Introduction
Epithelial to mesenchymal transition (EMT), which is
defined by the loss of epithelial characteristics and the
acquisition of mesenchymal properties, has been found to contribute
to cancer progression and metastasis in multiple types of cancer
including gastric cancer (1–3). EMT
phenotype in cancers has also been associated with poor clinical
outcome (2,4). Moreover, it has been proposed that
signaling pathways involved in metastasis are shared by EMT
(5,6). Therefore, elucidation of the signaling
pathways that govern EMT may advance our understanding of the
mechanisms of tumor metastasis.
EMT is believed to be governed by signals from the
tumor microenvironment including a variety of cytokines and growth
factors, such as epidermal growth factor (EGF) and transformation
growth factor-β (TGF-β) (7). An
in vivo study by Goswami et al (8) has suggested that macrophages express
EGF, which promotes the formation of elongated protrusions and cell
invasion of carcinoma cells. The capacity of EGF to induce EMT was
previously reported in various cell models including gastric cancer
cells (9,10). However, the molecular mechanisms
underlying the induction of EMT by EGF are still not well
characterized.
The urokinase plasminogen activator receptor (uPAR),
a glycosyl phosphatidylinositiol-anchored receptor, has been
implicated in EGF signaling and cancer invasion (11–13).
It has been demonstrated that an increased level of uPAR was
essential to the induction of EMT, and this increase was correlated
to tumor progression and aggressiveness (14,15).
The synthesis of uPAR was increased by diverse growth factors
including EGF (11,16,17).
Mounting evidence has suggested that extracellular signal-regulated
kinase 1/2 (ERK1/2) is a potent modulator of uPAR expression in
cancer cells (18,19). In addition, a study by Tushir and
DSouza-Schorey (20) also revealed
that ERK1/2 regulated uPAR expression during HGF-induced tubule
development. Furthermore, previous studies have indicated that uPAR
played a pivotal role in promoting EGF-induced tumor invasion
(16,19). Based on the aforementioned findings
and the lack of mechanistic studies in establishing the role of
EGF-induced upregulation of uPAR with respect to the acquisition of
EMT and tumor cell aggressiveness, we used gastric cancer cells as
a preclinical model for the present study. The results in the
present study indicated that EGF-induced EMT involved a cascade of
signaling events including activation of ERK1/2 signaling and
subsequent upregulation of uPAR.
Materials and methods
Cell culture and treatment
Human gastric cancer cell lines, BGC-823 and
SGC-7901, were purchased from the Chinese Academy of Sciences Cell
Bank (Shanghai, China); all cell lines were maintained at 37°C in a
5% CO2 incubator in Dulbeccos modified Eagles medium
(DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 µg/ml streptomycin. EGF was added to DMEM
supplemented with 1% FBS at a final concentration of 10 ng/ml.
Cells were made quiescent by serum starvation overnight followed by
EGF treatment for 7 days before experiments were conducted.
Wound healing assay
Cells were treated with or without EGF for 7 days.
Then, the cells were plated into a 96-well plate. When cells were
95–100% confluent, wounding was performed by scraping the cell
monolayer with a 10-µl pipette tip. Wound closure was monitored by
visual examination under an inverted microscope with an 100X
objective, at time-point zero and after 24 h.
Matrigel invasion assay
Cells were treated with or without EGF for 7 days.
Then 5×103 cells in DMEM with 1% phosphate-buffered
saline (PBS) were seeded onto the upper chamber, which were coated
with Matrigel (Sigma-Aldrich, St. Louis, MO, USA). As a
chemoattractant, DMEM with 10% FBS was added into the lower
compartment. After incubation for 24 h, the cells were fixed in
methanol for 20 min and stained with 0.1% crystal violet for 20
min. The cells on the upper surface of the filter were wiped off
with a cotton swab and the number of cells that had migrated out to
the lower surface of the membranes was counted in 5 randomly
selected fields. The experiment was repeated at least 3 times
independently.
Colony formation assay
Cells were treated with or without EGF for 7 days.
Subsequently, the cells were plated at a density of
2×103 cells in 6-well plates. Then the cells were
incubated at 37°C for 14 days. Next, colonies were stained with 2%
crystal violet, and the number of colonies that consisted of >20
cells was counted.
Small interfering RNA (siRNA)
transfection
siRNA-specific for uPAR was purchased from
GenePharma (Shanghai, China). As a non-specific control siRNA,
scrambled siRNA duplex was used which was also purchased from
GenePharma. The sequences of siRNA for uPAR were: 1,
5′-GGUGACGCCUUCAGCAUGAdTdT-3′; 2, 5′-GCCGUUACCUCGAAUGCAUdTdT-3′; 3,
5′-CACCACCAAAUGCAACGAGdTdT-3′; and for the scrambled sequence:
5′-UUCUCCGAACGUGUCACGUTT-3′. Transfection was carried out using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the
manufacturer's instructions. Silencing of uPAR was assayed at the
mRNA and protein expression level at 48 h after transfection.
Western blotting
Cells were harvested after the indicated treatment.
Protein was extracted in RIPA lysis buffer. Fifty micrograms of
protein was loaded on an SDS-PAGE gel, followed by protein
separation and electroblotting onto a polyvinylidene difluoride
(PVDF) membrane. The membrane was labeled with the following
primary antibodies: mouse anti-E-cadherin, mouse anti-vimentin and
goat anti-uPAR (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
rabbit anti-ERK1/2 and anti-phospho-ERK1/2 (Thr202/Tyr204) (Cell
Signaling Technology, Boston, MA, USA), and mouse anti-GAPDH
antibody (Chemicon, Temecula, CA, USA). HRP-conjugated secondary
antibodies were incubated in 5% BSA in Tris-buffered saline with
Tween-20 (TBST) buffer for 2 h at room temperature.
Immunoreactivity was detected using an enhanced chemiluminescence
detection system (Pierce, Rockford, IL, USA).
RT-PCR
Total RNAs were isolated using TRIzol reagent
according to the manufacturer's protocol (Invitrogen). Then, cDNA
was synthesized using the SuperScript First Strand Synthesis System
(Invitrogen), and amplified by polymerase chain reaction (PCR)
using the following primers: GAPDH, 5′-TGAACGGGAAGCTCACTGG-3′
(sense) and 5′-TCCACCACCCTGTTGCTGTA-3′ (antisense); E-cadherin,
5′-AGGATGGCTGAAGGTGACAGAG-3′ (sense) 5′-TGGCCTCAAAATCCAAGCCC-3′
(antisense); vimentin, 5′-GATGTGGATGTTTCCAAGCC-3′ (sense)
5′-ACCAGAGGGAGTGAATCCAG-3′ (antisense); uPAR,
5′-TTACCGAGGTTGTGTGTGGG-3′ (sense) 5′-GGGCATGTTGGCACATTGAG-3′
(antisense). The PCR for GAPDH was performed in 26 cycles at 95°C
for 30 sec, 55°C for 30 sec, and 72°C for 30 sec, for E-cadherin in
28 cycles at 95°C for 30 sec, 56°C for 30 sec, and 72°C for 40 sec,
for vimentin in 28 cycles at 95°C for 30 sec, 55°C for 30 sec, and
72°C for 40 sec, and for uPAR in 28 cycles at 95°C for 30 sec, 55°C
for 30 sec, and 72°C for 30 sec. The PCR products were resolved by
electrophoresis on 1% agarose.
Immunofluorescence staining
Cells were treated with or without EGF for 7 days
and grown on cover slips for 24 h. The cells on the slips were
washed with PBS, fixed with 4% paraformaldehyde, and permeabilized
with 0.2% Triton X-100. The cells were then incubated with
anti-phospho-ERK1/2 (Thr202/Tyr204) antibodies for 2 h followed by
PBS washes. Subsequently, the cells were incubated with
Rhodamine-conjugated anti-rabbit antibody for 1.5 h. The cells were
finally mounted with anti-fade mounting medium and viewed using a
Leica DM2500 fluorescence microscope (Leica, Wetzlar, Germany).
Statistical analysis
Statistical analysis was carried out using the SPSS
software (SPSS, Inc., Chicago, IL, USA). Student's t-test was used
to analyze the differences between 2 groups. When comparisons
between multiple groups were carried out, one-way ANOVA followed by
SNK tests were employed. Statistical significance was considered at
p<0.05.
Results
EGF induces EMT in gastric cancer
cells
EGF is one of the most abundant growth factors found
in the tumor microenvironment and induces EMT in multiple types of
cancer cells (21–23). In an attempt to recapitulate the
in vivo situation where cells are chronically exposed to EGF
in the tumor microenvironment, we exposed SGC-7901 and BGC-823
cells to 10 ng/ml EGF for up to 1 week. Following 7 days of
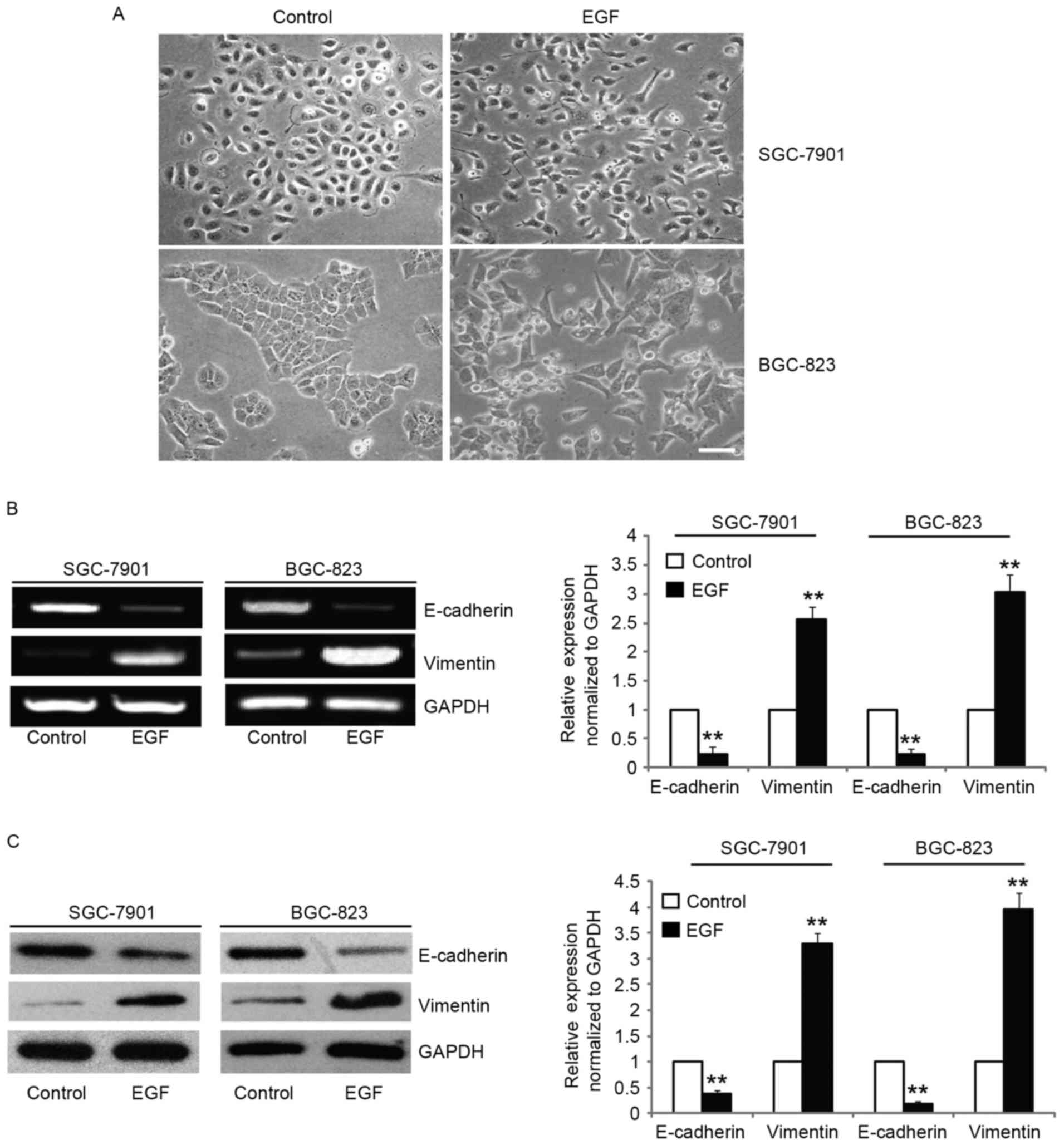
exposure to EGF, the morphologies of SGC-7901 and BGC-823 cells
were found to be completely changed to a mesenchymal phenotype,
with elongated and disseminated appearances (Fig. 1A). To confirm the mesenchymal
phenotype, we assessed the expression of molecular markers of EMT
such as vimentin and found that the mRNA and protein levels were
increased after EGF treatment (Fig. 1B
and C). In addition, the expression of E-cadherin, an
epithelial marker, was downregulated after EGF treatment (Fig. 1B and C). Collectively, these data
revealed that EGF induced the SGC-7901 and BGC-823 cells to undergo
EMT-like phenotypic changes.
Cell migration and invasive
characteristics are increased in gastric cancer cells after EGF
treatment
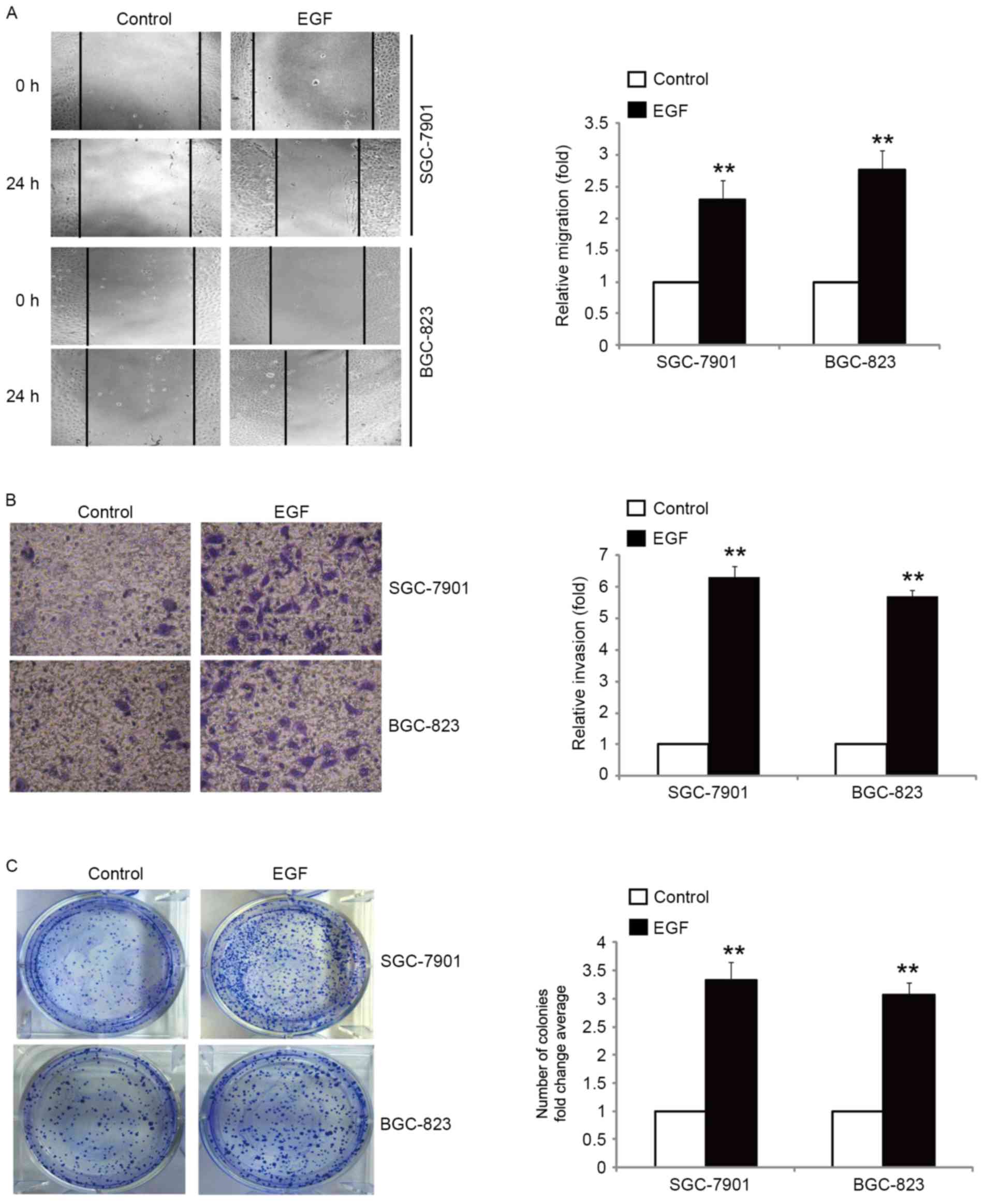
It is well known that tumor cells with an EMT
phenotype are more motile and invasive (24). Therefore, we examined the migratory
and invasive capacity of SGC-7901 and BGC-823 cells in response to
EGF treatment. Our results revealed that the migration rate was
increased with the treatment of cells with EGF, as compared with
the control cells (Fig. 2A). Using
a Boyden chamber invasion assay with Matrigel-coated polycarbonate
membranes, we found that more cells incubated with EGF had migrated
through the membrane than the control cells (Fig. 2B). We next determined whether EGF
promoted the clonogenic growth of SGC-7901 and BGC-823 cells by
colony formation assay. Treatment of cells with EGF resulted in a
significant promotion of clonogenic growth (Fig. 2C). Collectively, these results
demonstrated that EGF-induced EMT was accompanied by enhanced cell
migration, invasion and clonogenic growth.
EGF induces EMT via activation of the
ERK1/2 pathway
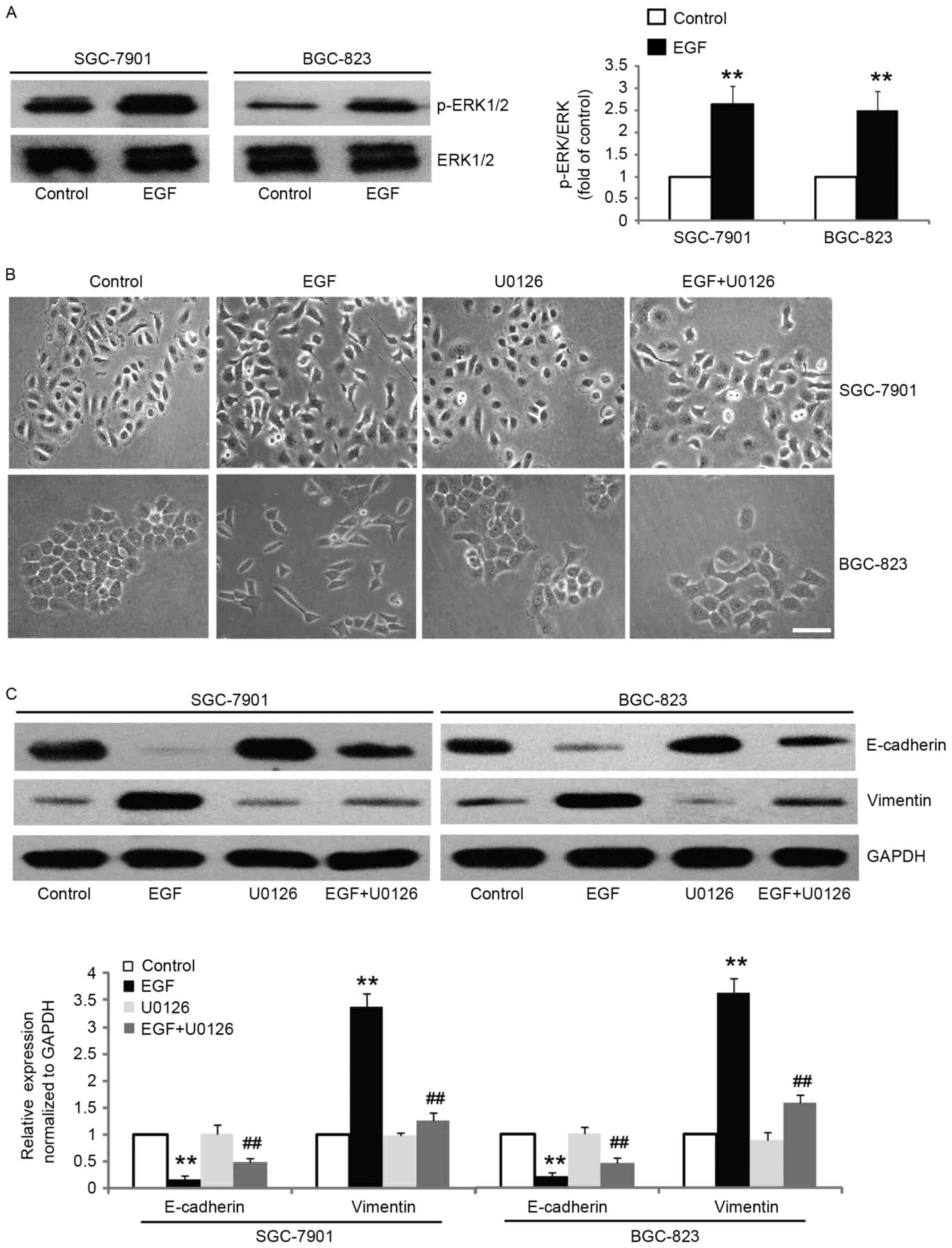
In order to assess the mechanism by which EGF
treatment induced EMT, we focused our investigation on ERK1/2
signaling since it has been implicated in EMT induction, metastasis
and invasion (25–27). We found that the level of
phospho-ERK1/2 was significantly increased after EGF stimulation,
whereas the total protein level of ERK1/2 remained unaltered
(Fig. 3A). The results revealed
that pretreatment of SGC-7901 and BGC-823 cells with ERK1/2
inhibitor prior to treatment with EGF maintained epithelial
morphology, while cells treated with EGF revealed transformation to
mesenchymal morphology (Fig. 3B).
Moreover, treatment of SGC-7901 and BGC-823 cells with ERK1/2
inhibitor exhibited partial reversal, where we observed incomplete
attenuation of EMT phenotype, as documented by the decreased
expression of vimentin, and increased expression of epithelial
marker E-cadherin (Fig. 3C). These
results revealed that EGF-induced EMT is mediated by the activation
of ERK1/2.
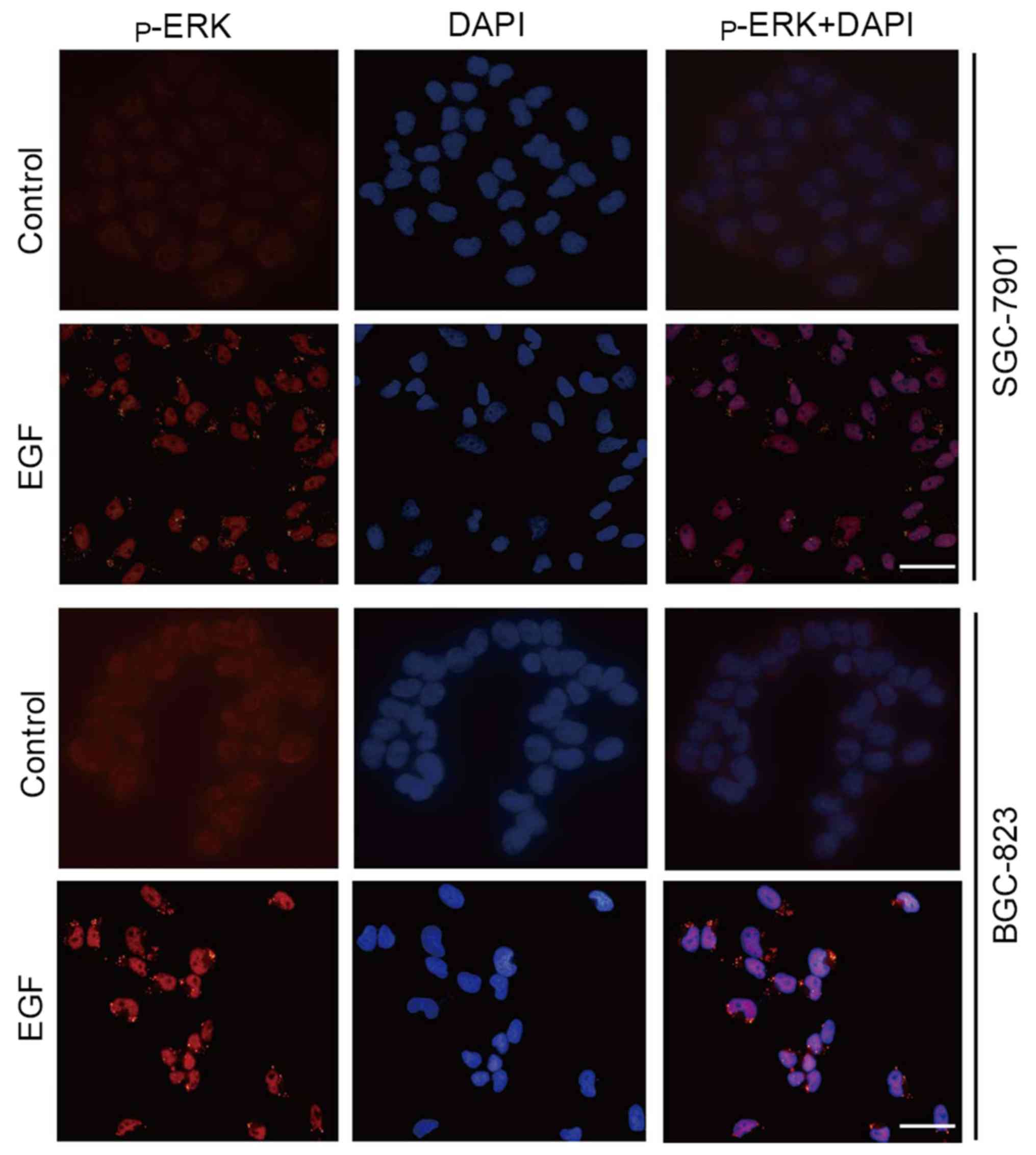
Since ERK1/2 regulates EMT not only depending on its
phosphorylation status, but also on its redistribution to the
nucleus and plasma membrane (28),
we also examined p-ERK1/2 localization in cultured cells after EGF
treatment. Immunofluorescence staining revealed that p-ERK1/2 was
weak and localized in the cytoplasm and nucleus of control cells.
However, p-ERK1/2 abundance was obviously increased in the nucleus
and periphery of the cells after exposure to EGF (Fig. 4).
uPAR is required for EGF-induced
EMT
Accumulating evidence has indicated that uPAR in
cancer cells promote EMT (14,29).
We then wished to examine whether uPAR is required for EGF-induced
EMT in gastric cancer cells. Notably, we found a dramatic increase
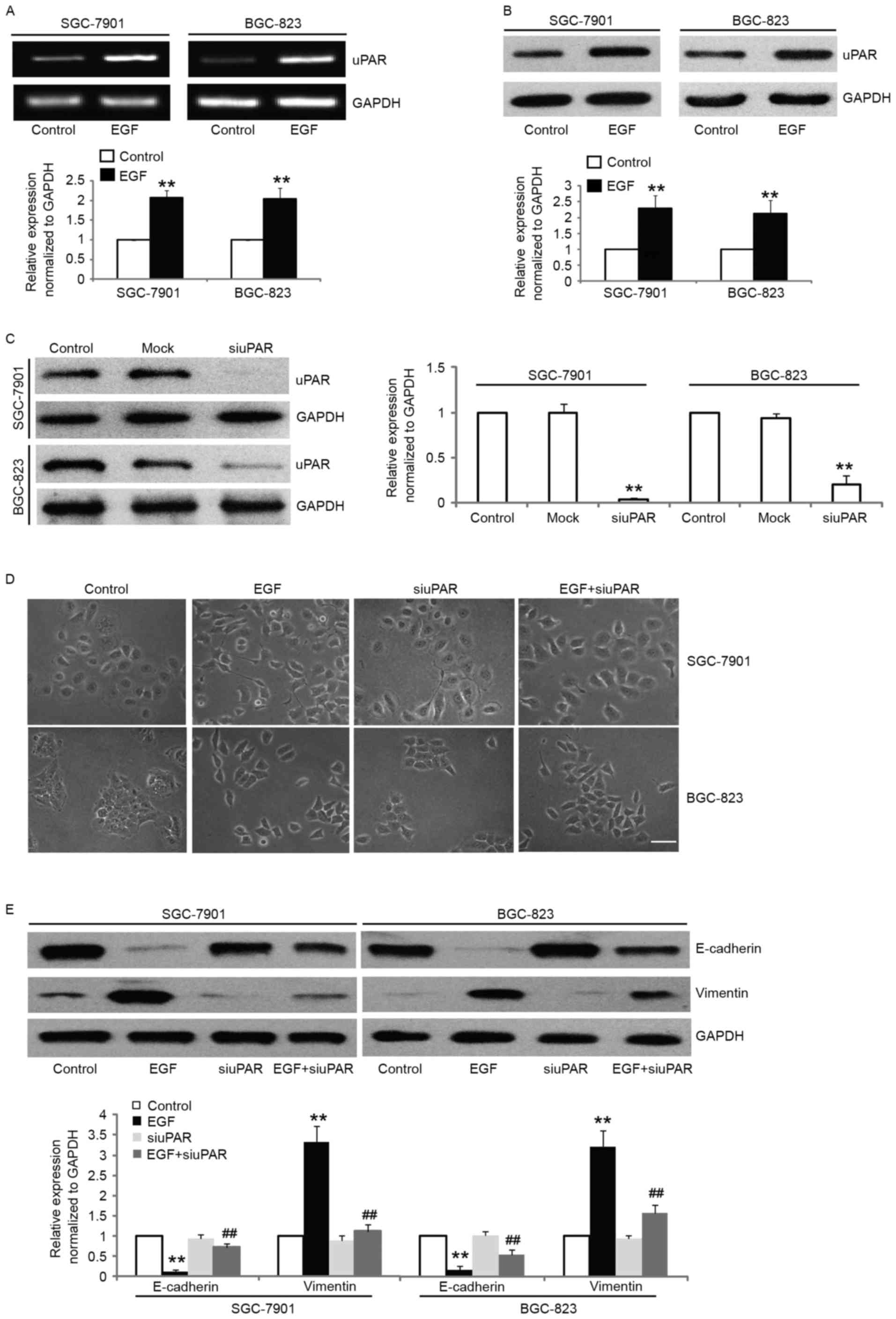
in the expression of uPAR both at the mRNA and protein levels in
SGC-7901 and BGC-823 cells after EGF stimulation (Fig. 5A and B). In order to further
investigate the role of uPAR in EGF-induced EMT in gastric cancer
cells, we knocked down the expression of uPAR protein in SGC-7901
and BGC-823 cells by uPAR-specific siRNA. The uPAR-siRNA
transfection resulted in significant knockdown of uPAR expression
as shown by western blotting (Fig.
5C) and significantly attenuated EGF-induced EMT, which was
confirmed morphologically (Fig.
5D). The transfection of SGC-7901 and BGC-823 cells with
uPAR-siRNA led to the partial reversal of the EMT phenotype as
documented by the decreased expression of vimentin, and the
increased expression of E-cadherin (Fig. 5E). These results demonstrated that
uPAR upregulation by EGF is mechanistically linked with EGF-induced
EMT in gastric cancer cells.
uPAR acts as a downstream target of
ERK1/2 and mediates EGF-induced EMT
It has been well documented that ERK1/2 regulates
growth factor-induced uPAR expression in cancer cells (16,19).
To determine whether the induced expression of uPAR by EGF observed
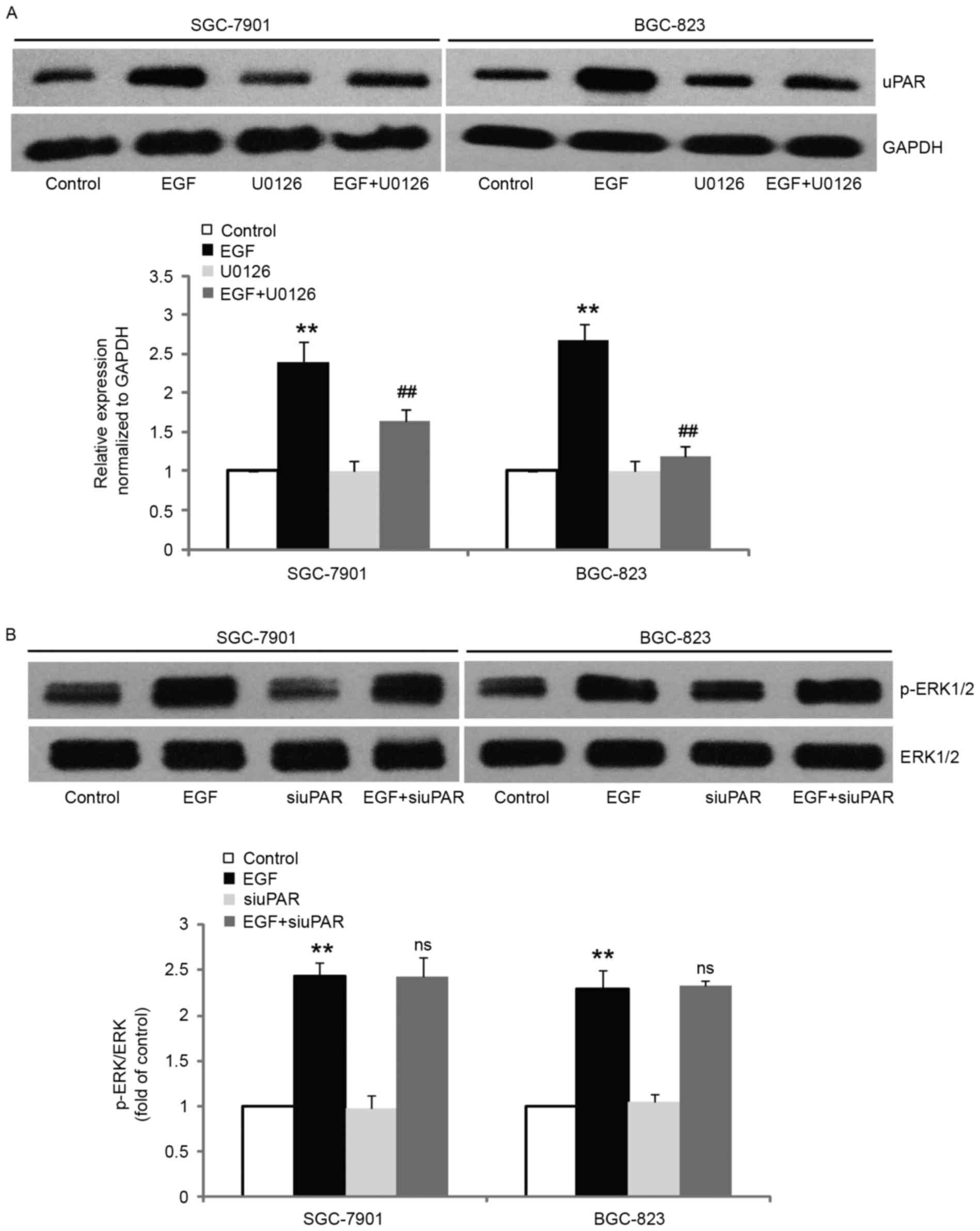
in our systems was ERK1/2-dependent, we blocked ERK1/2 activity by
pretreating the cells with U0126 and examined uPAR expression after
stimulation with EGF. The results revealed that pretreatment with
10 µM U0126 significantly inhibited EGF-induced uPAR expression in
comparison with control cells (Fig.
6A). However, inhibition of uPAR expression did not alter the
EGF-induced increase of ERK1/2 phosphorylation (Fig. 6B). These results revealed that uPAR
acts as a downstream molecule of ERK1/2 and mediates EGF-induced
EMT.
Discussion
EGF, which can be directly produced by
tumor-associated stroma cells, is one of the most abundant growth
factors found in tumor microenvironment and acts in a paracrine
fashion to cause EMT in different types of solid tumors, including
gastric cancer (28,30–32).
EGF treatment has also been demonstrated to increase cultured
cancer cell migration, invasion and proteolytic activity (33–35).
In an attempt to recapitulate the in vivo situation where
cells are chronically exposed to EGF in the tumor microenvironment,
we exposed gastric cancer cells to EGF for up to one week. In
addition, in order to understand the principal effects of EGF in
the absence of other growth factors, we intended to use serum-free
medium for gastric cancer cells to exclude other unnecessary growth
factors in the serum. However, this starvation culture induces
apoptosis of SGC-7901 and BGC-823 cells after 3–4 days (data not
shown). Therefore, EGF was added to the medium supplemented with 1%
FBS, which did not cause apoptosis within one week. In our system,
gastric cancer cell lines (SGC-7901 and BGC-823) underwent EMT
phenotypic changes after chronic exposure to EGF, which was
consistent with the decreased expression of an epithelial marker
concomitant with the increased expression of mesenchymal markers.
In order to further characterize these cells, we assessed the cell
migration, invasion and tumorigenic potential of EGF-treated cells
compared to the control cells. Our data revealed increased cell
migration, invasion and tumorigenic potential of EGF-treated cells
compared to the control cells. Therefore, EGF induced EMT in
gastric cancer cells, which is a critical step for tumor invasion
and metastasis. Based on this, the signaling mechanisms underlying
the effect of EGF on the induction of EMT were investigated.
There is increasing evidence that the activation of
ERK1/2 signaling contributes to cancer invasion and metastasis
(36,37). ERK1/2 signaling has also been
implicated in EMT induced by growth factors, such as TGF-β1 and FGF
(38,39). Similar to these findings, our
results revealed that the activity of ERK1/2 was increased after
EGF treatment. Inhibition of ERK activity by U0126 significantly
prevented EGF-induced EMT, suggesting that EGF-induced ERK
activation was responsible for the induction of EMT.
Increased expression of uPAR in human types of
cancer is associated with metastasis, whereas in low-grade cancer,
forced expression of uPAR promotes tumor metastasis (40,41).
Previous studies have revealed that endogenous uPAR plays an
important regulatory role in EMT and that EGF-induced cell invasion
is mediated by the upregulation of uPAR expression in gastric
cancer cells (14,16). In the present study, we found that
EGF-induced EMT was associated with an increase in uPAR expression.
Knockdown of uPAR by uPAR specific siRNA significantly attenuated
EMT induction by EGF treatment. These results revealed that
EGF-induced EMT was mediated by upregulation of uPAR.
In the present study, our data demonstrated that in
gastric cancer cells, ERK1/2 and uPAR mediated the EGF-induced EMT.
In some types of cells, uPAR is a downstream target of the ERK1/2
signaling cascade and inhibition of ERK1/2 was sufficient to
suppress uPAR expression (19,42,43).
In the present study, we demonstrated that blocking ERK1/2 activity
significantly prevented EGF-induced uPAR expression. Previous
studies have demonstrated that ERK1/2 activity was regulated by
uPAR (44,45). We found that specific downregulation
of uPAR in gastric cancer cells did not alter EGF-induced ERK1/2
activation. Therefore, it is possible that EGF induces uPAR
expression via the ERK1/2 pathway and, in turn, stimulates
initiation of EMT. The different results obtained by different
study groups may be due to the different cell systems used and
receptor-coupled signaling in these studies.
In summary, the present study demonstrated that
treatment with EGF induced cell migration and invasion by
activating EMT in gastric cancer cells. EGF treatment can lead to
the activation of the ERK1/2/uPAR cascade in gastric cancer cells
and contribute to EMT. These findings elucidate a molecular pathway
linking EGF signaling with uPAR in governing EMT, cell motility and
invasiveness, which may represent a rational molecular target for
manipulating gastric cancer.
References
|
1
|
Brabletz T: EMT and MET in metastasis:
Where are the cancer stem cells? Cancer Cell. 22:699–701. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Franco-Chuaire ML, Carolina Magda SC and
Chuaire-Noack L: Epithelial-mesenchymal transition (EMT):
Principles and clinical impact in cancer therapy. Invest Clin.
54:186–205. 2013.PubMed/NCBI
|
|
3
|
Fantozzi A, Gruber DC, Pisarsky L, Heck C,
Kunita A, Yilmaz M, Meyer-Schaller N, Cornille K, Hopfer U,
Bentires-Alj M, et al: VEGF-mediated angiogenesis links EMT-induced
cancer stemness to tumor initiation. Cancer Res. 74:1566–1575.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
No authors listed: An oncogenic splice
variant drives EMT and metastasis in breast cancer. Cancer Discov.
3:OF162013. View Article : Google Scholar
|
|
5
|
Chang RM, Xu JF, Fang F, Yang H and Yang
LY: MicroRNA-130b promotes proliferation and EMT-induced metastasis
via PTEN/p-AKT/HIF-1α signaling. Tumour Biol. 37:10609–10619. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sannino G, Armbruster N, Bodenhöfer M,
Haerle U, Behrens D, Buchholz M, Rothbauer U, Sipos B and Schmees
C: Role of BCL9L in transforming growth factor-β (TGF-β)-induced
epithelial-to-mesenchymal-transition (EMT) and metastasis of
pancreatic cancer. Oncotarget. 7:73725–73738. 2016.PubMed/NCBI
|
|
7
|
Gao D, Vahdat LT, Wong S, Chang JC and
Mittal V: Microenvironmental regulation of epithelial-mesenchymal
transitions in cancer. Cancer Res. 72:4883–4889. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goswami S, Sahai E, Wyckoff JB, Cammer M,
Cox D, Pixley FJ, Stanley ER, Segall JE and Condeelis JS:
Macrophages promote the invasion of breast carcinoma cells via a
colony-stimulating factor-1/epidermal growth factor paracrine loop.
Cancer Res. 65:5278–5283. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo BH, Xiong F, Wang JP, Li JH, Zhong M,
Liu QL, Luo GQ, Yang XJ, Xiao N, Xie B, et al: Epidermal growth
factor-like domain-containing protein 7 (EGFL7) enhances EGF
receptor-AKT signaling, epithelial-mesenchymal transition, and
metastasis of gastric cancer cells. PLoS One. 9:e999222014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Muthusami S, Prabakaran DS, Yu JR and Park
WY: EGF-induced expression of Fused Toes Homolog (FTS) facilitates
epithelial-mesenchymal transition and promotes cell migration in
ME180 cervical cancer cells. Cancer Lett. 351:252–259. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henic E, Noskova V, Høyer-Hansen G,
Hansson S and Casslén B: Estradiol attenuates EGF-induced rapid
uPAR mobilization and cell migration via the G-protein-coupled
receptor 30 in ovarian cancer cells. Int J Gynecol Cancer.
19:214–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu J, Jo M, Cavenee WK, Furnari F,
VandenBerg SR and Gonias SL: Crosstalk between the urokinase-type
plasminogen activator receptor and EGF receptor variant III
supports survival and growth of glioblastoma cells. Proc Natl Acad
Sci USA. 108:15984–15989. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu J, Muller KA, Furnari FB, Cavenee WK,
VandenBerg SR and Gonias SL: Neutralizing the EGF receptor in
glioblastoma cells stimulates cell migration by activating
uPAR-initiated cell signaling. Oncogene. 34:4078–4088. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gupta R, Chetty C, Bhoopathi P, Lakka S,
Mohanam S, Rao JS and Dinh DE: Downregulation of uPA/uPAR inhibits
intermittent hypoxia-induced epithelial-mesenchymal transition
(EMT) in DAOY and D283 medulloblastoma cells. Int J Oncol.
38:733–744. 2011.PubMed/NCBI
|
|
15
|
Ashour AA, Gurbuz N, Alpay SN, Abdel-Aziz
AA, Mansour AM, Huo L and Ozpolat B: Elongation factor-2 kinase
regulates TG2/β1 integrin/Src/uPAR pathway and
epithelial-mesenchymal transition mediating pancreatic cancer cells
invasion. J Cell Mol Med. 18:2235–2251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baek MK, Kim MH, Jang HJ, Park JS, Chung
IJ, Shin BA, Ahn BW and Jung YD: EGF stimulates uPAR expression and
cell invasiveness through ERK, AP-1, and NF-κB signaling in human
gastric carcinoma cells. Oncol Rep. 20:1569–1575. 2008.PubMed/NCBI
|
|
17
|
Smith HW and Marshall CJ: Regulation of
cell signalling by uPAR. Nat Rev Mol Cell Biol. 11:23–36. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bessard A, Frémin C, Ezan F, Coutant A and
Baffet G: MEK/ERK-dependent uPAR expression is required for
motility via phosphorylation of P70S6K in human hepatocarcinoma
cells. J Cell Physiol. 212:526–536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu Z, Xu R, Liu J, Zhang Y, Du J, Li W,
Zhang W, Li Y, Zhu Y and Gu L: GEP100 regulates epidermal growth
factor-induced MDA-MB-231 breast cancer cell invasion through the
activation of Arf6/ERK/uPAR signaling pathway. Exp Cell Res.
319:1932–1941. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tushir JS and D'Souza-Schorey C:
ARF6-dependent activation of ERK and Rac1 modulates epithelial
tubule development. EMBO J. 26:1806–1819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Shan F, Xiong G, Chen X, Guan X,
Wang JM, Wang WL, Xu X and Bai Y: EGF-induced C/EBPβ participates
in EMT by decreasing the expression of miR-203 in esophageal
squamous cell carcinoma cells. J Cell Sci. 127:3735–3744. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Lin Z, Sun L, Fan S, Huang Z,
Zhang D, Yang Z, Li J and Chen W: Akt/Ezrin Tyr353/NF-κB pathway
regulates EGF-induced EMT and metastasis in tongue squamous cell
carcinoma. Br J Cancer. 110:695–705. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cordonnier T, Bishop JL, Shiota M, Nip KM,
Thaper D, Vahid S, Heroux D, Gleave M and Zoubeidi A: Hsp27
regulates EGF/β-catenin mediated epithelial to mesenchymal
transition in prostate cancer. Int J Cancer. 136:E496–E507. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Savagner P: Epithelial-mesenchymal
transitions: From cell plasticity to concept elasticity. Curr Top
Dev Biol. 112:273–300. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ha GH, Park JS and Breuer EK: TACC3
promotes epithelial-mesenchymal transition (EMT) through the
activation of PI3K/Akt and ERK signaling pathways. Cancer Lett.
332:63–73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen B, Zeng X, He Y, Wang X, Liang Z, Liu
J, Zhang P, Zhu H, Xu N and Liang S: STC2 promotes the
epithelial-mesenchymal transition of colorectal cancer cells
through AKT-ERK signaling pathways. Oncotarget. 7:71400–71416.
2016.PubMed/NCBI
|
|
27
|
Wang Z, Qu L, Deng B, Sun X, Wu S, Liao J,
Fan J and Peng Z: STYK1 promotes epithelial-mesenchymal transition
and tumor metastasis in human hepatocellular carcinoma through
MEK/ERK and PI3K/AKT signaling. Sci Rep. 6:332052016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Y, Du J, Zheng J, Liu J, Xu R, Shen
T, Zhu Y, Chang J, Wang H, Zhang Z, et al: EGF-reduced Wnt5a
transcription induces epithelial-mesenchymal transition via
Arf6-ERK signaling in gastric cancer cells. Oncotarget.
6:7244–7261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Laurenzana A, Biagioni A, Bianchini F,
Peppicelli S, Chillà A, Margheri F, Luciani C, Pimpinelli N, Del
Rosso M, Calorini L, et al: Inhibition of uPAR-TGFβ crosstalk
blocks MSC-dependent EMT in melanoma cells. J Mol Med. 93:783–794.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clapéron A, Mergey M, Nguyen Ho-Bouldoires
TH, Vignjevic D, Wendum D, Chrétien Y, Merabtene F, Frazao A,
Paradis V, Housset C, et al: EGF/EGFR axis contributes to the
progression of cholangiocarcinoma through the induction of an
epithelial-mesenchymal transition. J Hepatol. 61:325–332. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grassi ML, Palma CS, Thomé CH, Lanfredi
GP, Poersch A and Faça VM: Proteomic analysis of ovarian cancer
cells during epithelial-mesenchymal transition (EMT) induced by
epidermal growth factor (EGF) reveals mechanisms of cell cycle
control. J Proteomics. 151:2–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu Q, Zhang Q, Ishida Y, Hajjar S, Tang X,
Shi H, Dang CV and Le AD: EGF induces epithelial-mesenchymal
transition and cancer stem-like cell properties in human oral
cancer cells via promoting Warburg effect. Oncotarget. 8:9557–9571.
2017.PubMed/NCBI
|
|
33
|
Han J, Xie Y, Lan F, Yu Y, Liu W, Chen J,
Zheng F, Ouyang X, Lin X, Lin Y, et al: Additive effects of EGF and
IL-1β regulate tumor cell migration and invasion in gastric
adenocarcinoma via activation of ERK1/2. Int J Oncol. 45:291–301.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
da Rosa MR, Falcão AS, Fuzii HT, da Silva
Kataoka MS, Ribeiro AL, Boccardo E, de Siqueira AS, Jaeger RG, de
Jesus Viana Pinheiro J and de Melo Alves Júnior S: EGFR signaling
downstream of EGF regulates migration, invasion, and MMP secretion
of immortalized cells derived from human ameloblastoma. Tumour
Biol. 35:11107–11120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo B, Gao J, Zhan J and Zhang H:
Kindlin-2 interacts with and stabilizes EGFR and is required for
EGF-induced breast cancer cell migration. Cancer Lett. 361:271–281.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu XX, Hu Z, Shen X, Dong LY, Zhou WZ and
Hu WH: IL-33 promotes gastric cancer cell invasion and migration
via ST2-ERK1/2 pathway. Dig Dis Sci. 60:1265–1272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hong H, Jiang L, Lin Y, He C, Zhu G, Du Q,
Wang X, She F and Chen Y: TNF-alpha promotes lymphangiogenesis and
lymphatic metastasis of gallbladder cancer through the
ERK1/2/AP-1/VEGF-D pathway. BMC Cancer. 16:2402016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kong B, Michalski CW, Hong X, Valkovskaya
N, Rieder S, Abiatari I, Streit S, Erkan M, Esposito I, Friess H,
et al: AZGP1 is a tumor suppressor in pancreatic cancer inducing
mesenchymal-to-epithelial transdifferentiation by inhibiting
TGF-β-mediated ERK signaling. Oncogene. 29:5146–5158. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shirakihara T, Horiguchi K, Miyazawa K,
Ehata S, Shibata T, Morita I, Miyazono K and Saitoh M: TGF-β
regulates isoform switching of FGF receptors and
epithelial-mesenchymal transition. EMBO J. 30:783–795. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Shen Y, Miao Y, Luan Y, Sun B and
Qiu X: Co-expression of uPAR and CXCR4 promotes tumor growth and
metastasis in small cell lung cancer. Int J Clin Exp Pathol.
7:3771–3780. 2014.PubMed/NCBI
|
|
41
|
Pavón MA, Arroyo-Solera I, Céspedes MV,
Casanova I, León X and Mangues R: uPA/uPAR and SERPINE1 in head and
neck cancer: Role in tumor resistance, metastasis, prognosis and
therapy. Oncotarget. 7:57351–57366. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
LaRusch GA, Mahdi F, Shariat-Madar Z,
Adams G, Sitrin RG, Zhang WM, McCrae KR and Schmaier AH: Factor XII
stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to
initiate angiogenesis. Blood. 115:5111–5120. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zheng D, Hu Z, He F, Gao C, Xu L, Zou H,
Wu Z, Jiang X and Wang J: Downregulation of galectin-3 causes a
decrease in uPAR levels and inhibits the proliferation, migration
and invasion of hepatocellular carcinoma cells. Oncol Rep.
32:411–418. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ahmed N, Oliva K, Wang Y, Quinn M and Rice
G: Downregulation of urokinase plasminogen activator receptor
expression inhibits Erk signalling with concomitant suppression of
invasiveness due to loss of uPAR-beta1 integrin complex in colon
cancer cells. Br J Cancer. 89:374–384. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Raghu H, Gondi CS, Dinh DH, Gujrati M and
Rao JS: Specific knockdown of uPA/uPAR attenuates invasion in
glioblastoma cells and xenografts by inhibition of cleavage and
trafficking of Notch-1 receptor. Mol Cancer. 10:1302011. View Article : Google Scholar : PubMed/NCBI
|