested that a prolonged KIT life-span rather
than an increase in de novo synthesis caused the observed
overexpression of KIT. Moreover, this prolonged life-span of KIT
was caused by a decrease in KIT ubiquitination that would result in
the retardation of its degradation in rCoMS1 cells. The decrease in
KIT ubiquitination in rCoMS1 cells may also explain why KIT is
overexpressed on the surface of rCoMS1 cells. Following activation,
cell surface KIT is ubiquitinated and subsequently targeted for
endocytosis and degradation in the lysosomes (20). Thus, a decrease in KIT
ubiquitination and subsequent retarded degradation of KIT may
result in its accumulation and overexpression on the surface of
rCoMS1 cells.
Regarding the overexpression of KIT in rCoMS1 cells,
we found that: i) KIT overexpression in rCoMS1 cells was decreased
by withdrawal of imatinib (rCoMS1-IM14d cells) and KIT was
re-overexpressed by re-treatment with imatinib (rCoMS1-IM14d+IM48h
cells); ii) KIT life-span was shortened in rCoMS1-IM14d cells
compared to rCoMS1 cells but its life-span increased again in
rCoMS1-IM14d+IM48h cells; and iii) KIT was ubiquitinated in
rCoMS1-IM14d cells but its ubiquitination level was markedly
decreased in rCoMS1-IM14d+IM48h cells. These findings confirmed
that the following series of events was induced in response to
imatinib: a decrease in KIT ubiquitination, a prolongation of KIT
life-span, and the resulting overexpression of KIT in rCoMS1 cells.
These findings also indicated that imatinib-induced overexpression
of KIT in rCoMS1 cells is not a permanently acquired feature but is
a reversible response of the cells. Moreover, imatinib failed to
induce overexpression of KIT in rCoMS1-IM14d cells in the presence
of the pan deubiquitinating enzyme inhibitor PR619 (Fig. 3E). This finding suggested that the
imatinib-induced decrease in KIT ubiquitination could be mediated
by upregulation and/or activation of deubiquitinating enzyme(s). In
the PR619 experiment, PR619 did not completely suppress KIT
expression. The residual KIT could be an inactivated form of KIT
since, although activated KIT undergoes ubiquitin-mediated
degradation, inactivated KIT is degraded via an
ubiquitin-independent process (21). Since a deubiquitinating enzyme
specific for ubiquitinated KIT has not yet been identified, we were
unable to confirm the role of such an enzyme in imatinib modulation
of KIT by using a KIT-specific deubiquitinating enzyme inhibitor.
However, in a recent study, it was shown that aberrantly activated
USP8, an enzyme that deubiquitinates several receptor tyrosine
kinases such as EGFR, Frizzled, ERBB, Met and Smoothened (18), induces the cell surface
overexpression of the EGFR by switching the endocytic trafficking
pathway of the EGFR from the endosome-lysosome degradation pathway
to the recycling pathway that recycles EGFR back to the plasma
membrane (19). Collectively, this
finding and our results might suggest that upregulation and/or
activation of a KIT deubiquitinating enzyme by imatinib could
induce a similar pathway switching of KIT that induces cell surface
overexpression of KIT on CoMS cells.
Dysregulation of ubiquitination pathways that
underlies resistance to kinase inhibitors has also been reported in
human melanomas (22). The
ubiquitin ligase, RNF125, is downregulated in BRAF
inhibitor-resistant melanomas, resulting in increased expression of
its substrate, JAK1, with a concomitant increase in EGFR expression
(22). This overexpression of JAK1
and EGFR was suggested to underlie the resistance of melanoma cells
to BRAF inhibitors. In contrast with BRAF inhibitor-resistant
melanomas, downregulation of an ubiquitin ligase such as Cbl, which
is an enzyme that can ubiquitinate KIT (23), would not be involved in imatinib
resistance in rCoMS1 cells because: i) overexpression of KIT was
clearly inhibited by a deubiquitinating enzyme inhibitor in
rCoMS1-IM14d cells and, ii) overexpression of KIT due to
downregulation of a ubiquitin ligase would not be prevented by a
deubiquitinating enzyme inhibitor.
The development of KIT-overexpressing
imatinib-resistant rCoMS1 cells required >2 months culture in
the presence of imatinib, whereas imatinib-sensitive rCoMS1-IM14d
cells re-overexpressed KIT and acquired resistance to imatinib in a
short period (~24–48 h) after re-treatment with imatinib. Although
the mechanisms that would explain this difference are currently
unclear, it may be possible that CoMS cells gradually became
committed to a steady state high expression level of the KIT
deubiquitinating enzyme during their continuous long-term exposure
to imatinib. Once the CoMS cells had acquired this property then
the addition of imatinib could directly or indirectly trigger
activation of this enzyme, resulting in the immediate
re-overexpression of KIT and the acquisition of resistance to
imatinib.
In conclusion, we have shown that a decrease in KIT
ubiquitination by imatinib treatment induces the overexpression of
KIT and subsequent imatinib resistance in imatinib sensitive canine
MCT cells. Moreover, this decrease in KIT ubiquitination was
considered to be associated with the activation of a
deubiquitinating enzyme by imatinib. Since canine MCT cells share
similar properties with KIT mutation-driven human tumors in terms
of sensitivity and the ability to acquire resistance against
imatinib, it may be possible that a similar mechanism of KIT
overexpression underlies the acquisition of imatinib resistance in
some human tumors that are driven by KIT mutation. For a possible
transfer of our findings to the human setting, further analysis
using canine clinical cases and human cell lines will be
needed.
Acknowledgements
This study was supported in part by a Grant-in-Aid
for Scientific Research (no. 15H04601) from the Ministry of
Education, Culture, Sports, Science and Technology of Japan.
References
|
1
|
Blay JY, Casali PG, Tos Dei AP, Le Cesne A
and Reichardt P: Management of gastrointestinal stromal tumour:
Current practices and visions for the future. Oncology. 89:1–13.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alvarez-Twose I, Matito A, Morgado JM,
Sánchez-Muñoz L, Jara-Acevedo M, García-Montero A, Mayado A, Caldas
C, Teodósio C, Muñoz-González JI, et al: Imatinib in systemic
mastocytosis: A phase IV clinical trial in patients lacking exon 17
KIT mutations and review of the literature. Oncotarget. Jul
19–2016.(Epub ahead of print). doi: 10.18632/oncotarget.10711.
|
|
3
|
Hodi FS, Corless CL, Giobbie-Hurder A,
Fletcher JA, Zhu M, Marino-Enriquez A, Friedlander P, Gonzalez R,
Weber JS, Gajewski TF, et al: Imatinib for melanomas harboring
mutationally activated or amplified KIT arising on mucosal, acral,
and chronically sun-damaged skin. J Clin Oncol. 31:3182–3190. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gramza AW, Corless CL and Heinrich MC:
Resistance to tyrosine kinase inhibitors in gastrointestinal
stromal tumors. Clin Cancer Res. 15:7510–7518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miranda C, Nucifora M, Molinari F, Conca
E, Anania MC, Bordoni A, Saletti P, Mazzucchelli L, Pilotti S,
Pierotti MA, et al: KRAS and BRAF mutations predict primary
resistance to imatinib in gastrointestinal stromal tumors. Clin
Cancer Res. 18:1769–1776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gounder MM and Maki RG: Molecular basis
for primary and secondary tyrosine kinase inhibitor resistance in
gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 67
Suppl 1:S25–S43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Debiec-Rychter M, Cools J, Dumez H, Sciot
R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, et
al: Mechanisms of resistance to imatinib mesylate in
gastrointestinal stromal tumors and activity of the PKC412
inhibitor against imatinib-resistant mutants. Gastroenterology.
128:270–279. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miselli FC, Casieri P, Negri T, Orsenigo
M, Lagonigro MS, Gronchi A, Fiore M, Casali PG, Bertulli R, Carbone
A, et al: c-KIT/PDGFRA gene status alterations possibly related to
primary imatinib resistance in gastrointestinal stromal tumors.
Clin Cancer Res. 13:2369–2377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lai S, Wang G, Cao X, Luo X, Wang G, Xia
X, Hu J and Wang J: KIT over-expression by p55PIK-PI3K leads to
Imatinib-resistance in patients with gastrointestinal stromal
tumors. Oncotarget. 7:1367–1379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Irie M, Takeuchi Y, Ohtake Y, Suzuki H,
Nagata N, Miyoshi T, Kagawa Y and Yamagami T: Imatinib mesylate
treatment in a dog with gastrointestinal stromal tumors with a
c-KIT mutation. J Vet Med Sci. 77:1535–1539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kobayashi M, Kuroki S, Ito K, Yasuda A,
Sawada H, Ono K, Washizu T and Bonkobara M: Imatinib-associated
tumour response in a dog with a non-resectable gastrointestinal
stromal tumour harbouring a c-KIT exon 11 deletion mutation. Vet J.
198:271–274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
London CA, Galli SJ, Yuuki T, Hu ZQ,
Helfand SC and Geissler EN: Spontaneous canine mast cell tumors
express tandem duplications in the proto-oncogene c-KIT. Exp
Hematol. 27:689–697. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bostock DE: Neoplasms of the skin and
subcutaneous tissues in dogs and cats. Br Vet J. 142:1–19. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bonkobara M: Dysregulation of tyrosine
kinases and use of imatinib in small animal practice. Vet J.
205:180–188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
London CA, Malpas PB, Wood-Follis SL,
Boucher JF, Rusk AW, Rosenberg MP, Henry CJ, Mitchener KL, Klein
MK, Hintermeister JG, et al: Multi-center, placebo-controlled,
double-blind, randomized study of oral toceranib phosphate
(SU11654), a receptor tyrosine kinase inhibitor, for the treatment
of dogs with recurrent (either local or distant) mast cell tumor
following surgical excision. Clin Cancer Res. 15:3856–3865. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kobayashi M, Kuroki S, Tanaka Y, Moriya Y,
Kozutumi Y, Uehara Y, Ono K, Tamura K, Washizu T and Bonkobara M:
Molecular changes associated with the development of resistance to
imatinib in an imatinib-sensitive canine neoplastic mast cell line
carrying a KIT c.1523A>T mutation. Eur J Haematol. 95:524–531.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ishiguro T, Kadosawa T, Mori K, Takagi S,
Okumura M and Fujinaga T: Establishment and characterization of a
new canine mast cell tumor cell line. J Vet Med Sci. 63:1031–1034.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin WL, Mao XY and Qiu GZ: Targeting
deubiquitinating enzymes in glioblastoma multiforme: Expectations
and challenges. Med Res Rev. 37:627–661. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reincke M, Sbiera S, Hayakawa A,
Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T,
Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, et al: Mutations in the
deubiquitinase gene USP8 cause Cushing's disease. Nat Genet.
47:31–38. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masson K, Heiss E, Band H and Rönnstrand
L: Direct binding of Cbl to Tyr568 and Tyr936 of the stem cell
factor receptor/c-KIT is required for ligand-induced
ubiquitination, internalization and degradation. Biochem J.
399:59–67. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yee NS, Hsiau CW, Serve H, Vosseller K and
Besmer P: Mechanism of down-regulation of c-KIT receptor. Roles of
receptor tyrosine kinase, phosphatidylinositol 3′-kinase, and
protein kinase C. J Biol Chem. 269:31991–31998. 1994.PubMed/NCBI
|
|
22
|
Kim H, Frederick DT, Levesque MP, Cooper
ZA, Feng Y, Krepler C, Brill L, Samuels Y, Hayward NK, Perlina A,
et al: Downregulation of the ubiquitin ligase RNF125 underlies
resistance of melanoma cells to BRAF inhibitors via JAK1
deregulation. Cell Rep. 11:1458–1473. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeng S, Xu Z, Lipkowitz S and Longley JB:
Regulation of stem cell factor receptor signaling by Cbl family
proteins (Cbl-b/c-Cbl). Blood. 105:226–232. 2005. View Article : Google Scholar : PubMed/NCBI
|