Introduction
Breast cancer is one of the major causes of
cancer-related death in women. Moreover, the incidence of breast
cancer continues to rise in the world. In the treatment of breast
cancer, Taxol/Paclitaxel is still regarded as one of the most
effective drugs (1), which could
prolong the survival of breast cancer patients in combination with
other chemotherapy drugs (2).
Unfortunately, continuous use of Taxol results in the acquired
resistance of tumor cells to this drug. However, the molecular
mechanisms of resistance to Taxol in breast cancer are not fully
understood (3–5).
Aurora kinase has been observed highly expressed and
amplified in breast cancer, which is related to survival and
tumorigenesis (6,7). Moreover, overexpression of Aurora A
induces chemoresistance in breast cancer cells and ovarian cancer
cells (8). In the clinical trials
for breast cancer, the expression level of Aurora A was related
with the resistance of cancer cells to Taxol. The exact role of
Aurora A in Taxol-resistant breast cancer remains unclear. A
previous report has shown that inhibition of Aurora A expression
could result in the enhancement of Taxol-induced apoptosis in a
variety of cancer cell lines, such as head and neck, esophageal and
breast cancer cell lines (9,10).
It has been established that Taxol inhibits signal
pathways related to cell survival and growth, such as Ras/ERK and
Akt kinase pathway (11–13). However, the Ras/ERK and Akt pathways
were overexpressed in drug-resistant ovarian and colorectal cancers
(14,15). Furthermore, the overexpression of
Akt and Ras/Raf/ERK pathways might be associated with the
phosphorylation of SRC (16). As
Aurora A was reported to promote proliferation and invasion through
activation of the SRC and upregulate downstream pathways such as
Ras/Raf/ERK and Akt/mTOR pathway (17,18),
we speculated that activated Aurora A might induce Taxol-resistance
of breast cancer cells through SRC/ERK or SRC/Akt pathway. In the
present study, by using our MCF-7/T cell and xenograft tumor
models, we aimed to determine whether inhibition of Aurora A
expression in Taxol-resistant breast cancer could downregulate the
expression of SRC and downstream signaling pathways such as Ras/ERK
and Akt and would suppress the tumor cell growth and restore cancer
cell chemosensitivity to Taxol.
Materials and methods
Cell lines
MCF-7 cells were obtained from the Cell Center of
CAMS & PUMC (Beijing, China). The cells were maintained in
Dulbeccos modified Eagles medium (DMEM; Invitrogen, Carlsbad CA,
USA) (19) supplemented with 10%
fetal bovine serum (FBS; Invitrogen), 100 IU/ml penicillin and 100
µg/ml streptomycin (Invitrogen) in a humidified incubator
containing 5% CO2 at 37°C.
Establishment of the Taxol-resistant
MCF-7 model in vitro and in vivo
In the present study, MCF-7 resistant cell line
MCF-7/T was developed through continued administration of
sub-lethal concentrations of Taxol to the cells. A highly invasive
and low colony was obtained from the MCF-7/T cells. In brief,
MCF-7/T colonies were selected after 18 pulse drug treatments with
Taxol. The majority of the cells were dead following 24 h of
exposure to Taxol. The survived cells were then washed with 0.01
mol/l phosphate-buffered saline (PBS) and cultured in Taxol growth
medium. After 1–2 days, the dead cells were washed out with PBS and
fresh Taxol medium was added. Once the cells reached 70–80%
confluence, they were preserved for further study. The
Taxol-resistant cell line was stabilized for ~6 months after
treatment initiation and the resistant phenotype was developed. For
the maintenance of Taxol-resistant cells, the MCF-7/T cells were
grown in the presence of 0.001 µmol/l Taxol. Prior to
experimentation, MCF-7/T cells were maintained in a Taxol-free
culture medium and subcultured for at least three generations. The
drug-resistant characteristics of the MCF-7/T cells were tested
using various concentrations of Taxol.
Taxol-resistant cells lose the
resistant characteristics after injection into nude mice
To obtain an in vivo Taxol-resistant MCF-7
xenograft model (MCF-7/T) for studying resistance mechanisms
frequently observed in a human therapeutic setting, MCF-7 cells
were injected subcutaneously into the left flanks of the athymic
nude mice (Balb/c-nu/nu females, 6–8 weeks old) which were
purchased from Vital River Laboratory Animal Technology, Co., Ltd.
(Beijing, China) and housed in the controlled environment at 25°C
on a 12-h light/dark cycle. Mice were maintained following the
rules of the National Institute of Health Guide for the Care and
Use of Laboratory Animals. Taxol-resistant MCF-7 xenograft tumors
were achieved after ten passages of Taxol treatment. For each
passage, mice were treated with 30.0 mg/kg Taxol 24 h before
sacrifice. Then, xenograft tumors were collected and transplanted
into the new athymic nude mice. After ten passages (~12 months),
drug-resistant characteristics of the xenografts were determined by
the absence of tumor regression after treatment of Taxol.
Tissue culture from MCF-7 and
Taxol-resistant MCF-7 xenograft model
Tumor tissues from a Taxol-resistant MCF-7 and a
parent MCF-7 xenograft model were cut into small pieces with
surgical scissors and minced with sterile razor blades until the
explants size was <1 mm3. The explants were
transferred to flasks, trypsinized (Invitrogen) for 30 min, covered
with complete medium and incubated in an atmosphere of 5%
CO2 and 95% air at 37°C. The medium was replaced after
48 h.
Generation of the stable knockdown
Aurora A in the MCF-7 and MCF-7/T cell
MCF-7 and MCF-7/T cell was seeded at a density of
1×105 cells/well in 6-well plates and incubated
overnight or until cells reached 50% confluence. They were then
transfected with either the AurA microRNAs, or control microRNA
vector (BLOCK-iT™ Pol II miR RNAi Expression Vector kit with EmGFP,
purchased from Invitrogen) through Lipofectamine 2000 (Invitrogen)
following the manufacturer's protocol. The transfected cells were
initially selected in DMEM medium containing 8 µg/ml Blasticidin S
HCl (Invitrogen). Selective pressure was maintained in a medium
containing 8 µg/ml Blasticidin S HCl for two weeks. Clones with
green fluorescence were collected for further culture in regular
media. Then, cells were harvested for western blot analysis of
Aurora A expression. Two stable transfected cell clones with AurA
microRNAs, were designated as MCF-7/T/AurA1 and MCF-7/T/AurA2.
Stable transfected cells with control microRNA were designated as
MCF-7/C and MCF-7/T/C (10). Since
Aurora A-miRNA silencing constructs express GFP (BLOCK-iT™ Pol II
miR RNAi Expression Vector kit with EmGFP) which would interfere
with the flow cytometry (Annexin V-FITC) and TUNEL assay, we did
not use flow cytometry (Annexin V-FITC) and TUNEL assay to detect
apoptosis in the following experiments.
Analysis of cell proliferation and
viability
MCF-7/C, MCF-7/T/C, MCF-7/T/AurA1 and MCF-7/T/AurA2
cells were seeded in 96-well plates in DMEM medium supplemented
with 10% fetal bovine serum (FBS). The proliferation of the cells
was monitored by CCK-8 assay every day for 14 days. MCF-7/C,
MCF-7/T/C, MCF-7/T/AurA1 and MCF-7/T/AurA2 cells were seeded in
96-well plates in DMEM medium supplemented with 10% FBS. Cells were
treated with dimethyl sulfoxide (DMSO) or Taxol for 72 h and then
cell viability was measured with CCK-8 assay.
Colony formation assay
MCF-7/C, MCF-7/T/C, MCF-7/T/AurA1 and MCF-7/T/AurA2
cells were trypsinized to single-cell suspensions. Then, cells were
diluted in DMEM culture medium containing 10% FBS, and 300–600
cells were plated in each well of the 6-well plates. Cells were
incubated with 5% CO2 at 37°C for 14 days, and colonies
were washed with PBS, fixed and stained with 0.005% crystal violet
in methanol. Numbers of colonies were manually counted. Experiments
were performed in triplicate and were repeated thrice.
Cell death and cell cycle
analysis
MCF-7/C, MCF-7/T/C, MCF-7/T/AurA1 and MCF-7/T/AurA2
cells were treated with either Taxol, or 0.1% DMSO for 72 h, washed
in PBS, and fixed with ice-cold 70% ethanol overnight. Cells were
then suspended in PBS containing RNase A (100 µg/ml) and propidium
iodide (50 µg/ml) and 0.1% Triton X-100, and incubated in the dark
for at least 1 h (20). Cell cycle
profiles and death population were determined by flow cytometric
(FCM) analysis.
In vitro invasion assay
Invasion was determined using a variation of the
Boyden chamber assay as previously described (21). Briefly, cells were trypsinized and
counted; next, 1×106 cells (MCF-7/C, MCF-7/T/C,
MCF-7/T/AurA1 and MCF-7/T/AurA2) and cells cultured from tumor
tissue (MCF-7, MCF-7/T xenograft model) suspended in 200 µl of DMEM
containing 0.1% BSA. The cells were seeded into the upper
compartment (Costar, Costar, Cambridge, MA, USA) coated
polycarbonate filter with a pore size of 8.0 µm in a 24-well plate.
Each polycarbonate filter was coated with 10 µl of 0.5% Matrigel
before the addition of cells. DMEM medium (500 µl) containing 10%
FBS was added to the lower compartment as a chemoattractant. After
18 h of incubation at 37°C with 5% CO2, the cells on the
lowerside of the filter were fixed to the membrane using methanol
for 10 min. Filters were stained with hematoxylin and eosin
(H&E) at room temperature. Cells in the upper compartment were
removed using a cotton swab, leaving only the cells on the
underside of the filter, representing those cells that had
successfully invaded across the Matrigel-coated filter. The
chambers were then photographed to compare the amount of invasive
cells on the underside of the membrane. Five visual fields were
photographed in every membrane and nuclear-stained cells were
manually counted. All samples were run in triplicate.
Cell lysates and western blot
analysis
Lysates (from MCF-7/C, MCF-7/T/C, MCF-7/T/AurA1 and
MCF-7/T/AurA2 cells) were prepared following the method previously
described. Portion of three randomly selected tumors from each
group (MCF-7 and MCF-7/T) were homogenized for lysate preparation
as previously described (22). For
western blot analysis, samples were transferred to a nitrocellulose
membrane by semi-wet electrophoresis (Invitrogen), and incubated
with primary antibodies (rabbit anti-Aurora A, rabbit
anti-phosphorylated Aurora A (Thr288), rabbit anti-phosphorylated
SRC (Tyr416), rabbit anti-SRC, rabbit anti-phosphorylated Akt
(Ser473), rabbit anti-Akt, rabbit anti-phosphorylated mTOR, rabbit
anti-mTOR, rabbit anti-phosphorylated p70s6 (Thr389), rabbit
anti-phosphorylated c-Raf (Ser259), rabbit anti-c-Raf, rabbit
anti-phosphorylated MEK, rabbit anti-MEK, rabbit
anti-phosphorylated p-ERK, rabbit anti-ERK, mouse anti-cyclin B,
mouse anti-cdc2, rabbit anti-Bcl-2, rabbit anti-c-caspase 3, rabbit
anti-c-PARP, rabbit anti-MMP9, rabbit anti-MMP2 and rabbit
anti-β-actin) overnight at 4°C, detected with horseradish
peroxidase-conjugated anti-rabbit or anti-mouse IgG (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) and developed using an ECL
Western blot detection and analysis system (Applygen Technologies,
Inc., Beijing, China). Membranes were tested for equal loading by
probing for β-actin.
ELISA assay
Lysates (portion of three randomly selected tumors
from MCF-7 and MCF-7/T) were mixed and homogenized for lysate
preparation as previously described (22). For ELISA assay, the protocol of
Phospho-Aurora A (Thr288) sandwich ELISA kit (#7114; Cell Signaling
Technology, Danvers, MA, USA) was followed.
Statistical analysis
All values were expressed as mean ± SD. Values were
compared using the Student's t-test. P<0.05 was considered
significant.
Results
Activation of Aurora A and SRC in
Taxol-resistant MCF-7 model (MCF-7/T) in vitro and in vivo
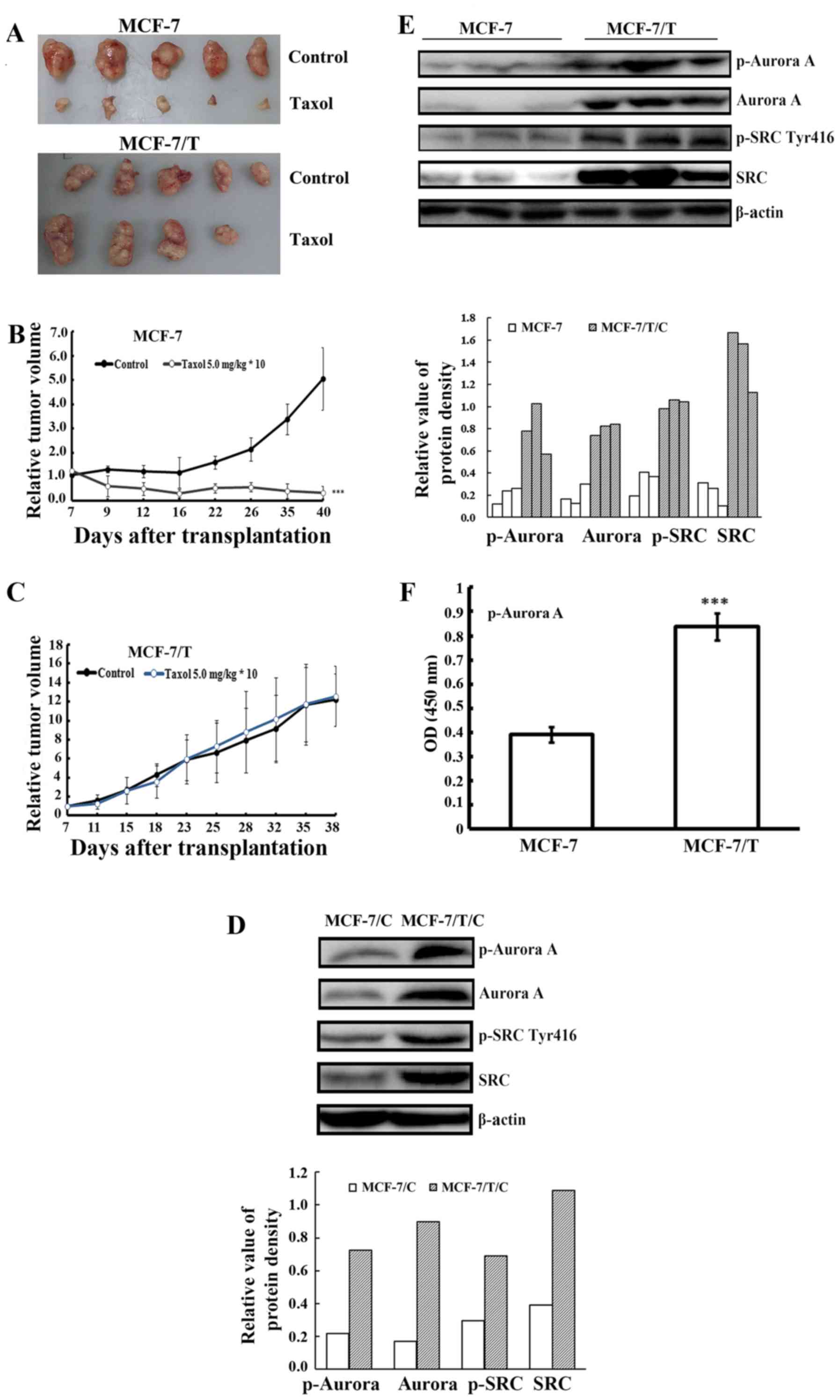
To examine whether Aurora A activation was related
to the resistance of breast cancer to Taxol, we established a
Taxol-resistant breast cancer cell clone MCF-7/T in vitro
and developed two xenograft tumor models, the Taxol-resistant
MCF-7/T and the Taxol-sensitive parental MCF-7. When a Taxol dose
of 5.0 mg/kg was adopted, the parental Taxol-sensitive MCF-7 tumors
exhibited significant growth inhibition while the MCF-7/T
xenografts presented resistance to Taxol. The relative tumor volume
(RTV) of MCF-7 was 5.06±2.46 (control) vs. 0.34±0.22 (Taxol 5.0
mg/kg) (Table I and Fig. 1A and B), whereas the RTV of MCF-7/T
was 12.19±2.78 (control) vs. 12.59±3.17 (Taxol 5.0 mg/kg) (Table II and Fig. 1A and C). The results indicated that
MCF-7/T tumors had developed resistance to Taxol in vivo and
was suitable for further study.
 | Table I.Effects of Taxol on MCF-7 tumors in
athymic mice. |
Table I.
Effects of Taxol on MCF-7 tumors in
athymic mice.
|
|
|
| Body weight
(g) | Tumor size | Tumor weight |
|---|
|
|
|
|
|
|
|
|---|
|
| Dose (mg/kg) | No. of animals
(n) | Start | End | Volume
(mm3) | RTV | T/C (%) | (g) | Inhibition (%) |
|---|
| Control |
| 5/5 | 22.6±0.6 | 26.0±1.2 | 1163.8±503.2 | 5.06±2.46 |
| 1.02±0.46 |
| Taxol | 5.0 | 5/5 | 21.0±1.6 | 22.8±2.3 |
68.3±32.5a |
0.34±0.22a | 6.75 |
0.04±0.02a | 96.36 |
| Table II.Effects of Taxol on MCF-7/T tumors in
athymic mice. |
Table II.
Effects of Taxol on MCF-7/T tumors in
athymic mice.
|
|
|
| Body weight
(g) | Tumor size | Tumor weight |
|---|
|
|
|
|
|
|
|
|---|
|
| Dose (mg/kg) | No. of animals
(n) | Start | End | Volume
(mm3) | RTV | T/C (%) | (g) | Inhibition (%) |
|---|
| Control |
| 5/5 | 18.0±1.4 | 18.6±2.4 | 1373.5±363.1 | 12.19±2.78 |
| 1.26±0.24 |
|
| Taxol | 5.0 | 5/4 | 16.8±1.9 | 17.5±2.1 | 1871.0±981.5 | 12.59±3.17 | 103.3 | 1.36±0.48 | – |
We further examined the expression of Aurora A in
MCF-7/C and MCF-7/T/C cell lines, as well as MCF-7 and MCF-7/T
xenograft tumors. Western blot analysis revealed that both p-Aurora
A and Aurora A were highly expressed in Taxol-resistant MCF-7/T
cells and xenograft tumors analyzed (Fig. 1D and E). ELISA further confirmed
that the Aurora A was activated in MCF-7/T xenograft tumors
(Fig. 1F). Given previous report
(23) suggested that Aurora A might
function through phosphorylation of SRC at tyrosine 416, we herein
validated that the level of p-SRC and SRC was upregulated in
MCF-7/T cells and tumors as well (Fig.
1D and E). These results implied that the activation of Aurora
A and SRC might be linked to the resistance of the breast cancer to
Taxol.
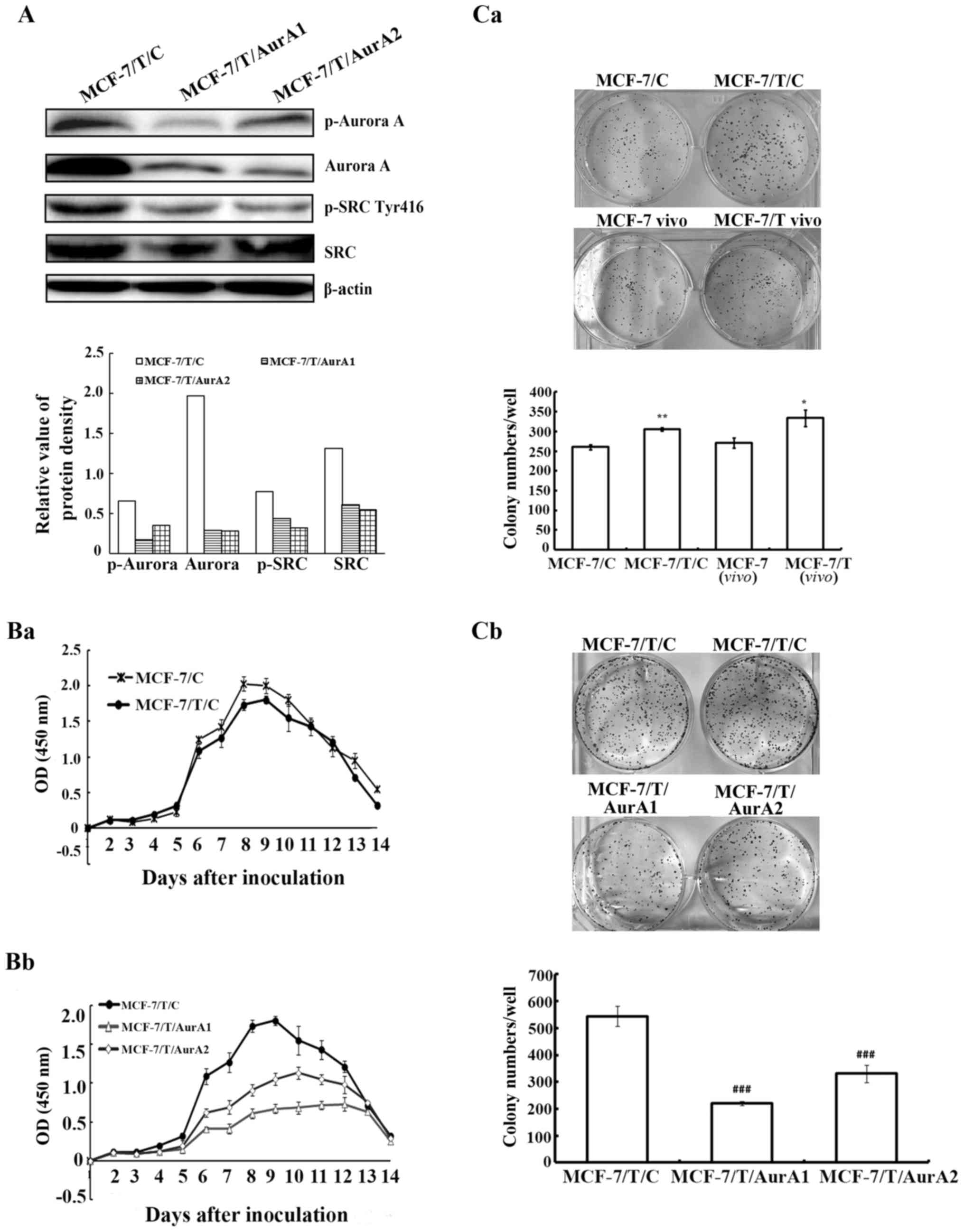
Specific silencing of Aurora A in
MCF-7/T cells led to down-regulation of SRC phosphorylation
To further determine the partnership between Aurora
A and SRC in resistance to Taxol, MCF-7/T cells were transfected
with either vector-containing human-specific Aurora A microRNAs or
a vector-containing control microRNA. Two Aurora A microRNA
stable-expressing clones (named as MCF-7/T/AurA1 and MCF-7/T/AurA2)
were chosen for subsequent experiments. Our western blot analyses
displayed that Aurora A as well as p-Aurora A were downregulated
significantly in MCF-7/T/AurA1 and MCF-7/T/AurA2 cells compared to
control microRNA-expressing cells (MCF-7/T/C) (Fig. 2A). Concurrently, SRC phosphorylation
significantly decreased while the total of SRC was not influenced
(Fig. 2A). These data indicated
that SRC activity might be regulated by Aurora A in MCF-7/T
cells.
Knockdown of Aurora A inhibits MCF-7/T
cell proliferation
To explore whether high expression of Aurora A
contributes to cell proliferation, the MCF-7/T/AurA1 and
MCF-7/T/AurA2 cells were monitored in vitro by CCK-8 assay
for 14 days. While there was no significant cell viability
difference between MCF-7/C and MCF-7/T/C (Fig. 2Ba), the MCF-7/T cell proliferation
significantly decreased upon silencing of Aurora A (MCF-7/T/AurA1
and MCF-7/T/AurA2) in a time-dependent manner compared with the
negative MCF-7/T/C controls, and the highest inhibitory rates were
65.1 and 47.5% (Fig. 2Bb) on the
8th day, respectively. In addition, our colony formation assay
indicated that the number of colonies in the MCF-7/T/C cells was
higher than in MCF-7/C cells (average colony number, 305.7±4.0 and
260.3±6.4, respectively) (Fig.
2Ca), whereas the number of colonies in the MCF-7/T/AurA1 and
MCF-7/T/AurA2 cells (average colony number, 221.3±31.9 and
330.7±10.07, respectively) were significantly decreased compared to
the MCF-7/T/C control cells (average colony number, 544.7±7.6)
(Fig. 2Cb).
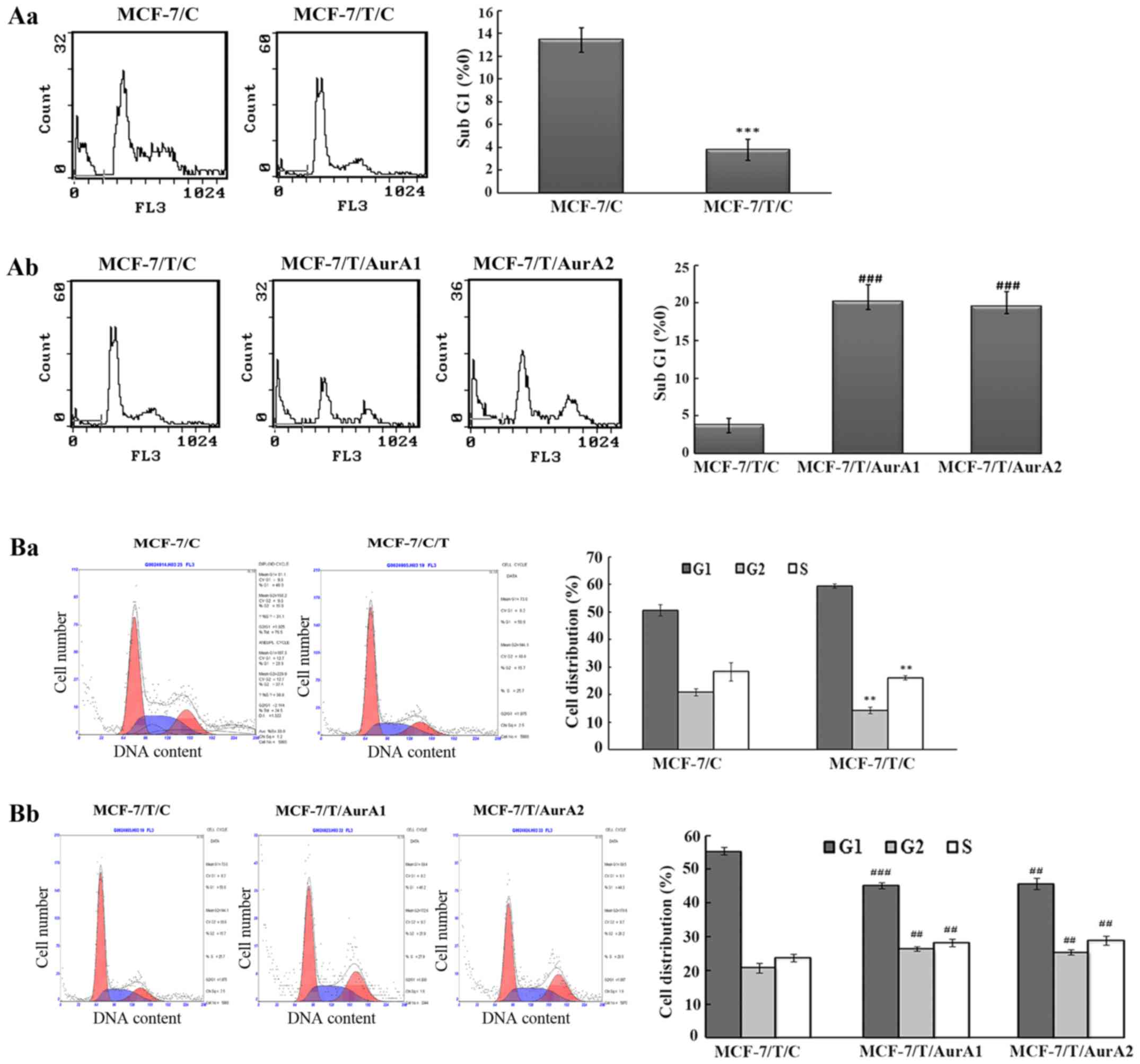
Silencing of Aurora A induces MCF-7/T
cells death
To study the impact of Aurora A expression on cell
death in Taxol resistant breast cancer cells, we examined the
percentage of SubG1 phase in MCF-7/T sublines (MCF-7/T/C,
MCF-7/T/AurA1 and MCF-7/T/AurA2) by FCM. The results revealed, that
when both MCF-7/C and MCF-7/T/C cells were cultured for 72 h, the
percentage of MCF-7/T/C cells in the SubG1 (3.8±0.9%) was lower
than the parental MCF-7/C population (13.4±1.1%) (Fig. 3Aa). Moreover, in the silenced
MCF-7/T/AurA1 and MCF-7/T/AurA2 cells, the SubG1 phase percentage
was increased by 20.2±2.3 and 19.5±2.0% compared with control
MCF-7/T/C cells (3.8±0.9%) (Fig.
3Ab). These results demonstrated that downregulation of Aurora
A could induce MCF-7/T cell death.
Knockdown of Aurora A causes G2/M
arrest in MCF-7/T cells
As Aurora A could exert positive effects on the cell
cycle in several cell lines in a dose-dependent manner, we further
examined whether knockdown of Aurora A would lead to cell cycle
arrest in MCF-7/T. The results showed that the percentage of G2/M
phase in the Taxol-resistant MCF-7/T/C cells was lower than that in
the MCF-7/C control cells. The percentages of the G2/M cells were
14.3±1.3% for MCF-7/C and 20.9±1.3 for MCF-7/T/C (Fig. 3Ba). We observed that inhibition of
Aurora A by microRNAs led to cell G2/M-phase accumulation (Fig. 3Bb) and G1-phase decrease in the
Taxol-resistant cells. The cell percentages of G2/M phase in
MCF-7/T/AurA1 and MCF-7/T/AurA2 cells increased by 26.4±0.8 and
25.4±0.7%, respectively, compared with 20.8±1.5% in MCF-7/T/C
control cells (Fig. 3Bb).
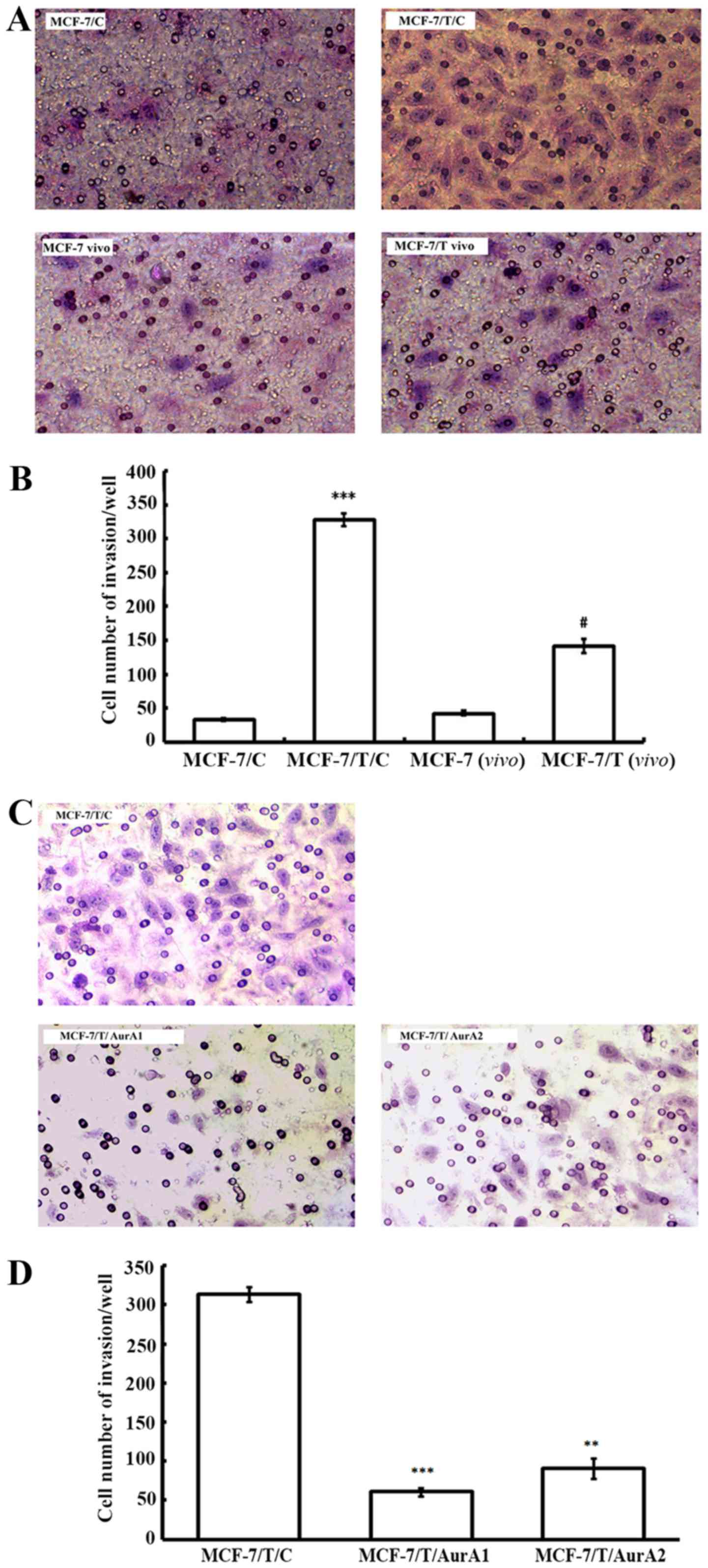
Silencing of Aurora A inhibits MCF-7/T
cell invasion in vitro
To determine the effect of elevated Aurora A on
MCF-7/T invasive capacity, we further conducted invasion assay. The
MCF-7/T sublines and MCF-7/C control cells were allowed to invade
through a membrane coated with Matrigel, toward a chemo-attractant
(10% FBS) for 18 h. While only 32.5±2.1/well MCF-7/C cells
exhibited invasion, there were 327.5±9.2/well MCF-7/T/C cells
invaded through the membrane, indicating MCF-7/T/C cells presented
an enhanced invasive ability compared with MCF-7/C cells (Fig. 4A and B). Moreover, the
xenograft-originated Taxol-resistant MCF-7 cells [MCF-7/T(vivo)]
were more invasive than the parent xenograft-originated MCF-7 cells
[MCF-7(vivo)], [MCF-7/T(vivo), 141.0±9.9 vs. MCF-7(vivo), 42.5±3.5]
(Fig. 4A and B). After knockdown of
Aurora A by microRNAs, the invaded cells decreased by 80.7%
(MCF-7/T/AurA1, 60.5±4.9/well) and 71.0% (MCF-7/T/AurA2,
91.0±12.7/well) compared to the untreated control MCF-7/T/C cells
(313.5±9.20/well) (Fig. 4C and D),
suggesting that inhibition of Aurora could considerably reduce the
invasive ability of MCF-7/T cells.
Suppression of Aurora A downregulates
the Raf/ERK and Akt pathway in MCF-7/T cells
Aurora A interacts with several proteins related to
proliferation, survival, cell cycle, apoptosis, and invasive
signaling pathways such as Raf/ERK and Akt (24–26).
We examined whether knockdown of Aurora A would downregulate the
ERK and Akt pathways in the MCF-7 and MCF-7/T subline cells and
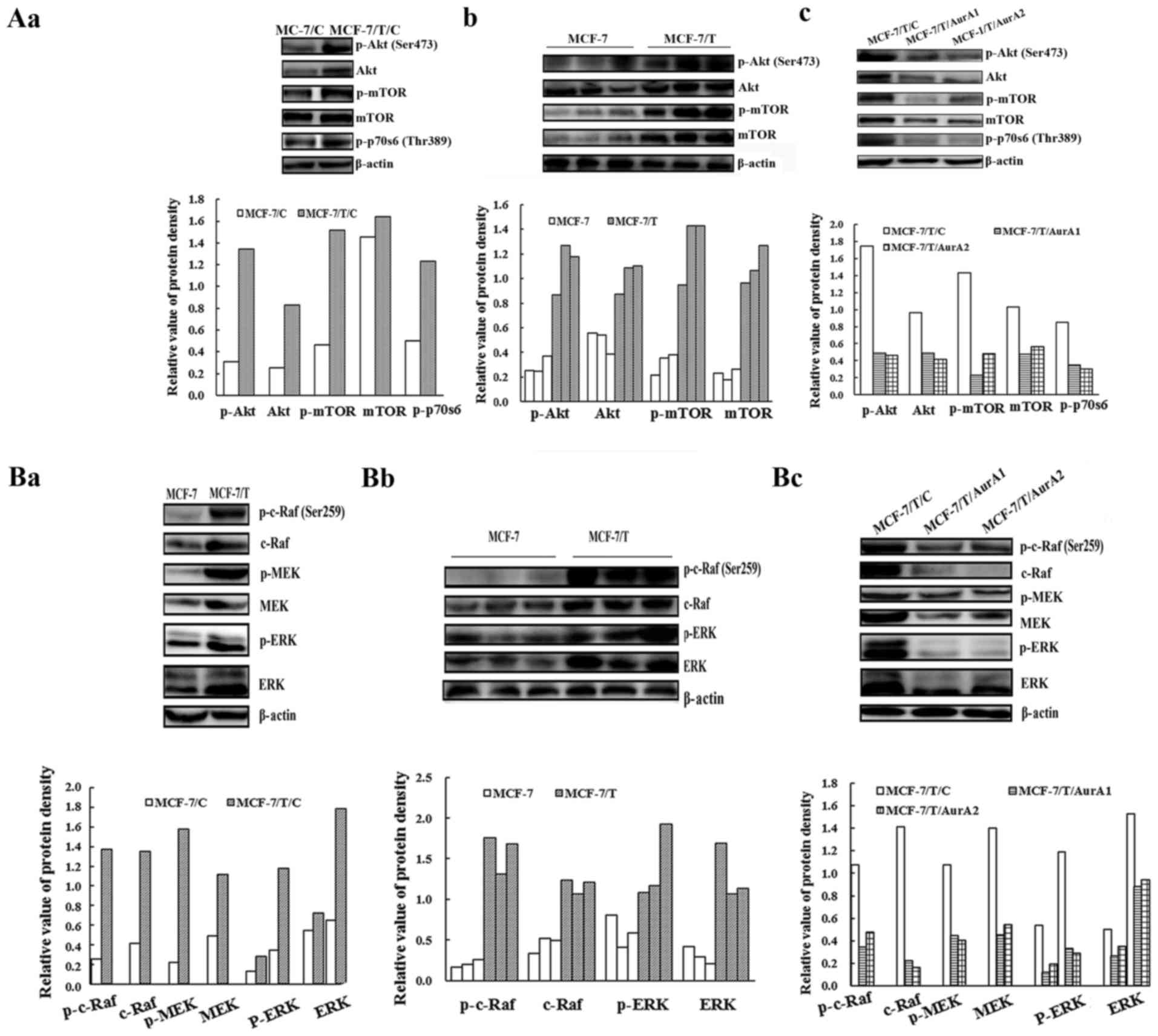
xenograft tumors. As shown in Fig. 5Aa,
5Ab, 5Ba and 5Bb, Akt, mTOR, p70s6, c-Raf, MEK and ERK were
highly phosphorylated in the Taxol resistant MCF-7 cells and
tumors. Whereas, the phosphorylation and basal levels of these
proteins were remarkably reduced in MCF-7/T/AurA1 and MCF-7/T/AurA2
cells compared with the negative control MCF-7/T/C (Fig. 5Ac and 5Bc), indicating inhibition of
Aurora A significantly downregulated the Ras/Raf/ERK and Akt
pathway.
Since cyclin B/cdc2 complex regulates cell cycle
(27), we herein found that the
cyclin B and cdc2 were overexpressed in MCF-7/T cells and xenograft
tumors (Fig. 5Ca and 5Cb).
Nevertheless, the expressions of cyclin B and cdc2 were decreased
in the MCF-7/T/AurA1 and MCF-7/T/AurA2 cells though the decrease of
cyclin B was not significant in the MCF-7/T/AurA1 cells (Fig. 5Cc), suggesting that Aurora A
regulates cell cycle through cyclin B/cdc2 in MCF-7/T cells. In
addition, knockdown of Aurora A downregulated Bcl-2 and induced the
cleavage of caspase-3 and PARP in MCF-7/T/AurA1 and MCF-7/T/AurA2
cells, suggesting that knockdown of Aurora A might enhance
apoptosis (Fig. 5Cc).
MMPs play an important role in degrading
extracellular matrix components and promoting invasion of tumor
cells (28). We observed that the
expression of MMP2 and MMP9 was much higher in MCF-7/T/C cells than
that in the MCF-7/T/AurA1 and MCF-7/T/AurA2 cells (Fig. 5Da and Db), implying that silencing
Aurora A might inhibit the invasion of MCF-7/T cells.
Inhibition of Aurora A restores the
sensitivity of MCF-7/T/C to Taxol in vitro
To investigate the chemosensitivity of MCF-7/T/C
cells to Taxol after knockdown of Aurora A, we performed
dose-response experiments to measure IC50 values with
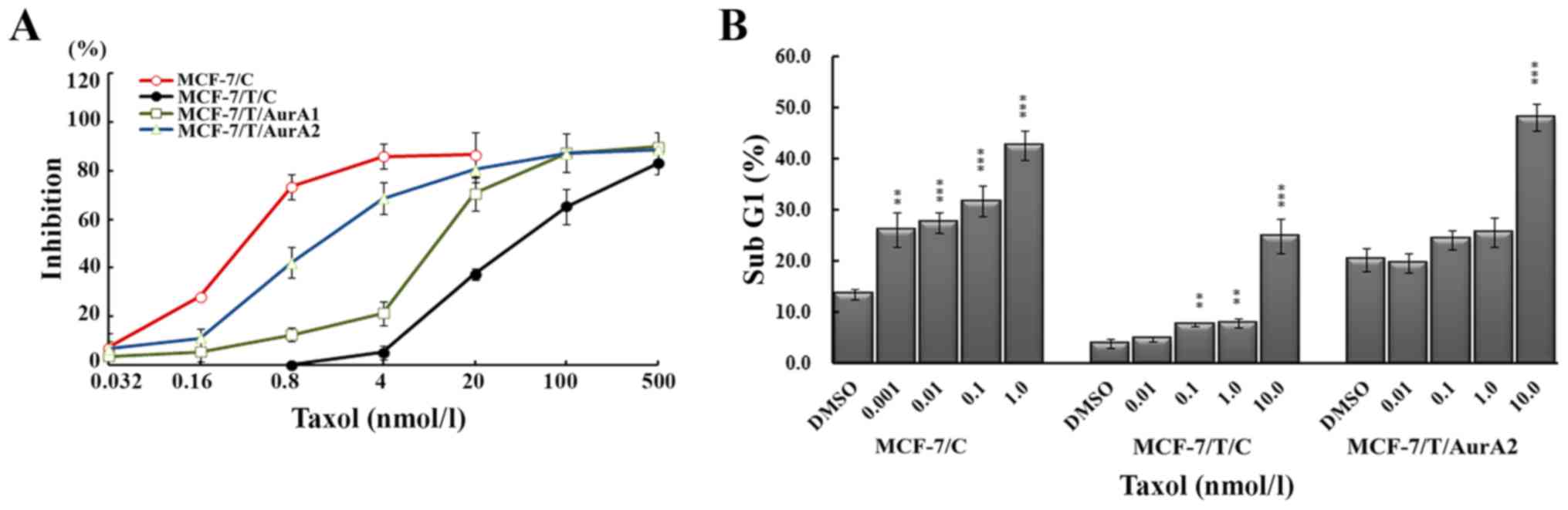
Taxol treatment for 72 h. As expected, the MCF-7/T/C cells showed
lower sensitivity to Taxol (IC50=85.72±3.41 nmol/l) than
MCF-7/C cells (IC50=0.43±0.022 nmol/l) (Fig. 6A). In contrast, inhibition of Aurora
A increased the susceptibility of MCF-7/T/C to Taxol, the
IC50 values for MCF-7/T/AurA1 and MCF-7/T/AurA2 reduced
to 12.67±0.20 and 3.25±0.77 nmol/l, respectively. The sensitivity
to Taxol advanced 6.8- and 26.4-fold in MCF-7/T/AurA1 and
MCF-7/T/AurA2 cells, respectively, as compared with that in the
MCF-7/T/C cells (Fig. 6A). These
results indicated that inhibition of Aurora A resulted in enhanced
chemosensitivity in Taxol-resistant breast cancer cells.
Inhibition of Aurora A enhances the
Taxol-induced MCF-7/T cell death
The MCF-7/T cells were administered with Taxol for
72 h and cell death (SubG1) was evaluated after PI staining by FCM.
Taxol induced death in a dose-dependent manner. When the cells were
treated with 1.0 nmol/l Taxol, the percentages of death cells in
MCF-7/C and Taxol-resistant MCF-7/T/C cells were 42.53±2.87 and
7.78±0.92%, respectively. The percentage of dead MCF-7/T/C cells
elevated to 24.83±3.33% at a dose of 10.0 nmol/l of Taxol (Fig. 6B). Whereas, the MCF-7/T/AurA2
knockdown cells presented much higher dead cell percentages after
Taxol treatment, 25.57±2.82% (1.0 nmol/l) and 48.03±2.58% (10.0
nmol/l), suggesting that inhibition of Aurora A in MCF-7/T cells
could enhance Taxol-mediated death.
Discussion
Taxol resistance is a major obstacle to successful
chemotherapy in cancer patients. In our previous study, we detected
that p-Aurora A, along with p-gp, was highly expressed in MX-1/T
and MCF-7/T cells, and demonstrated that kinase Aurora A was
related with tumor resistance in Taxol resistance MX-1/T partly
through p-ERK/P-gp (10). However,
that study focused on the three triple-negative breast cancer
cells, and the mechanism of drug-resistance was only involved in
the P-gp and p-ERK. As is known, besides p-gp, Taxol resistance
might be attributed to the upregulation of several factors
including the Class III β-tubulin (TUBB3) in lung cancer (19,29),
PI3K/Akt/mTOR (30–33) in breast and renal tumors, NF-κB,
FGFR2 and CHK2 (34) in gastric
neoplasia (35,36) and SRC in ovarian carcinomas
(16,37). In addition, overexpression of Aurora
A has been identified in the chemo-resistant breast and ovarian
cancer cells (8,38). Thus, we want to further elicit the
function of Aurora A in Taxol-resistant ER-positive breast cancer,
and investigate the mechanism of resistance related with Aurora A
except p-gp.
In this study, we demonstrated that overexpressed
Aurora A was associated with the proliferation, invasion, and
Taxol-resistance of the breast cancer cell line MCF-7/T in
vitro and in vivo. Although both Aurora A and p-Aurora A
may play a role in the chemoresistance of MCF-7/T cells, p-Aurora A
is the dominate factor because the pathway is supposed to be
activated through phosphorylation of Aurora A, and phosphorylated
Aurora A plays a more active role. Knockdown of the Aurora A
concomitantly decreases the p-Aurora A in the same cell. In this
study, silencing of Aurora A by microRNA could efficiently inhibit
MCF-7/T growth, colony formation, and invasion. The knockdown of
the Aurora A led to a significant accumulation of G2/M phase cells
and death in vitro, which further confirmed that Aurora A
played an important part in the Taxol-resistance of breast cancer
cells. In addition, inhibition of Aurora A significantly enhanced
the chemosensitivity of the resistant breast cancer cells to Taxol
by increasing cell death. These results further elucidated that
Aurora A contributed to the Taxol-resistance of breast cancer
cells.
Although it has been established that Taxol inhibits
cell survival and growth by downregulating Ras/ERK and Akt kinase
pathway (11–13), several studies have demonstrated
that the Ras/ERK and Akt pathways were overexpressed in
drug-resistant ovarian and colorectal cancers (14,15),
and the overexpression of Akt and Ras/Raf/ERK pathways might be
related to the phosphorylation of SRC (16). Following these previous findings, we
performed similar experiments and found that Aurora A, along with
SRC, Ras/Raf/ERK and Akt pathway, was highly upregulated in the
Taxol-resistant breast cancer cells and xenograft models. Silencing
of Aurora A significantly reduced SRC phosphorylation and
downregulated the Ras/Raf/ERK and Akt pathway in the
Taxol-resistant MCF-7/T cells. Studies indicated that inhibition of
SRC tyrosine kinase could enhance the chemosensitivity to Taxol
through downregulating several pathways such as Ras/Raf/ERK and
PI3K/Akt (39,40), thus, re-sensitize resistant ovarian
cancer cells to Taxol (41,42). Consistent with reported findings in
ovarian cancer cells, our results showed that Aurora A was related
to the Taxol-resistance of breast cancer cells through SRC, and
these pathways might regulate the chemoresistance in the
Taxol-resistant breast cancer cells. SRC might be a potent
molecular target for the therapy of Taxol resistant breast
cancer.
In addition, studies have revealed that the growth
inhibition was more effective by blocking both Ras/Raf/ERK and
PI3K/Akt/mTOR pathways simultaneously, which reduced Taxol
resistance in several types of cancer (25,26,43).
Likewise, Taxol-resistant MCF-7/T cells displayed simultaneous
activation of SRC, Akt and ERK accompanied with high expression of
Aurora A. Silencing Aurora A expression in MCF-7/T cells using
microRNA targeting Aurora A led to the decrease of p-SRC Tyr416.
Moreover, the silenced MCF-7/T cells showed not only significant
low level of activated ERK and Akt, but also low malignancy and
high sensitivity to Taxol.
Aurora kinase was the main regulator of cell cycle
(44), and inhibition of Aurora A
could induce G2/M phase cells. Our results showed the percentage of
the G2/M phase in MCF-7/T cells significantly increased after
silencing Aurora A. It has been proposed that inhibition of Aurora
A could increase cleavage of procaspase-3 and reduce Bcl-2
expression through inhibition of Akt (45). Similarly, we observed that the
cleaved caspase-3 and PARP proteins significantly increased while
Bcl-2 and p-Akt decreased in silenced Aurora A MCF-7/T cells.
Therefore, we confirmed that silencing of Aurora A led to
Akt-related tumor cell growth inhibition and cell death.
In addition, Ras/Raf/ERK and Akt/mTOR pathways have
been reported to mediate critical signals for the regulation of MMP
secretion and expression (28,32,46,47).
In this study, we demonstrated that MMP2 and MMP9 were highly
expressed in Taxol-resistant breast cancer cells, and inhibition of
Aurora A caused the decrease of MMP2 and MMP9, accompanied with the
downregulation of ERK and Akt activity.
In summary, our data demonstrated that the activated
Aurora A plays a critical role in Akt and ERK-mediated survival in
Taxol-resistant human MCF-7 cells through p-SRC. Inhibition of
Aurora A could enhance the sensitivity of drug-resistant MCF-7/T
cells to Taxol. The findings provided evidence that Aurora A might
provide an additional and effective target for small molecule
chemotherapeutic intervention in drug-resistant breast cancer. In
addition, pathways such as Ras/Raf/ERK and Akt/mTOR have been
well-studied and related inhibitors are available (25,32,33),
targeting Aurora A and Ras/Raf/ERK, and Akt/mTOR simultaneously may
improve the sensitivity of breast cancer cells to Taxol, which
could be a promising strategy for the treatment of patients with
Taxol-resistant breast cancer. The insights into the mechanisms of
Taxol resistance and sensitization would represent a useful basis
for further development of strategies to circumvent Taxol-related
chemoresistance in breast cancer clinical practice.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (Approval no. 81102025) and the
CAMS Initiative for Innovative Medicine (Approval no.
2016-12-M-1-008).
References
|
1
|
Schettini F, Giuliano M, De Placido S and
Arpino G: Nab-paclitaxel for the treatment of triple-negative
breast cancer: Rationale, clinical data and future perspectives.
Cancer Treat Rev. 50:129–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wan Y, Huang A, Yang Y, Xie G, Chen X, Hu
J, Chen X, Yang L, Li J, Chen L, et al: A vector-based short
hairpin RNA targeting Aurora A inhibits breast cancer growth. Int J
Oncol. 36:1121–1128. 2010.PubMed/NCBI
|
|
3
|
Chen S, Dong Q, Hu S, Cai J, Zhang W, Sun
J, Wang T, Xie J, He H, Xing J, et al: Proteomic analysis of the
proteins that are associated with the resistance to paclitaxel in
human breast cancer cells. Mol Biosyst. 10:294–303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J, Tan M, Huang WC, Li P, Guo H, Tseng
LM, Su XH, Yang WT, Treekitkarnmongkol W, Andreeff M, et al:
Mitotic deregulation by survivin in ErbB2-overexpressing breast
cancer cells contributes to Taxol resistance. Clin Cancer Res.
15:1326–1334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lai D, Ho KC, Hao Y and Yang X: Taxol
resistance in breast cancer cells is mediated by the hippo pathway
component TAZ and its downstream transcriptional targets Cyr61 and
CTGF. Cancer Res. 71:2728–2738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lens SM, Voest EE and Medema RH: Shared
and separate functions of polo-like kinases and aurora kinases in
cancer. Nat Rev Cancer. 10:825–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mountzios G, Terpos E and Dimopoulos MA:
Aurora kinases as targets for cancer therapy. Cancer Treat Rev.
34:175–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zou Z, Yuan Z, Zhang Q, Long Z, Chen J,
Tang Z, Zhu Y, Chen S, Xu J, Yan M, et al: Aurora kinase A
inhibition-induced autophagy triggers drug resistance in breast
cancer cells. Autophagy. 8:1798–1810. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Terakawa T, Miyake H, Kumano M and
Fujisawa M: Growth inhibition and enhanced chemosensitivity induced
by down-regulation of Aurora-A in human renal cell carcinoma Caki-2
cells using short hairpin RNA. Oncol Lett. 2:713–717.
2011.PubMed/NCBI
|
|
10
|
Li Y, Tang K, Zhang H, Zhang Y, Zhou W and
Chen X: Function of Aurora kinase A in Taxol-resistant breast
cancer and its correlation with P-gp. Mol Med Rep. 4:739–746.
2011.PubMed/NCBI
|
|
11
|
Bergstralh DT, Taxman DJ, Chou TC,
Danishefsky SJ and Ting JP: A comparison of signaling activities
induced by Taxol and desoxyepothilone B. J Chemother. 16:563–576.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li W, Liu J, Jackson K, Shi R and Zhao Y:
Sensitizing the therapeutic efficacy of taxol with shikonin in
human breast cancer cells. PLoS One. 9:e940792014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Q, Si S, Schoen S, Chen J, Jin XB
and Wu G: Suppression of autophagy enhances preferential toxicity
of paclitaxel to folliculin-deficient renal cancer cells. J Exp
Clin Cancer Res. 32:992013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang NN, Zhao LJ, Wu LN, He MF, Qu JW,
Zhao YB, Zhao WZ, Li JS and Wang JH: Mechanistic analysis of
taxol-induced multidrug resistance in an ovarian cancer cell line.
Asian Pac J Cancer Prev. 14:4983–4988. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu R, Nakano K, Iwasaki H, Kumagai M,
Wakabayashi R, Yamasaki A, Suzuki H, Mibu R, Onishi H and Katano M:
Dual blockade of phosphatidylinositol 3-kinase and
mitogen-activated protein kinase pathways overcomes
paclitaxel-resistance in colorectal cancer. Cancer Lett.
306:151–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Le XF and Bast RC Jr: Src family kinases
and paclitaxel sensitivity. Cancer Biol Ther. 12:260–269. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liao CH, Sang S, Ho CT and Lin JK:
Garcinol modulates tyrosine phosphorylation of FAK and subsequently
induces apoptosis through down-regulation of Src, ERK, and Akt
survival signaling in human colon cancer cells. J Cell Biochem.
96:155–169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qin B, Ariyama H, Baba E, Tanaka R, Kusaba
H, Harada M and Nakano S: Activated Src and Ras induce gefitinib
resistance by activation of signaling pathways downstream of
epidermal growth factor receptor in human gallbladder
adenocarcinoma cells. Cancer Chemother Pharmacol. 58:577–584. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gan PP, Pasquier E and Kavallaris M: Class
III beta-tubulin mediates sensitivity to chemotherapeutic drugs in
non small cell lung cancer. Cancer Res. 67:9356–9363. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Tang K, Zhang L, Li C, Niu F, Zhou
W, Yang H, Feng Z and Chen X: The molecular mechanisms of a novel
multi-kinase inhibitor ZLJ33 in suppressing pancreatic cancer
growth. Cancer Lett. 356:392–403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Zhou W, Wei L, Jin J, Tang K, Li C,
Teh BT and Chen X: The effect of Aurora kinases on cell
proliferation, cell cycle regulation and metastasis in renal cell
carcinoma. Int J Oncol. 41:2139–2149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Zhang ZF, Chen J, Huang D, Ding Y,
Tan MH, Qian CN, Resau JH, Kim H and Teh BT: VX680/MK-0457, a
potent and selective Aurora kinase inhibitor, targets both tumor
and endothelial cells in clear cell renal cell carcinoma. Am J
Transl Res. 2:296–308. 2010.PubMed/NCBI
|
|
23
|
Do TV, Xiao F, Bickel LE, Klein-Szanto AJ,
Pathak HB, Hua X, Howe C, OBrien SW, Maglaty M, Ecsedy JA, et al:
Aurora kinase A mediates epithelial ovarian cancer cell migration
and adhesion. Oncogene. 33:539–549. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mendiola M, Barriuso J, Mariño-Enríquez A,
Redondo A, Domínguez-Cáceres A, Hernández-Cortés G, Pérez-Fernández
E, Sánchez-Navarro I, Vara JA, Suárez A, et al: Aurora kinases as
prognostic biomarkers in ovarian carcinoma. Hum Pathol. 40:631–638.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kawaguchi W, Itamochi H, Kigawa J,
Kanamori Y, Oishi T, Shimada M, Sato S, Shimogai R, Sato S and
Terakawa N: Simultaneous inhibition of the mitogen-activated
protein kinase kinase and phosphatidylinositol 3-kinase pathways
enhances sensitivity to paclitaxel in ovarian carcinoma. Cancer
Sci. 98:2002–2008. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Knuefermann C, Lu Y, Liu B, Jin W, Liang
K, Wu L, Schmidt M, Mills GB, Mendelsohn J and Fan Z:
HER2/PI-3K/Akt activation leads to a multidrug resistance in human
breast adenocarcinoma cells. Oncogene. 22:3205–3212. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li JP, Yang YX, Liu QL, Pan ST, He ZX,
Zhang X, Yang T, Chen XW, Wang D, Qiu JX, et al: The
investigational Aurora kinase A inhibitor alisertib (MLN8237)
induces cell cycle G2/M arrest, apoptosis, and autophagy via p38
MAPK and Akt/mTOR signaling pathways in human breast cancer cells.
Drug Des Devel Ther. 9:1627–1652. 2015.PubMed/NCBI
|
|
28
|
Wang X, Lu N, Niu B, Chen X, Xie J and
Cheng N: Overexpression of Aurora-A enhances invasion and matrix
metalloproteinase-2 expression in esophageal squamous cell
carcinoma cells. Mol Cancer Res. 10:588–596. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gan PP, McCarroll JA, Pouha ST, Kamath K,
Jordan MA and Kavallaris M: Microtubule dynamics, mitotic arrest,
and apoptosis: Drug-induced differential effects of
betaIII-tubulin. Mol Cancer Ther. 9:1339–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Almhanna K, Cubitt CL, Zhang S, Kazim S,
Husain K, Sullivan D, Sebti S and Malafa M: MK-2206, an Akt
inhibitor, enhances carboplatinum/paclitaxel efficacy in gastric
cancer cell lines. Cancer Biol Ther. 14:932–936. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Clark AS, West K, Streicher S and Dennis
PA: Constitutive and inducible Akt activity promotes resistance to
chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol
Cancer Ther. 1:707–717. 2002.PubMed/NCBI
|
|
32
|
Chen J, Huang D, Rubera I, Futami K, Wang
P, Zickert P, Khoo SK, Dykema K, Zhao P, Petillo D, et al:
Disruption of tubular Flcn expression as a mouse model for renal
tumor induction. Kidney Int. 88:1057–1069. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu M, Si S, Li Y, Schoen S, Xiao GQ, Li X,
Teh BT, Wu G and Chen J: Flcn-deficient renal cells are tumorigenic
and sensitive to mTOR suppression. Oncotarget. 6:32761–32773. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gutiérrez-González A, Belda-Iniesta C,
Bargiela-Iparraguirre J, Dominguez G, Alfonso García P, Perona R
and Sanchez-Perez I: Targeting Chk2 improves gastric cancer
chemotherapy by impairing DNA damage repair. Apoptosis. 18:347–360.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haruki K, Shiba H, Fujiwara Y, Furukawa K,
Iwase R, Uwagawa T, Misawa T, Ohashi T and Yanaga K: Inhibition of
nuclear factor-κB enhances the antitumor effect of paclitaxel
against gastric cancer with peritoneal dissemination in mice. Dig
Dis Sci. 58:123–131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Qiu H, Yashiro M, Zhang X, Miwa A and
Hirakawa K: A FGFR2 inhibitor, Ki23057, enhances the
chemosensitivity of drug-resistant gastric cancer cells. Cancer
Lett. 307:47–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
George JA, Chen T and Taylor CC: SRC
tyrosine kinase and multidrug resistance protein-1 inhibitions act
independently but cooperatively to restore paclitaxel sensitivity
to paclitaxel-resistant ovarian cancer cells. Cancer Res.
65:10381–10388. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ding YH, Zhou ZW, Ha CF, Zhang XY, Pan ST,
He ZX, Edelman JL, Wang D, Yang YX, Zhang X, et al: Alisertib, an
Aurora kinase A inhibitor, induces apoptosis and autophagy but
inhibits epithelial to mesenchymal transition in human epithelial
ovarian cancer cells. Drug Des Devel Ther. 9:425–464.
2015.PubMed/NCBI
|
|
39
|
Puls LN, Eadens M and Messersmith W:
Current status of SRC inhibitors in solid tumor malignancies.
Oncologist. 16:566–578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wheeler DL, Iida M and Dunn EF: The role
of Src in solid tumors. Oncologist. 14:667–678. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pengetnze Y, Steed M, Roby KF, Terranova
PF and Taylor CC: Src tyrosine kinase promotes survival and
resistance to chemotherapeutics in a mouse ovarian cancer cell
line. Biochem Biophys Res Commun. 309:377–383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen T, Pengetnze Y and Taylor CC: Src
inhibition enhances paclitaxel cytotoxicity in ovarian cancer cells
by caspase-9-independent activation of caspase-3. Mol Cancer Ther.
4:217–224. 2005.PubMed/NCBI
|
|
43
|
Kim SH, Juhnn YS and Song YS: Akt
involvement in paclitaxel chemoresistance of human ovarian cancer
cells. Ann N Y Acad Sci. 1095:82–89. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fu J, Bian M, Jiang Q and Zhang C: Roles
of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res.
5:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Long M, Yin G, Liu L, Lin F, Wang X, Ren
J, Wei J, Dong K and Zhang H: Adenovirus-mediated Aurora A shRNA
driven by stathmin promoter suppressed tumor growth and enhanced
paclitaxel chemotherapy sensitivity in human breast carcinoma
cells. Cancer Gene Ther. 19:271–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Marshall CJ: MAP kinase kinase kinase, MAP
kinase kinase and MAP kinase. Curr Opin Genet Dev. 4:82–89. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tseng YS, Lee JC, Huang CY and Liu HS:
Aurora-A overexpression enhances cell-aggregation of Ha-ras
transformants through the MEK/ERK signaling pathway. BMC Cancer.
9:4352009. View Article : Google Scholar : PubMed/NCBI
|