Introduction
Chemo- and radio-resistance are major clinical
problems in cancer treatment, and often lead to tumor recurrence
and even a fatal outcome. Increasing evidence has demonstrated that
the three hallmarks of tumor malignancy, metastasis, neoplastic
stem cells and chemo- and radio-resistance are not independent from
one another (1,2). Induction of epithelial-to-mesenchymal
transition (EMT) concomitantly results in the emergence of cancer
stem cells (CSCs) with enhanced tumor-initiating capacity, changes
to specific cell-surface markers, increased chemo- and
radio-resistance and resistance to apoptosis (3–5).
By suppressing the transcription of E-cadherin and
stemness-inhibiting microRNAs, or driving non-CSCs to enter the CSC
state in certain types of cancer cells, zinc finger E-box binding
homeobox 1 (ZEB1) acts as a key contributor to EMT activation and
stemness maintenance (6–8). Additionally, data has indicated that
ZEB1 is also crucial for chemoresistance and radioresistance
(9). The chemotherapeutic drug
gemcitabine has demonstrated a limited success rate in pancreatic
cancer; however, a previous study revealed that the stable
knockdown of ZEB1 was able to significantly sensitize pancreatic
cancer cells to gemcitabine treatment (7). The expression level of ZEB1 in
pancreatic cancer cells also correlates with the resistance of
these cells to 5-fluorouracil and cisplatin (10). With regard to radiotherapy, comet
assays have demonstrated that ZEB1-depleted SUM159 cells
experienced more DNA damage following ionizing radiation treatment
(11), while overexpression of the
major ZEB1 target microRNAs, miR-200c or miR-205, efficiently
sensitized various types of cancer cells to irradiation by
suppressing DNA repair protein, ubiquitin-conjugating enzyme E2N,
and stemness factors (12,13).
Although ZEB1 has a clear mechanism regarding its
effect on EMT and dedifferentiation, how it promotes chemo- and
radio-resistance remains to be elucidated. Other studies have
reported that by directly interacting with ubiquitin-specific
peptidase (USP)7 and enhancing its deubiquitylating activity on
checkpoint kinase 1 (CHK1) (11),
ZEB1 was positively correlated with the protein level of CHK1 and
was required for the clearance of DNA double-strand breaks (DSBs).
This indicates that ZEB1 plays a role in the DNA damage response
(DDR), because CHK1 is an important protein of the DDR that
regulates homologous recombination repair and prevents G2 to M
transition (14).
Although activation of DDR pathways may decrease
tumor development by acting as a barrier to unchecked proliferation
in its early stages (15), when
aberrantly activated, DDR has been demonstrated to be involved in
the development of cancer cell resistance to the lethal effects of
genotoxic agents (16). By
coordinating cell cycle arrest, apoptosis, senescence and DNA
repair pathways, DDR forms a complex network that is responsible
for various outcomes following DNA damage (17). However, due to the loss of p53, an
apoptosis-inducing DDR regulator, and other unknown mechanisms,
tumor cells rely heavily upon the DNA repair pathways and thus,
struggle to survive following DNA damage, with increased genome
instability as a consequence (18).
We hypothesized that, in addition to stabilizing
CHK1, the transcriptional repressor ZEB1 may be involved in an
unbalanced DDR by affecting its other key components. To further
understand the targets via which ZEB1 fulfills its regulation of
DDR, the present study mapped the target gene occupancy of ZEB1 in
colorectal cancer cells using a chromatin immunoprecipitation
(ChIP)-on-chip method (19), and
identified USP17-like family member 2 (DUB3), chromodomain helicase
DNA-binding protein 1-like (CHD1L) and double homeobox 4 (DUX4) as
three potential downstream genes of ZEB1. Additionally, the results
indicate that the suppression of the three genes by ZEB1 mediates a
dysregulated DDR, leading to chemoresistance and anti-apoptotic
processes.
Materials and methods
Cell culture
The human colorectal cancer cell line LoVo, which
was purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA), was cultured in F12K medium supplemented with
10% heat-inactivated fetal bovine serum (FBS), penicillin (100
U/ml) and streptomycin (100 g/ml), in a humidified 5%
CO2 incubator at 37°C.
ChIP
The ChIP was performed following the instructions of
an Enzymatic Chromatin IP kit (cat no. 9003; Cell Signaling
Technology, Inc., Danvers, MA, USA). Briefly, ~4×107
cells were prepared for each experiment. Formaldehyde (1%
concentration) was added to crosslink proteins to DNA for 10 min.
Following cell lysis and nuclei collection, the chromatin was
fragmented by partial digestion with micrococcal nuclease to obtain
chromatin fragments of 1–5 nucleosomes in size (150–900 bp). The
crosslinked chromatin preparation was then immunoprecipitated with
5 µg polyclonal ZEB1 antibody (cat no. sc-25388 X; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) or negative control normal
rabbit IgG (cat no. 2729; Cell Signaling Technology, Inc.) at 4°C
overnight. Elution of chromatin from the crosslinked complex using
Protein G magnetic beads was performed with KingFisher Flex
Magnetic Particle Processors. After reversing the crosslinks, DNA
was purified using the reagents and spin columns provided in the
Enzymatic Chromatin IP kit. In addition, a positive control histone
H3 rabbit monoclonal antibody (cat no. 4620; Cell Signaling
Technology, Inc.) and a primer for its known binding gene,
ribosomal protein L30, were also included in the experiment to
analyze IP efficiency. Three biological replicates were performed
and successful enrichment was validated in each experiment.
Array hybridization, staining and
scanning
ChIP DNA was amplified to 7.5 µg, using the
high-performance liquid chromatography-purified primers: A,
5′-GTTTCCCAGTCACGG TC(N)9-3′; and B,
5′-GTTTCCCAGTCACGGTC-3′. The dUTP-containing post-amplified samples
was then fragmented by uracil-DNA glycosylase and labeled by
terminal deoxynucleotidyl transferase (cat no. 72033) with the
biotin-like labeling reagent (cat no. 79015) (both from USB Corp.;
Affymetrix, Inc., Santa Clara, CA, USA). The labeled DNA was
hybridized to six Affymetrix GeneChip Human Promoter 1.0R arrays
(three biological replicates using ZEB1 antibody and three using
normal rabbit IgG) at 45°C for 16 h. The arrays were washed and
stained by R-phycoerythrin streptavidin (cat no. S-866; Molecular
Probes; Thermo Fisher Scientific, Inc., Waltham, MA, USA) using
Fluidics Station 450 and then scanned using GeneChip Scanner 3000
7G, which was controlled by Affymetrix GeneChip Command Console
software (Affymetrix, Inc.).
Array data analysis
Affymetrix Tiling Analysis software (Affymetrix,
Inc.) and Integrated Genome Browser were used to select the
positive intervals that satisfied the two conditions: that the
signal intensity of the ZEB1-binding DNA and the intensity ratio of
the ZEB1-binding DNA signal/normal rabbit IgG-binding DNA signal
were within the top 1% among all the intervals. The filter criteria
ruled out the false-positive regions with a high intensity ratio
that resulted from the division of two infinitely small numbers.
The positive intervals of each chromosome in bed files were
uploaded to the GALAXYP online platform (usegalaxyp.org) to produce a tail-to-head
concatenation and then submitted to the Cis-regulatory Element
Annotation System (CEAS) to produce nearby gene mapping and motif
identification. For each positive ChIP region, CEAS identified the
nearest RefSeq gene names and predicted locations within 300 kb
upstream or downstream of the gene. Regions within 1 kb upstream
from the gene 5′ start site were assumed to be proximal promoters;
those within 1 kb downstream from the gene 3′-end were reported to
be immediate downstream regulatory elements; while those
distributed >1 kb from the gene were assumed to be enhancers
(20).
Short hairpin RNA (shRNA) constructs
and transfection
The LoVo cell line was selected as a suitable host
for transfection. To avoid off-target effects, two different
sequences were used to knock down ZEB1. Using Endofectin™ Plus,
cells were transfected with plasmids (both from GeneCopoeia, Inc.,
Rockville, MD, USA) containing short hairpin RNA targeting ZEB1
HSH017963-4-LVRH1GP (OS241004) or HSH017963-5-LVRH1GP (OS397209).
Parallel transfection was performed using plasmids with
non-targeting shRNA CSHCTR001-1-LVRH1GP (OSNEG20) to generate
scrambled control clones. The cells were then selected with
puromycin and after 4 weeks, single colonies were analyzed for ZEB1
expression by western blotting assay. The results revealed that the
transfections had successfully induced a decrease in the protein
level of ZEB1. The shRNA targeted sequences were as follows:
ZEB1-knockdown-1, 5′-GGTCAACTATCACTAGTGT-3′; ZEB1-knockdown-2,
5′-TGATCAGCCTCAATCTGCA-3′; and scrambled control,
5′-GCTTCGCGCCGTAGTCTTA-3′. Knockdown is referred to as KD
hereinafter.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA from the cells was extracted and reverse
transcribed into cDNA according to the protocols of the Total RNA
kit I (Omega Bio-Tek, Inc., Norcross, GA, USA) and First Strand
cDNA Synthesis kit (GeneCopoeia, Inc.). The cDNA template (100 ng)
was amplified using 0.2 µM primers and the 20 µl SYBR-Green-based
qPCR reaction system (GeneCopoeia, Inc.). A Bio-Rad IQ 5 instrument
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used to perform
the reaction and detect the fluorescent signals. Standard curves
were created to confirm the amplification efficiency of each gene,
while melting curves ensured the specificity of the amplification.
Normalization to the reference gene (β-actin) was performed for
each sample. Fold changes between the mRNA level of the ZEB1-KD
groups and the scrambled control group were calculated according to
the 2−ΔΔCq method (21).
The sequences of primers were as follows (5′-3′): USP17 forward,
AGGTGAGTGGCAGTTCAACC and reverse, GGAAGCTTCTTCCTGGGAGC; CHD1L
forward, ACTAGCATTCCTGTATTCTGGGG and reverse,
CACGCTCATAGCTGTAGCCTC; DUX4 forward, CGATGGCCCTCCCGACA and reverse,
GGCGTGACCTCTCATTCTGA; and β-actin forward, CCTAGAAGCATTTGCGGTGG and
reverse, GAGCTACGAGCTGCCTGACG.
Cell proliferation assay
Cells were seeded into 96-well plates at 5,000
cells/200 µl/well, treated with increasing concentrations of the
DNA-damaging agents cisplatin (cat no. P4394) and etoposide (cat
no. E1383) (both from Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany), and then cultured for 24 h. Cell proliferation was
assessed using Cell Counting Kit-8 (Boster Biological Technology,
Ltd., Wuhan, China), and IC50 values were
calculated.
Flow cytometric analysis
Cells were seeded in 6-well plates at
5×105 cells/2.5 ml/well and treated with DNA-damaging
agents at the indicated concentrations for 24 h. For cell cycle
analysis, cell suspensions were harvested, washed with PBS, and
resuspended in propidium iodide (PI) stain buffer [0.1% (v/v)
Triton X-100, 10 µg/ml PI and 100 µg/ml DNase-free RNaseA;
Sigma-Aldrich; Merck Millipore] for 30 min in the dark. The DNA
contents of the cells were analyzed using a BD FACSCalibur (BD
Biosciences, Franklin Lakes, NJ, USA). For apoptosis analysis, the
cells were stained with PI and allophycocyanin-conjugated Annexin V
and the FACSCalibur was used to detect early-stage apoptotic
cells.
Statistical analysis
Data are presented as the mean ± standard deviation
from at least three independent experiments. When data followed a
normal distribution, the statistical significance between
experimental values was assessed by unpaired Student's t-test using
SPSS software, and P<0.05 was considered to indicate a
statistically significant difference.
Results
USP17, CHD1L and DUX4 as putative
downstream targets of ZEB1
The Human promoter 1.0R array is tiled with
>25,500 human promoter regions. Each region covers ~7.5 kb
upstream to 2.45 kb downstream of the 5′ transcription start sites
of >1,300 cancer-associated genes. The DNA fractions that
co-immunoprecipitated with ZEB1 were hybridized to the array, in
order to identify putative downstream targets of ZEB1 (normal
rabbit IgG-binding DNA was used as the background).
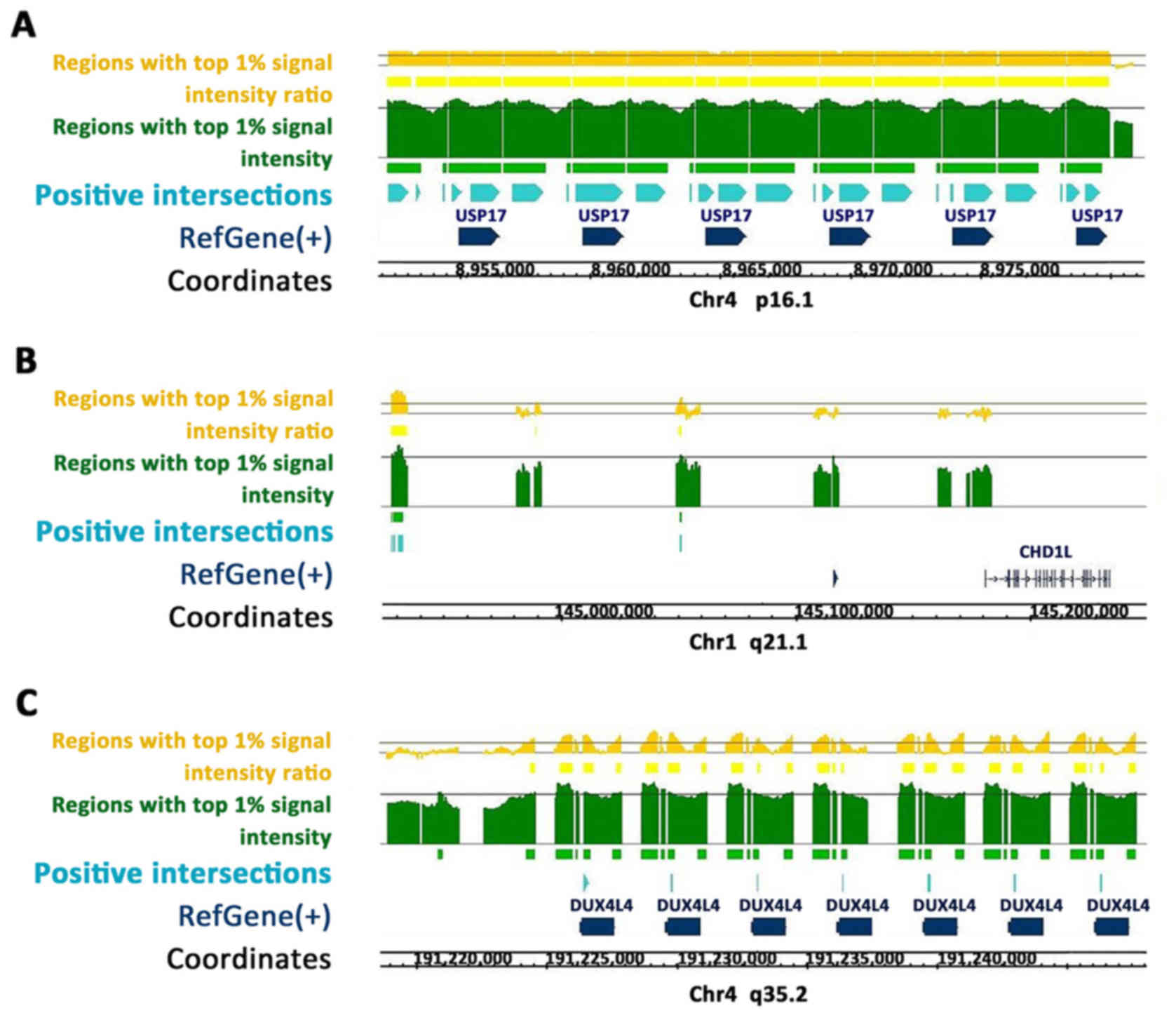
Using the Homo sapiens hg18 genome (Refgene;
NCBI Build 36.1; March 2006), continuous positive regions enriched
along the USP17 (Fig. 1A), CHD1L
(Fig. 1B) and DUX4 (Fig. 1C) gene sequences were identified.
CEAS predicted ZEB1 binding sites located at the enhancer, promoter
and immediate downstream regions of the USP17 gene; at the
enhancer, intron and exon regions of the DUX4 gene; and at the
enhancer regions of the CHD1L gene.
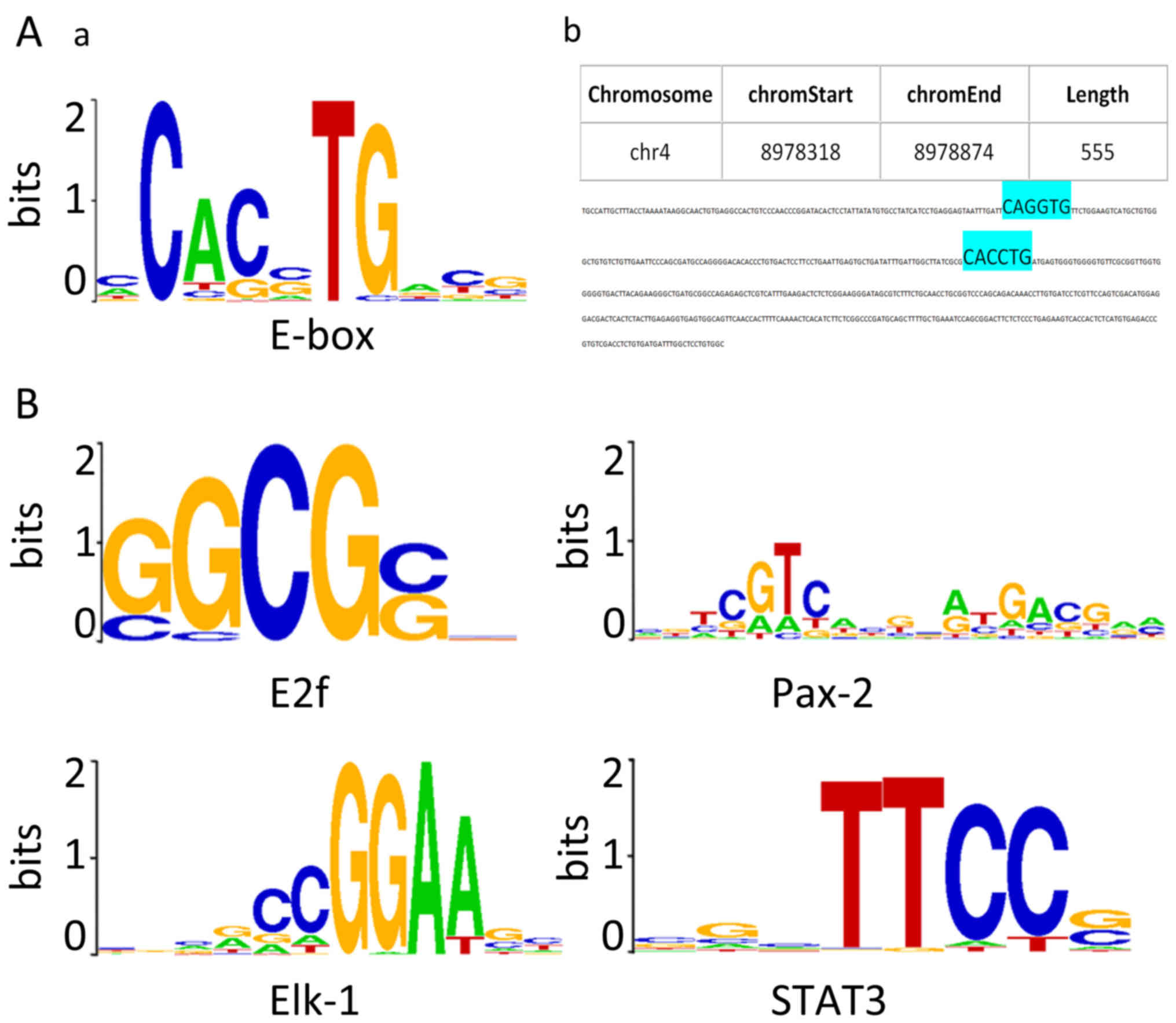
E-box elements and predicted
ZEB1-binding motifs are present within the positive regions
As ZEB1 is a well-known E-box binding
transcriptional repressor, the sequences of each positive region
from the three putative target genes (in FASTA format) were
screened for the presence of E-box elements (5′-CANNTG-3′). As
shown in Fig. 2A-a, CACCTG, CACGTG,
CAGCTG, CAGGTG were the four most canonical E-box sequences.
Duplication of the sequences, particularly in an
AGGTG-Nn-CACCT structure, have been reported to
possess strong ZEB1-binding affinity (22,23).
In 24 positive intervals along the USP17 gene, 10 CACCTG sequences,
1 CAGGTG sequence and 12 CAGCTG sequences, and, notably, an
AGGTG-Nn-CACCT duplication were identified
(Fig. 2A). In 12 upstream positive
regions of the CHD1L gene, two CACCTG sequences were detected. The
7 positive regions along the DUX4 gene were identified to contain
CAGCTG sequences.
In addition to E-boxes, CEAS identified ~40
predicted ZEB1-binding motifs using the default algorithm, arranged
in ascending order of p-value. The top four ranked sequences
predicted to be ZEB1-interacting DNA motifs following CEAS analysis
are presented in Fig. 2B, namely
E2f, Pax-2, Elk-1 and STAT3 motifs. The FASTA files were also
searched for the de novo predicted ZEB1-binding elements,
and identified those consensus binding sites within the positive
regions along the three genes.
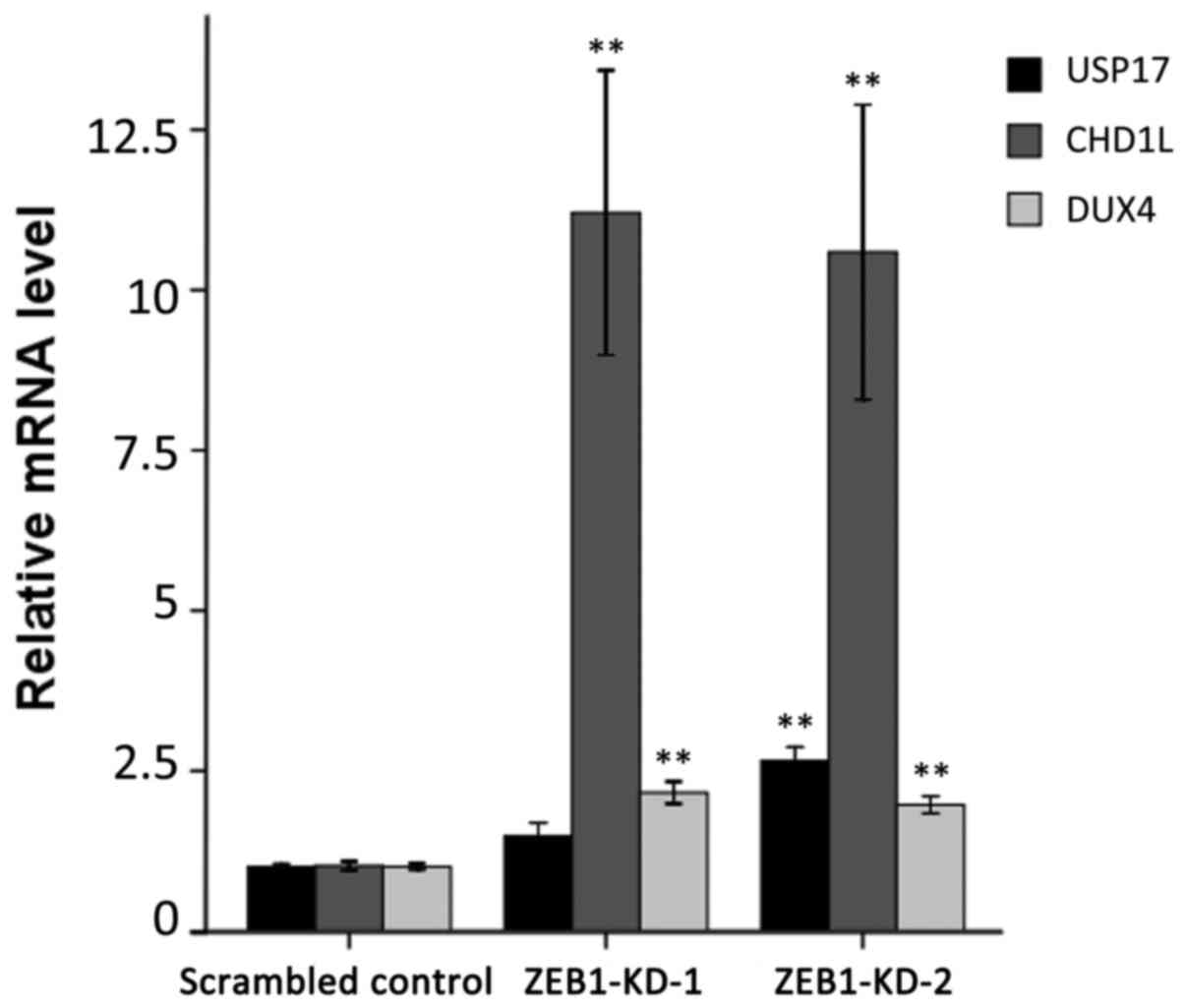
Stable depletion of ZEB1 enhances the
expression of the putative target genes
To confirm the ChIP-on-chip data, the mRNA level of
the three genes was examined for their responses to ZEB1 knockdown
by RT-qPCR. As demonstrated in Fig.
3, the three genes exhibited significantly enhanced expression
following ZEB1 depletion compared with the scrambled control group.
The relative expression levels of USP17, CHD1L and DUX4 reached
1.48-, 11.21- and 2.16-fold in ZEB1-KD-1 cells, and 2.66-, 10.59-
and 1.97-fold in ZEB1-KD-2 cells, respectively.
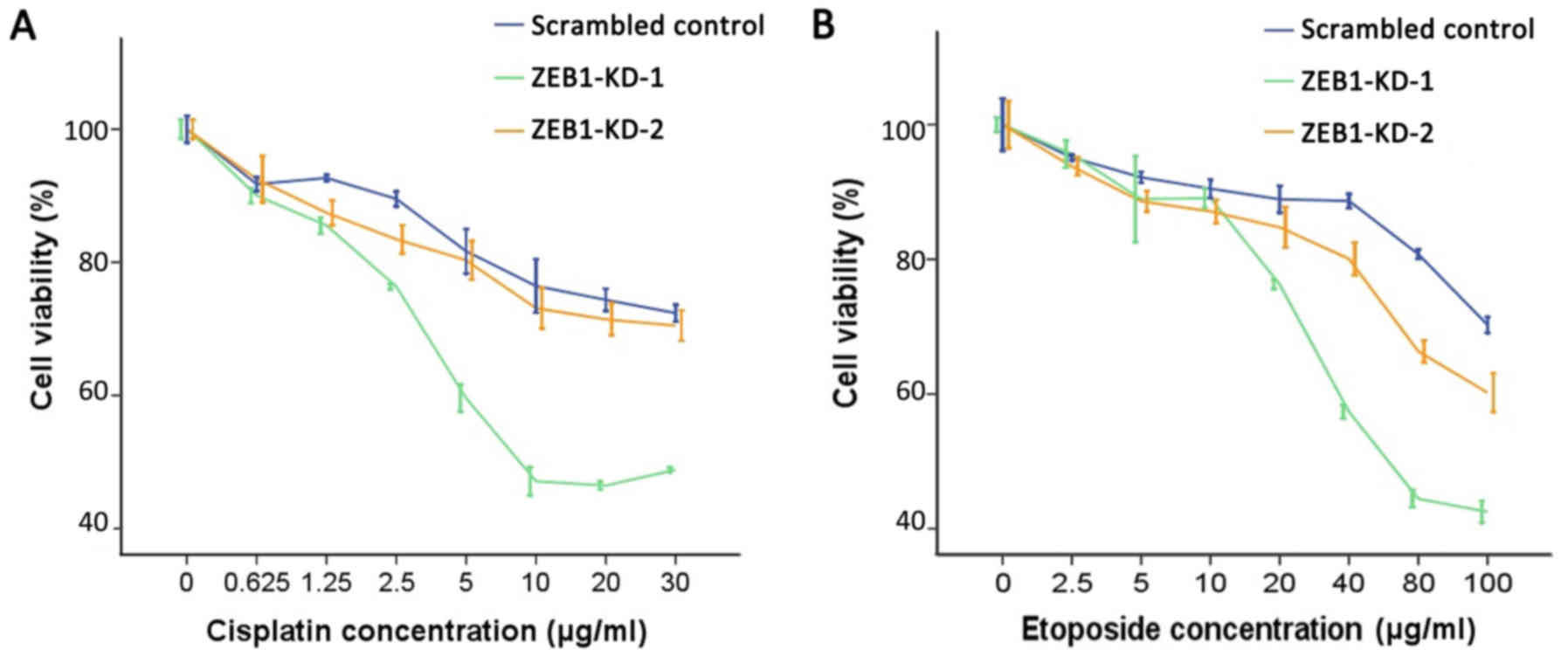
ZEB1 knockdown induces sensitivity to
genotoxic agents
Overexpression of USP17 and CHD1L was previously
reported to cause alterations to the DDR, resulting in increased
sensitivity to DNA-damaging agents (24,25).
Therefore, it was investigated whether silencing of ZEB1 may induce
similar effects. Data demonstrated that when ZEB1 was knocked down
by two specific shRNA constructs, cell proliferation was
significantly impaired and cells were more susceptible to cisplatin
and etoposide-induced DNA damage. As demonstrated in Fig. 4, ZEB1 knockdown enhanced the
inhibitory effect of cisplatin and etoposide on cell proliferation.
Compared with the scrambled control group, the two ZEB1-knockdown
LoVo cell lines exhibited decreased IC50 values for
cisplatin and etoposide as shown in Table I.
 | Table I.IC50 values for cisplatin
and etoposide in scrambled control and ZEB1-knockdown cells
(µg/ml). |
Table I.
IC50 values for cisplatin
and etoposide in scrambled control and ZEB1-knockdown cells
(µg/ml).
|
| Genotoxic
agents |
|---|
|
|
|
|---|
| Group | Cisplatin | Etoposide |
|---|
| Scrambled
control | 52.26 | 206.64 |
| ZEB1-KD-1 | 21.03 | 73.90 |
| ZEB1-KD-2 | 48.62 | 128.38 |
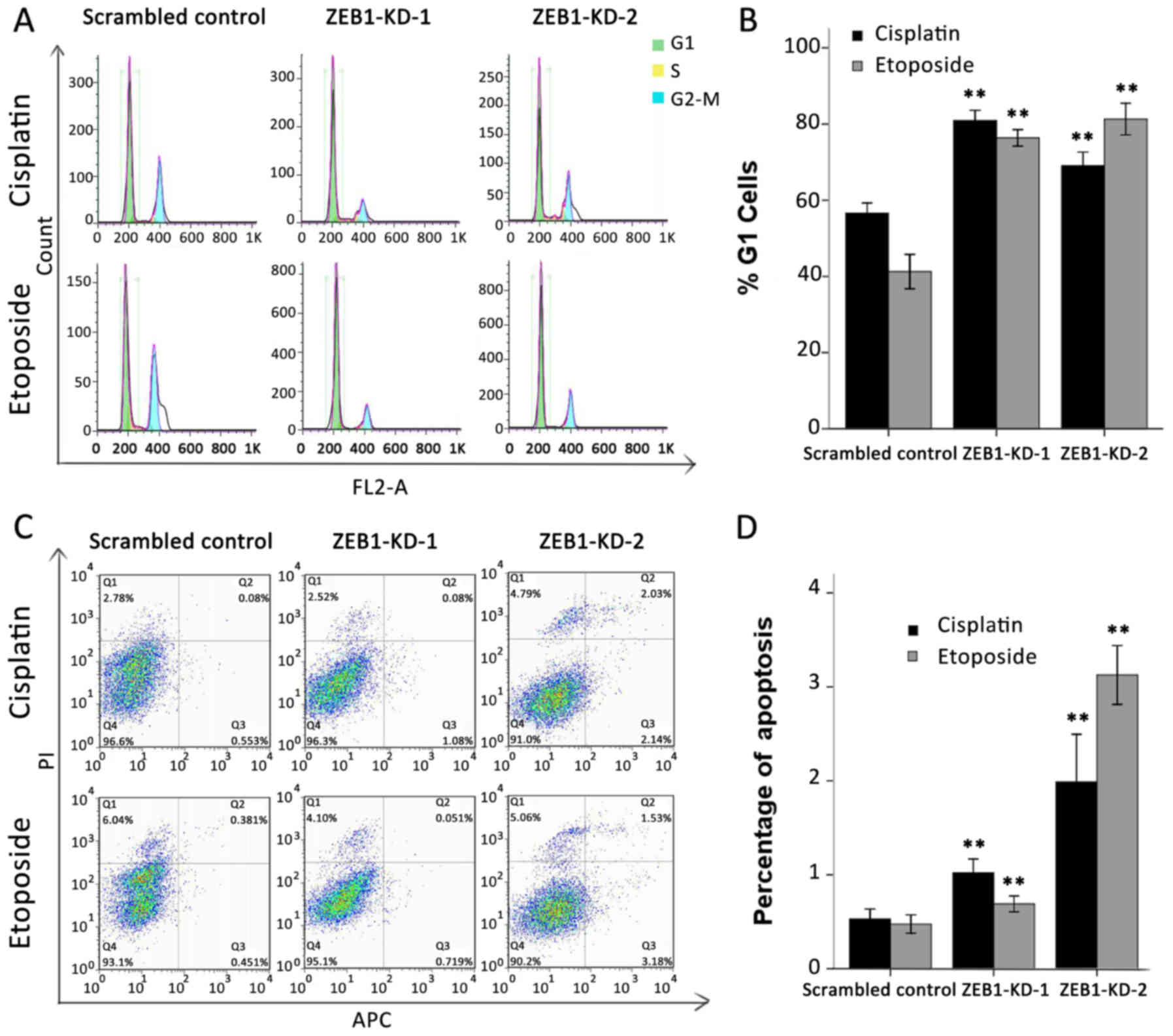
Silencing of ZEB1 blocks G1/S
transition and increases apoptosis
A disabled G1/S checkpoint, together with its
mediated anti-apoptotic effect, is another characteristic of an
unbalanced DDR network (26). It
has been previously reported that overexpression of USP17 or DUX4
may lead to G1/S arrest and a higher apoptotic rate (27,28).
Therefore, the present study evaluated whether ZEB1 silencing
impacted the cell cycle and apoptosis in a similar way. Cells were
synchronized in G1 by serum starvation, then treated with cisplatin
at 0.5 µg/ml or etoposide at 3 µg/ml for 24 h and analyzed using
flow cytometry. As expected, >70% of ZEB1-depleted cells failed
to exit G1 and progress into the S phase following drug treatment.
By contrast, only 57 and 42% of scrambled shRNA-transfected cells
accumulated in the G1 phase following cisplatin or etoposide
treatment, respectively (Fig. 5A).
The results of three independent experiments are summarized in
Fig. 5B. With regard to the
apoptosis assay, the proportion of cells undergoing early apoptosis
was also markedly upregulated in response to ZEB1 depletion.
Cisplatin and etoposide treatment induced early apoptosis in only
0.53 and 0.48% of scrambled control cells, respectively; however,
the same treatment induced early apoptosis in 1.02 and 0.69% of
ZEB1-KD-1 cells and 1.99 and 3.13% ZEB1-KD-2 cells (Fig. 5C). The results of three independent
experiments are summarized in Fig.
5D.
Discussion
To investigate the repertoire of ZEB1 in therapy
resistance, and particularly in the DDR process, the current study
mapped the DNA binding sites of ZEB1 in >1,300 cancer-associated
genes using the ChIP-on-chip method, and identified positive
intervals enriched along three DDR genes: USP17, CHD1L and DUX4. To
confirm that the potential targets were not detected as a result of
nonspecific binding of the ZEB1 antibody and cross-linked
protein-DNA complexes, each positive region was searched for E-box
elements, and changes in the mRNA expression of the three newly
identified ZEB1 downstream genes were analyzed following ZEB1
knockdown. Collectively, the E-boxes (particularly the
AGGTG-Nn-CACCT duplication) identified in the binding
regions, and the enhanced mRNA expression of these genes, supported
the prediction that USP17, CHD1L and DUX4 are ZEB1 target genes.
Besides, the top four ranked predicted ZEB1-interacting motifs
detected in the positive intervals along the three genes also
slightly supported them as ZEB1 downstream targets. The
ZEB1-binding activity of the predicted motifs will be confirmed
later by pull-down assay using recombinant ZEB1 protein and
oligonucleotides of the motifs, which will provide new insight into
transcriptional regulation by ZEB1.
In order to functionally validate the three ZEB1
downstream target genes, their detailed functions in the DDR were
reviewed, and whether the recovery of their expression by ZEB1
silencing may initiate similar biological effects was examined.
Generally, previous studies have reported that the three potential
targets of ZEB1 inhibit DNA repair by multiple mechanisms, and
induce apoptosis and cell cycle arrest. Considering that USP17
suppresses the repair of DSBs (24)
and DUX4 may inhibit interstrand crosslink (ICL) repair by
indirectly suppressing proliferating cell nuclear antigen (PCNA)
(28,29), the current study investigated the
results of treating ZEB1-silenced cells with two DSB-causing
agents, cisplatin (which creates DSBs as a result of inhibition of
the ICL repair process) (30) and
etoposide (31), and confirmed the
chemosensitization effect of ZEB1 knockdown. We speculate that this
sensitization effect was partially dependent on the subsequent
upregulation of the potential ZEB1 downstream targets, particularly
USP17. As a deubiquitylating enzyme, USP17 decreases the DNA
damage-induced monoubiquitination of H2A histone family member X
(H2AX), thus hindering the recruitment of the critical DNA repair
effector proteins (tumor protein p53-binding protein 1 and
ubiquitin interaction motif-containing 1/BRCA1) that promote the
two major repair ways (non-homologous end joining and homologous
recombination) to help cells overcome DSBs (24,32).
In the present study, it is possible that the upregulation of USP17
resulting from ZEB1 silencing increased the deubiquitylation of
H2AX, leading to impaired DSB repair.
In addition to USP17, the upregulation of CHD1L and
DUX4 were also likely to be involved in chemosensitization. CHD1L
(also termed ALC1) has long been defined as an oncogene as it
disrupts the cell death program and activates the AKT
serine/threonine kinase 1 pathway (33). In the DDR, CHD1L has a dual role. A
previous study demonstrated that CHD1L has PAR-dependent chromatin
remodeling activity that confers a more relaxed chromatin structure
which, on one hand, facilitates DNA repair by enhancing nucleosome
displacement and recruitment of DNA repair factors, while, on the
other hand, it also increases the interaction of genotoxic agents
with DNA, causing further damage. Thus, for chemotherapeutic drugs
that preferentially act on relaxed chromatin, CHD1L may intensify
their cytotoxic effect. In a previous study using cells with CHD1L
overexpression, phleomycin exposure increased H2AX phosphorylation
and increased comet tail lengths in a comet assay, implying an
increase in DNA damage (25). It is
not clear whether the increase in CHD1L expression in the current
study affected drug-DNA contact or DNA repair factor recruitment.
Further efforts will investigate how transient transfection of
CHD1L siRNA in ZEB1-knockdown cells affects their response to
cisplatin and etoposide.
Previous research regarding DUX4 has predominantly
focused on facioscapulohumeral muscular dystrophy, a progressive
disease involving the continuous degeneration of muscle cells and
tissue. DUX4, which is normally transcriptionally silenced, was
identified to express and encode a transcriptional activator of
several genes in the p53 pathway, including p21 and caspase-3,
inducing G1/S arrest and apoptosis in myoblasts (28). It is unclear whether DUX4 activates
these molecules in cancer cells in the same manner as it does in
myoblasts. If so, DUX4 may be a strong inhibitor of DNA repair, as
the downstream target p21 suppresses various DNA repair pathways,
predominantly by promoting PCNA degradation or by disrupting its
recruitment of DNA repair factors (34). Cisplatin is a well-established ICL
inducer and PCNA is an activator of the ICL repair pathway
(35); thus, it is reasonable to
speculate that the DUX4-p21-PCNA chain is involved in increasing
the susceptibility of ZEB1-knockdown cells to cisplatin. Future
efforts will be made to verify the DUX4-p21 interaction in cancer
cells.
Furthermore, the present study demonstrated an
increased apoptosis rate and G1/S arrest following cisplatin and
etoposide treatment in ZEB1-knockdown cells. This suggests that
USP17 and DUX4 are downstream targets of ZEB1, as both USP17 and
DUX4 have been previously reported to facilitate apoptosis and
induce G1 arrest (27,28). CHD1L was demonstrated to promote
cell proliferation, inhibit programmed cell death and accelerate
G1/S transition, and we hypothesize that the pro-apoptotic and cell
cycle-blocking effects of USP17 and DUX4 counteracted the influence
of CHD1L on the two processes, resulting in the final outcome
observed in the study. Intriguingly, in the DDR, cell cycle arrest
is often considered as a means of self-preservation that allows
time for DNA repair and cell survival (31). Indeed, either more active repair or
increased DNA damage that requires more time to be dealt with may
lead to cell cycle arrest. However, as apoptosis was increased and
the enhanced cytotoxic effect of genotoxic agents on ZEB1-knockdown
cells strongly indicated an increase in unrepaired DNA lesions, we
hypothesize that the G1 arrest indicated further DNA
impairment.
In conclusion, three DDR-associated genes were
identified as ZEB1 downstream targets and the present study
demonstrated that their suppression by ZEB1 may contribute to
ZEB1-mediated chemoresistance. This study provides a foundation for
a more detailed understanding of the regulation of DDR by ZEB1, and
suggests that inhibiting ZEB1 may be a promising treatment for
cancer, as it targets three vital malignancy-associated events
simultaneously: EMT, stemness and drug resistance.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81273636).
References
|
1
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitra A, Mishra L and Li S: EMT, CTCs and
CSCs in tumor relapse and drug-resistance. Oncotarget.
6:10697–10711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pattabiraman DR and Weinberg RA: Tackling
the cancer stem cells - what challenges do they pose? Nat Rev Drug
Discov. 13:497–512. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koren E and Fuchs Y: The bad seed: Cancer
stem cells in tumor development and resistance. Drug Resist Updat.
28:1–12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sánchez-Tilló E, Lázaro A, Torrent R,
Cuatrecasas M, Vaquero EC, Castells A, Engel P and Postigo A: ZEB1
represses E-cadherin and induces an EMT by recruiting the SWI/SNF
chromatin-remodeling protein BRG1. Oncogene. 29:3490–3500. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wellner U, Schubert J, Burk UC,
Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D,
zur Hausen A, et al: The EMT-activator ZEB1 promotes tumorigenicity
by repressing stemness-inhibiting microRNAs. Nat Cell Biol.
11:1487–1495. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chaffer CL, Marjanovic ND, Lee T, Bell G,
Kleer CG, Reinhardt F, D'Alessio AC, Young RA and Weinberg RA:
Poised chromatin at the ZEB1 promoter enables breast cancer cell
plasticity and enhances tumorigenicity. Cell. 154:61–74. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang P, Sun Y and Ma L: ZEB1: At the
crossroads of epithelial-mesenchymal transition, metastasis and
therapy resistance. Cell Cycle. 14:481–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y,
Zhang J, Yuan J, Wang M, Chen D, Sun Y, et al: ATM-mediated
stabilization of ZEB1 promotes DNA damage response and
radioresistance through CHK1. Nat Cell Biol. 16:864–875. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cortez MA, Valdecanas D, Zhang X, Zhan Y,
Bhardwaj V, Calin GA, Komaki R, Giri DK, Quini CC, Wolfe T, et al:
Therapeutic delivery of miR-200c enhances radiosensitivity in lung
cancer. Mol Ther. 22:1494–1503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang P, Wang L, Rodriguez-Aguayo C, Yuan
Y, Debeb BG, Chen D, Sun Y, You MJ, Liu Y, Dean DC, et al: miR-205
acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat
Commun. 5:56712014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Srivastava M and Raghavan SC: DNA
double-strand break repair inhibitors as cancer therapeutics. Chem
Biol. 22:17–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tian H, Gao Z, Li H, Zhang B, Wang G,
Zhang Q, Pei D and Zheng J: DNA damage response - a double-edged
sword in cancer prevention and cancer therapy. Cancer Lett.
358:8–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Skvortsov S, Debbage P, Lukas P and
Skvortsova I: Crosstalk between DNA repair and cancer stem cell
(CSC) associated intracellular pathways. Semin Cancer Biol.
31:36–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fokas E, Prevo R, Hammond EM, Brunner TB,
McKenna WG and Muschel RJ: Targeting ATR in DNA damage response and
cancer therapeutics. Cancer Treat Rev. 40:109–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Benada J and Macurek L: Targeting the
checkpoint to kill cancer cells. Biomolecules. 5:1912–1937. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weigelt K, Moehle C, Stempfl T, Weber B
and Langmann T: An integrated workflow for analysis of ChIP-chip
data. Biotechniques. 45:131–140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ji X, Li W, Song J, Wei L and Liu XS:
CEAS: Cis-regulatory element annotation system. Nucleic Acids Res.
34:(Web Server issue). W551–W554. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sekido R, Murai K, Funahashi J, Kamachi Y,
Fujisawa-Sehara A, Nabeshima Y and Kondoh H: The delta-crystallin
enhancer-binding protein delta EF1 is a repressor of
E2-box-mediated gene activation. Mol Cell Biol. 14:5692–5700. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Remacle JE, Kraft H, Lerchner W, Wuytens
G, Collart C, Verschueren K, Smith JC and Huylebroeck D: New mode
of DNA binding of multi-zinc finger transcription factors: deltaEF1
family members bind with two hands to two target sites. EMBO J.
18:5073–5084. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Delgado-Díaz MR, Martín Y, Berg A, Freire
R and Smits VA: Dub3 controls DNA damage signalling by direct
deubiquitination of H2AX. Mol Oncol. 8:884–893. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahel D, Horejsí Z, Wiechens N, Polo SE,
Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, et
al: Poly(ADP-ribose)-dependent regulation of DNA repair by the
chromatin remodeling enzyme ALC1. Science. 325:1240–1243. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Furgason JM and Bahassi M: Targeting DNA
repair mechanisms in cancer. Pharmacol Ther. 137:298–308. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burrows JF, McGrattan MJ, Rascle A,
Humbert M, Baek KH and Johnston JA: DUB-3, a cytokine-inducible
deubiquitinating enzyme that blocks proliferation. J Biol Chem.
279:13993–14000. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu H, Wang Z, Jin S, Hao H, Zheng L, Zhou
B, Zhang W, Lv H and Yuan Y: Dux4 induces cell cycle arrest at G1
phase through upregulation of p21 expression. Biochem Biophys Res
Commun. 446:235–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weber AM and Ryan AJ: ATM and ATR as
therapeutic targets in cancer. Pharmacol Ther. 149:124–138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aparicio T, Baer R and Gautier J: DNA
double-strand break repair pathway choice and cancer. DNA Repair
(Amst). 19:169–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng W, Su Y and Xu F: CHD1L: A novel
oncogene. Mol Cancer. 12:1702013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jung YS, Qian Y and Chen X: Examination of
the expanding pathways for the regulation of p21 expression and
activity. Cell Signal. 22:1003–1012. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rohleder F, Huang J, Xue Y, Kuper J, Round
A, Seidman M, Wang W and Kisker C: FANCM interacts with PCNA to
promote replication traverse of DNA interstrand crosslinks. Nucleic
Acids Res. 44:3219–3232. 2016. View Article : Google Scholar : PubMed/NCBI
|