Introduction
Prostate cancer (PCa) is the second leading cause of
cancer-related deaths in men in the United States (1). Despite the initial promise of androgen
deprivation therapy (ADT), relapse of aggressive and therapeutic
resistant tumors is a principal cause of death among patients
(1). This ADT resistant form of PCa
is referred to as hormone refractory or castration-resistant
prostate cancer (CRPC) (2), which
is associated with continuous androgen receptor (AR) signaling even
in the absence of androgen (3–7).
Various mechanisms underlying the constitutive AR signaling may
include AR gene amplification, ligand-independent AR activation by
cytokines or kinases, both intracrine and/or intratumoral androgen
production, overexpression of AR co-activators, and most
importantly, the expression of constitutively active AR splice
variants (AR-Vs) (8,9). These truncated forms of AR lack the
C-terminal ligand binding domain (LBD) but retain the
transactivating N-terminal domain (NTD), thus promoting
transcriptional activation of AR target genes despite castrated
hormone levels (8–10).
AR-V7 (also known as AR3) is a major splice variant
of full-length AR (AR-FL) that encodes a functional protein
(11–19). Increased AR-V7 levels are detected
in tumor specimens (11) and in
circulating tumor cells (18) from
CRPC patients. Survey of primary tumor tissues before and after
castration resistance clearly showed increased AR-V7 expression
following the outgrowth of CRPC tumors (11–17).
Furthermore, resistance to the potent second-generation
anti-androgens, e.g. enzalutamide (ENZ) and abiraterone acetate
(ABI) has been attributed to overexpression of AR-V7 (20,21).
Studies also demonstrated a crucial role of AR-FL in regulating
dimerization and transactivation function of AR-V7 (22), which is implicated in
castration-resistant cell growth (23,24).
Therefore, there is an urgent need to develop therapeutic
strategies that effectively suppress the constitutive tumor
promoting signals associated with AR-FL and AR-V7 action in
CRPC.
Sulforaphane (SFN), an isothiocyanate phytochemical
found in cruciferous vegetables (e.g. broccoli), is a promising
anticancer agent with multiple cellular targets (25–27).
Several studies have also implicated SFN as a promising agent for
metastatic CRPC, especially since it shows specific toxicity
towards transformed cells without significant adverse effect on
primary prostate epithelial cells (28–31).
At pharmacological doses, SFN has been shown to slow down the
progression of PCa (32–34). A recent study has also documented
the ability of SFN to target the cancer stem cell (CSC) phenotype
(35,36). Mechanistic studies have reported
SFN-induced cell death to be initiated by reactive oxygen species
(ROS) (37,38) and the release of hydrogen sulfide
(39). Therefore, SFN may partly
display its effect via epigenetic modifications of Nrf2 gene
leading to the activation of downstream
anti-oxidative/detoxification stress pathway and also by
suppression of Akt survival pathway (40–42).
SFN has also been shown to inhibit AR-FL levels by destabilization
of protein, primarily by inactivating HDAC6 and the subsequent
suppression of the chaperone function of heat-shock proteins
(Hsp90) (43–45). However, efficacy of SFN against CRPC
cells that express both AR-FL and AR-V7 has not been tested.
Our previous study using both androgen-dependent
(LNCaP) and androgen-independent (C4-2B) cell lines showed that SFN
can potentiate the efficacy of ENZ by rapidly decreasing AR-FL
levels (46). In the current study,
we show that SFN can suppress the levels of both AR-FL and AR-V7
proteins in the 22Rv1 cells. Mechanistic studies demonstrated the
efficacy of SFN in increasing both ubiquitination and proteasomal
activity in 22Rv1 cells. Since the multimodal actions of SFN are
known to decrease Hsp90 (43–45)
and increase Nrf2 (40,41), we tested whether combined exposure
to an Hsp90 inhibitor (ganetespib) and an Nrf2 activator
(bardoxolone-methyl) can be similarly effective. Our findings
showed that co-exposure to physiologically achievable doses of the
above clinically approved drugs decreases both AR-FL and AR-V7
levels and sensitizes 22Rv1 cells to the anticancer effects of
ENZ.
Materials and methods
Cell culture
CWR22Rv1 (an androgen-independent PCa cell line that
expresses both AR-FL and AR-V7) and LNCaP (an androgen-dependent
PCa cell line that expresses only AR-FL) were purchased from
American Type Culture Collection (ATCC; Rockville, MD, USA). The
C4-2B cell line (an androgen-independent sub-line of LNCaP that
expresses AR-FL and low levels of AR-Vs) was a kind gift from Dr.
Leland Chung (47). The cell lines
were maintained in RPMI-1640 media supplemented with 10% fetal
bovine serum (FBS) (Atlanta Biologicals; Lawrenceville, GA, USA)
and 1% penicillin/streptomycin (CellGro; Manassas, VA, USA) in a
humidified incubator containing 5% CO2 at 37°C. To mimic
steroid hormone deprived conditions, experiments were carried out
in phenol-red free media supplemented with 10% charcoal-stripped
FBS (CS-FBS) (Atlanta Biologicals).
Reagents
Sulforaphane (SFN) and MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Ganetespib (G)
and enzalutamide (ENZ) were obtained from ApexBio (Houston, TX,
USA). Cycloheximide (CHX) was purchased from Cayman chemicals (Ann
Arbor, MI, USA). MG132 was obtained from EMD Millipore (Billerica,
MA, USA). Bardoxolone methyl was purchased from Selleckchem
(Houston, TX, USA). All drugs were dissolved in 100% DMSO and
diluted in media immediately before use. The final DMSO
concentration used in experiments was less than 0.1%. The primary
antibodies including rabbit polyclonal anti-AR (N-20) (sc-816) and
mouse monoclonal anti-GAPDH (sc-47724) were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). A mouse monoclonal
antibody against ubiquitinated proteins (FK2) (BML-PW8810) was
obtained from Enzo Life Sciences (Farmingdale, NY, USA). The
horseradish peroxidase (HRP)-conjugated goat anti-rabbit (A0545)
and goat anti-mouse (A9044) secondary antibodies were purchased
from Sigma-Aldrich. The goat anti-rabbit secondary antibody tagged
with Texas red (T-2767) was purchased from Thermo Fisher Scientific
(Rockford, IL, USA).
MTT assay
MTT assays were performed to determine cell
viability post exposure to the drug(s). In brief, ~5,000 cells were
seeded in 96-well culture plates and allowed to adhere overnight.
Cells were then synchronized by overnight incubation in serum-free
medium, and treated with desired concentrations of drug(s), alone
or in different combinations for 24–72 h. Cell viability was
determined by adding MTT solution (5 mg/ml) and incubating for 3 h
at 37°C. The formazan crystals were then solubilized in DMSO and
optical density (O.D.) was measured at 540 nm by using a µQuant
spectrophotometric plate reader from Bio-Tek (Seattle, WA, USA). In
each individual experiment, changes in cell survival following drug
treatments are expressed as percent of untreated control.
Western immunoblot
Whole cell lysates were harvested at different time
points post-treatment(s) using RIPA lysis buffer from Santa Cruz
Biotechnology and total protein content was quantified using the
bicinchoninic acid (BCA) protein assay reagent from Thermo Fisher
Scientific. Briefly, 10 µg of protein lysate was electrophoresed in
10% SDS-PAGE gels followed by electrotransfer onto nitrocellulose
membrane. After blocking nonspecific binding using 5% casein in
TBS-T buffer (tris buffer saline with 0.1% Tween-20), membranes
were incubated overnight at 4°C with the primary antibodies against
AR (1:500 dilution), GAPDH (1:3,000 dilution) or ubiquitin (1:1,000
dilution). This was followed by incubation with the corresponding
HRP-conjugated secondary antibodies (1:2,000 dilution) for 1 h.
Membranes were developed using the SuperSignal West Femto Substrate
(Thermo Fisher Scientific). Immunoblots were scanned using the
ImageQuant LAS 500 scanner (GE Healthcare; Princeton, NJ, USA) and
band intensities were quantified using the ImageJ software from NIH
(Bethesda, MD, USA). Densitometric value for AR proteins (AR-FL and
AR-V7) was normalized to GAPDH levels.
Isolation of Triton-soluble and
-insoluble fractions
Cells were lysed in RIPA lysis buffer containing 1%
Triton X-100 on ice and centrifuged at 16,000 × g for 15 min at
4°C. The supernatant was collected as Triton-soluble (TS) fraction
and the pellets (Triton-insoluble fraction) (TI) were further
solubilized in SDS buffer (2% SDS in 50 mM Tris-HCl) followed by
boiling for 15 min (48).
Approximately 10 µg of protein from both TS and TI fractions was
electrophoresed in 10% SDS-PAGE gels and immunoblots were analyzed
for AR protein levels.
Proteasomal activity assay
A Proteasome Assay kit (Cayman chemicals; cat #
10008041) was used to measure proteasomal activity of control
(untreated) and SFN treated cells. The proteasome inhibitor, MG132
was used as a positive control. Briefly, cells were seeded in
96-well microtiter plates (1×105 cells per well) and
allowed to adhere overnight. After appropriate drug treatments,
plates were centrifuged at 500 × g for 5 min and culture media was
aspirated. Assay buffer (200 µl) was added to each well followed by
centrifugation at 500 × g for 5 min and the supernatant was
aspirated. The lysis buffer (100 µl) was then added to each well
followed by gentle shaking for 30 min. Plates were centrifuged at
1,000 × g for 10 min and 90 µl of the supernatant from each well
was transferred to corresponding wells in a black (opaque) 96-well
plate (Sigma-Aldrich). For sample activity measurements, assay
buffer (10 µl) was added to these wells followed by the addition of
proteasome substrate, SUC-LLVY-AMC (10 µl). Fluorescence intensity
was measured using an FLx-800 fluorimeter (Bio-Tek) with the
absorption (excitation) set at 360 nm and the emission set at 480
nm. Mean fluorescence intensities (MFI) were normalized to protein
content in each sample.
Wound-healing assay
Wound-healing assays were carried out to measure the
effect of drugs on the migratory phenotype of PCa cells, as
previously described (49).
Briefly, cells were seeded in 6-well plates (1×106 cells
per well) and grown until they formed a confluent monolayer. The
monolayers were scratched using a 200 µl pipette tip, wells were
washed with PBS and images of the wound (0-time point) were
captured using a Leica Microsystems microscope (Buffalo Grove, IL,
USA). Growth media (CS-FBS) was returned to each culture and
treatments were initiated. Change in wound width was captured after
48 h and cell migration (wound closure) was calculated by measuring
the distance between 4–5 random points within the wound edges.
Colony forming units assay
Cells (500 cells/dish) were seeded in 60-mm petri
dishes in 3 replicates and grown in medium supplemented with 2%
FBS. The drugs, alone or in combination, were added after 48 h and
replenished in the second week. After two weeks in culture,
colonies were fixed with 100% ethanol and stained with 0.2% crystal
violet in 20% methanol. The colony forming units (CFU) were
enumerated using ImageJ software (NIH). Change in total CFUs were
compared in both control (untreated) and drug-exposed cultures.
Immunofluorescence microscopy
Subcellular localization of AR post-treatment with
SFN was visualized by immunofluorescence microscopy (IFM). Briefly,
cells (3×104) were seeded in chamber slides (EMD
Millipore) and allowed to adhere overnight. After treatment, cells
were fixed in ice cold methanol followed by permeabilization with
0.1% Triton X-100 for 1 h. After blocking in 10% goat serum, slides
were incubated overnight at 4°C with the primary antibody (1:300
dilution) followed by incubation with the corresponding Texas Red
tagged secondary antibody (1:1,000 dilution) for 1 h. The
Vectashield (Burlingame, CA) mounting medium containing the nuclear
stain diamino-2-phenylindole (DAPI) was then added to the slides
and cover slips were mounted. Images were captured using a
fluorescent microscope from Leica Microsystems Inc.
Statistical analysis
Statistical analysis was carried out using GraphPad
Prism (version 6) Software (San Diego, CA, USA). Results were
expressed as the standard error of mean (± SEM). Significant
changes from controls were determined by a two-tailed Student's
t-test and P-values of <0.05 were considered significant. For
synergy determination, the CompuSyn software (ComboSyn, Inc.,
Paramus, NJ, USA) was used and combination index (CI) was
calculated based on the Chou-Talalay method, which quantitatively
determines additive (CI=1), synergistic (CI <1) or antagonistic
(CI >1) effects (50).
Results
The AR-V7 expressing 22Rv1 cells are
resistant to androgen deprivation conditions and enzalutamide
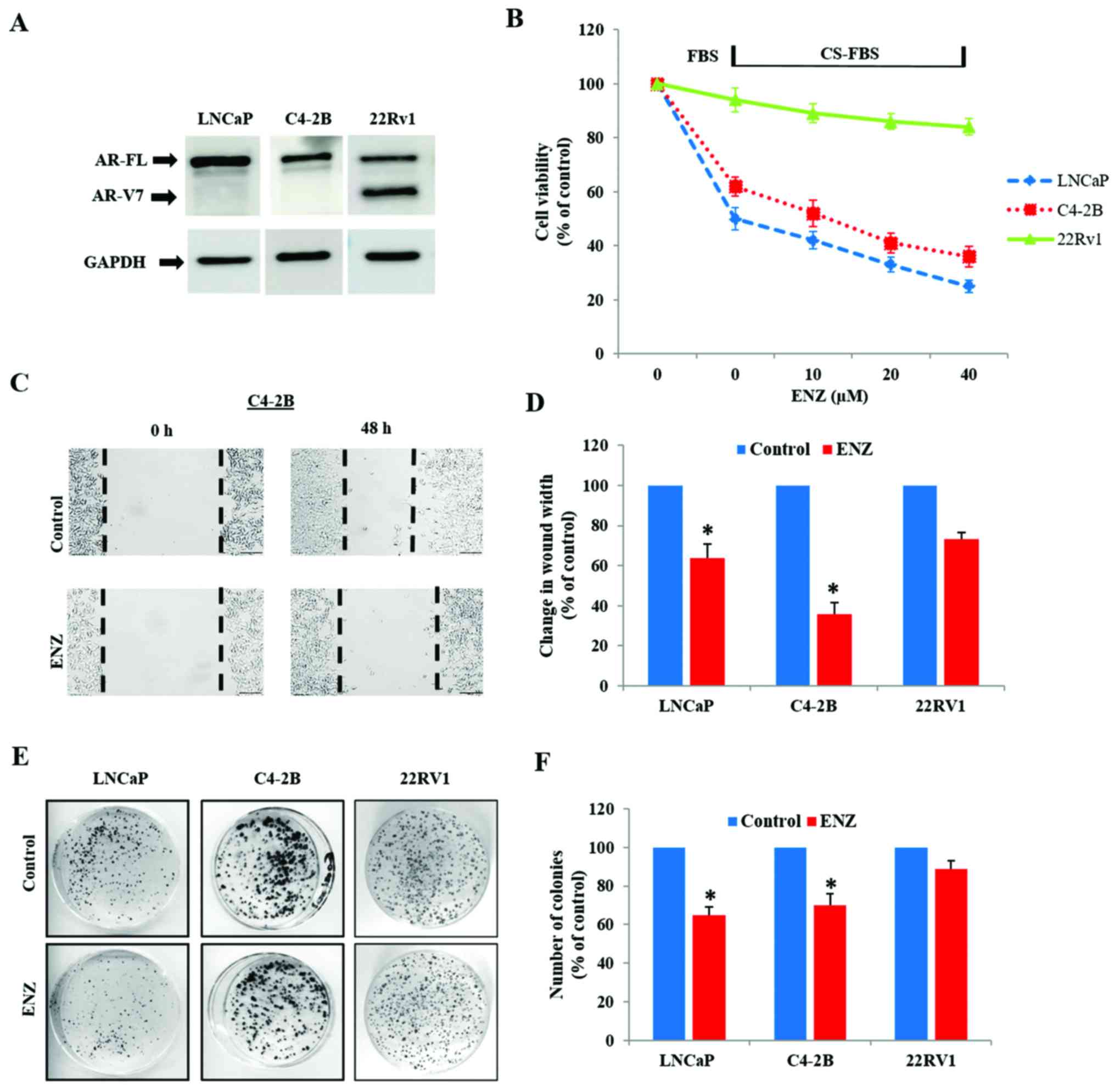
Immunoblot analysis showed that while the LNCaP and
C4-2B cells express only AR-FL protein (110 kDa), 22Rv1 cells
express AR-V7 (75 kDa) in addition to AR-FL (Fig. 1A). In order to confirm the inherent
resistance of 22Rv1 to androgen deprivation and anti-androgen
treatment, as compared to LNCaP and C4-2B, cells were cultured
under normal (FBS media) and androgen-depleted conditions (CS-FBS
media) in the absence or presence of enzalutamide (ENZ).
Differences in cell viability, migratory behavior and clonogenic
ability were compared in these three cell lines. In contrast to
LNCaP and C4-2B cells, androgen depletion (CS-FBS) had no impact on
the viability of 22Rv1 cells (Fig.
1B). Furthermore, although both LNCaP and C4-2B cells were
susceptible to ENZ (10–40 µM), only a slight suppression in 22Rv1
cell viability was seen following 72 h exposure to ENZ.
Wound-healing assays clearly showed that ENZ decreased the
wound-closure ability (migration potential) in both LNCaP and C4-2B
cells (Fig. 1C and D), but did not
affect the migratory behavior of 22Rv1 cells (Fig. 2C and D). In addition, colony-forming
unit (CFU) assays showed that long-term exposure to ENZ (0.4 µM)
showed less suppressive effect on the clonogenic ability of 22Rv1
cells, compared to LNCaP and C4-2B cells (Fig. 1E and F).
SFN increases the anticancer efficacy
of ENZ in 22Rv1 cells
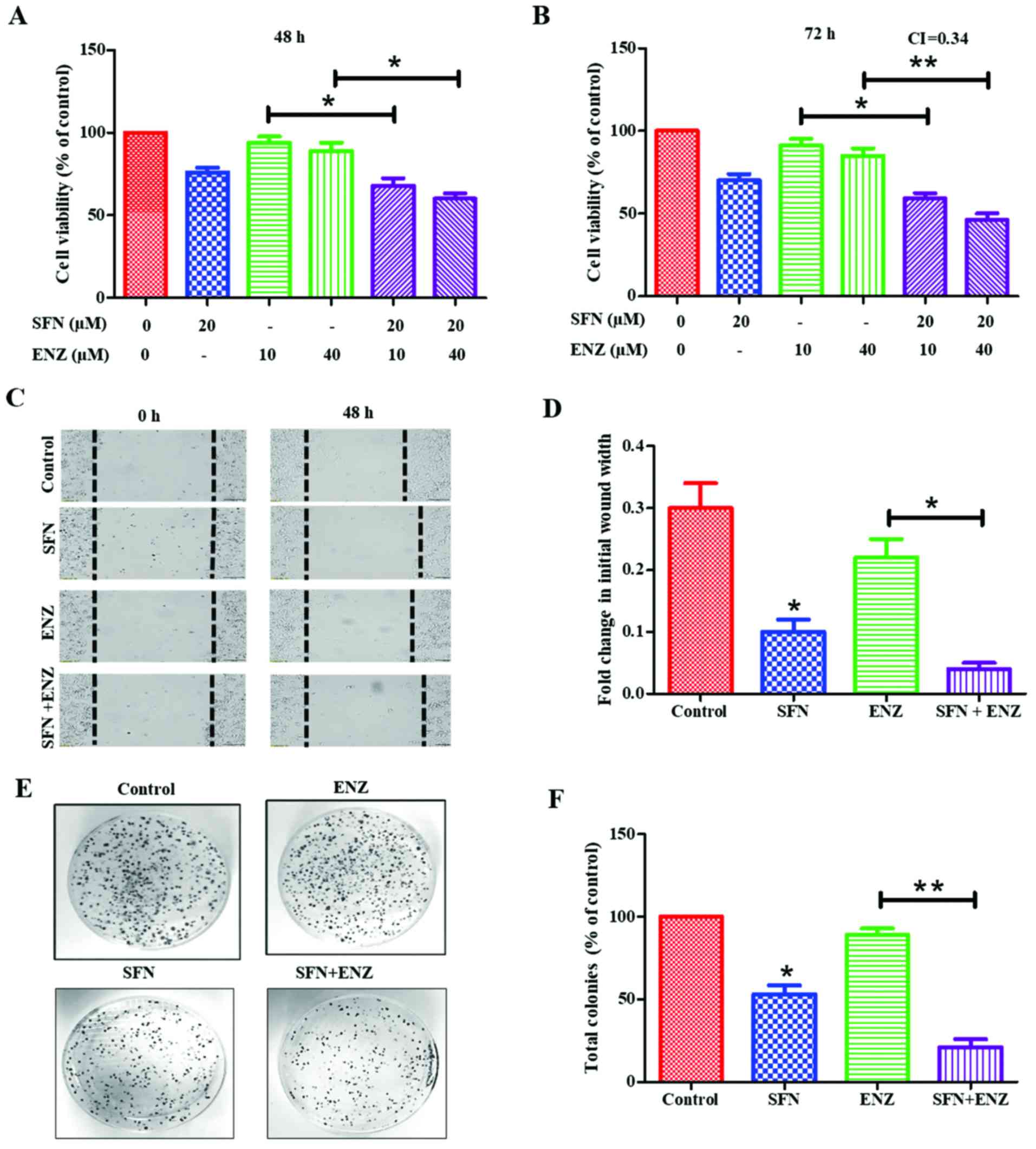
Exposure to SFN alone showed a dose- and
time-dependent suppressive effect on 22Rv1 proliferation, with ~40%
suppression following 72 h exposure to 30 µM of SFN (data not
shown). However, co-exposure to lower doses of SFN was able to
sensitize these cells to ENZ (Fig. 2A
and B). When ENZ (10 or 40 µM) was used in combination with
sub-IC50 dose of SFN (20 µM), a significant (P<0.05)
increase in cytotoxicity was observed within 48 h, which was more
evident at 72 h. Combination index (CI) calculations demonstrated
that this drug combination functions in a synergistic manner at 72
h post-exposure (CI=0.34) (Fig.
2B). Furthermore, co-exposure to SFN increased the ability of
ENZ to suppress migration/motility of 22Rv1 cells (Fig. 2C and D). Wound-healing assays were
carried out in the absence or presence of SFN (10 µM), alone and in
combination with ENZ (20 µM). In control cultures, the wound-width
decreased by approximately 30% after 48 h in culture, and exposure
to ENZ alone did not significantly decrease the wound closure.
However, exposure to SFN alone decreased wound-closure by almost
70% and co-exposure to SFN and ENZ decreased it by as much as 90%.
Our investigations also showed that long-term exposure to SFN can
suppress the clonogenic ability of 22Rv1 cells, and further
increase the efficacy of ENZ in suppressing CFUs (Fig. 2E and F). To optimize our combination
studies, we first determined the effect of each drug alone [data
not shown] and then used doses that caused <50% decrease in
CFUs. Although ENZ (0.4 µM) alone did not significantly decrease
CFUs, exposure to SFN (0.2 µM) caused ~50–55% decrease and cells
exposed to SFN and ENZ combination showed almost a complete
suppression of CFUs (P<0.01) (Fig.
2F). Thus, SFN potentiates the efficacy of ENZ in suppressing
the proliferation, migration and clonogenic ability of 22Rv1
cells.
SFN decreases both AR-FL and AR-V7
levels in 22Rv1 cells
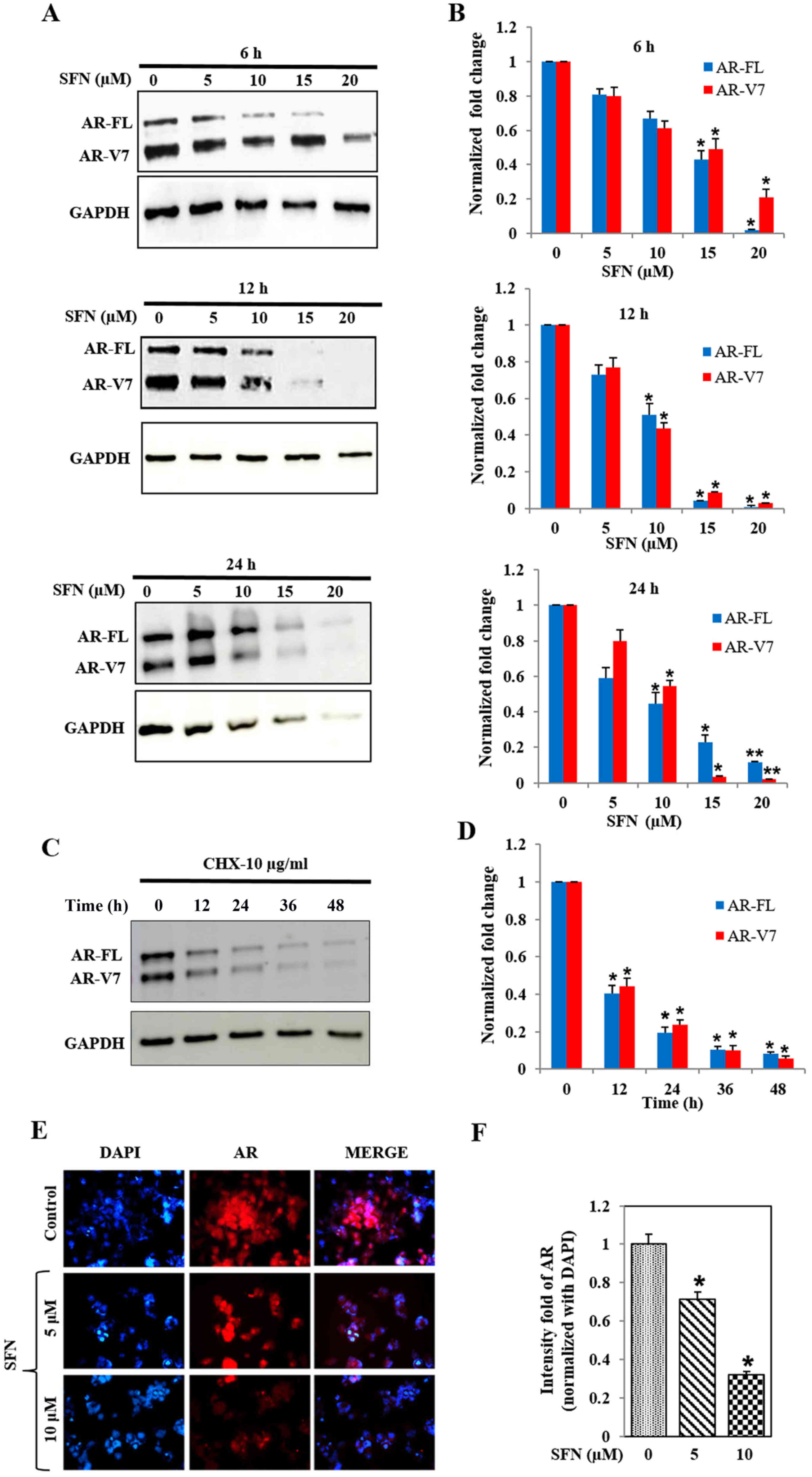
Immunoblot analysis showed that SFN treatment
reduced protein levels of both AR-FL and AR-V7 in 22Rv1 cells, in a
concentration- and time-dependent manner (Fig. 3A and B). High dose of SFN (20 µM)
abrogated AR protein levels within 6 h, whereas 50% reduction was
observed with 15 µM SFN at this time point. At 12 and 24 h,
significant (P<0.05) suppression of both AR-FL and AR-V7 was
evident even with the lower dose of SFN (10 µM). The reduction in
AR-FL protein levels was more pronounced than the concomitant
reduction in AR-V7 levels, suggesting that SFN may differentially
affect the stability of these two AR proteins. Studies using the
translation inhibitor, cycloheximide (10 µg/ml) clearly showed that
the inherent stability of these two AR proteins is much greater in
the 22Rv1 cells (Fig. 3C and D) and
clearly indicated that SFN increases the rate of degradation of
both AR-FL and AR-V7. Immunofluorescence microscopy (IFM) studies
demonstrated that the treatment of 22Rv1 cells with SFN (5 and 10
µM) can significantly reduce total AR protein levels within 24 h
(Fig. 3E and F). Comparative
analysis demonstrated that SFN suppresses both cytoplasmic and
constitutively expressed nuclear AR levels in 22Rv1 cells.
SFN increases proteasomal activity,
protein ubiquitination and aggregation of both AR-FL and AR-V7
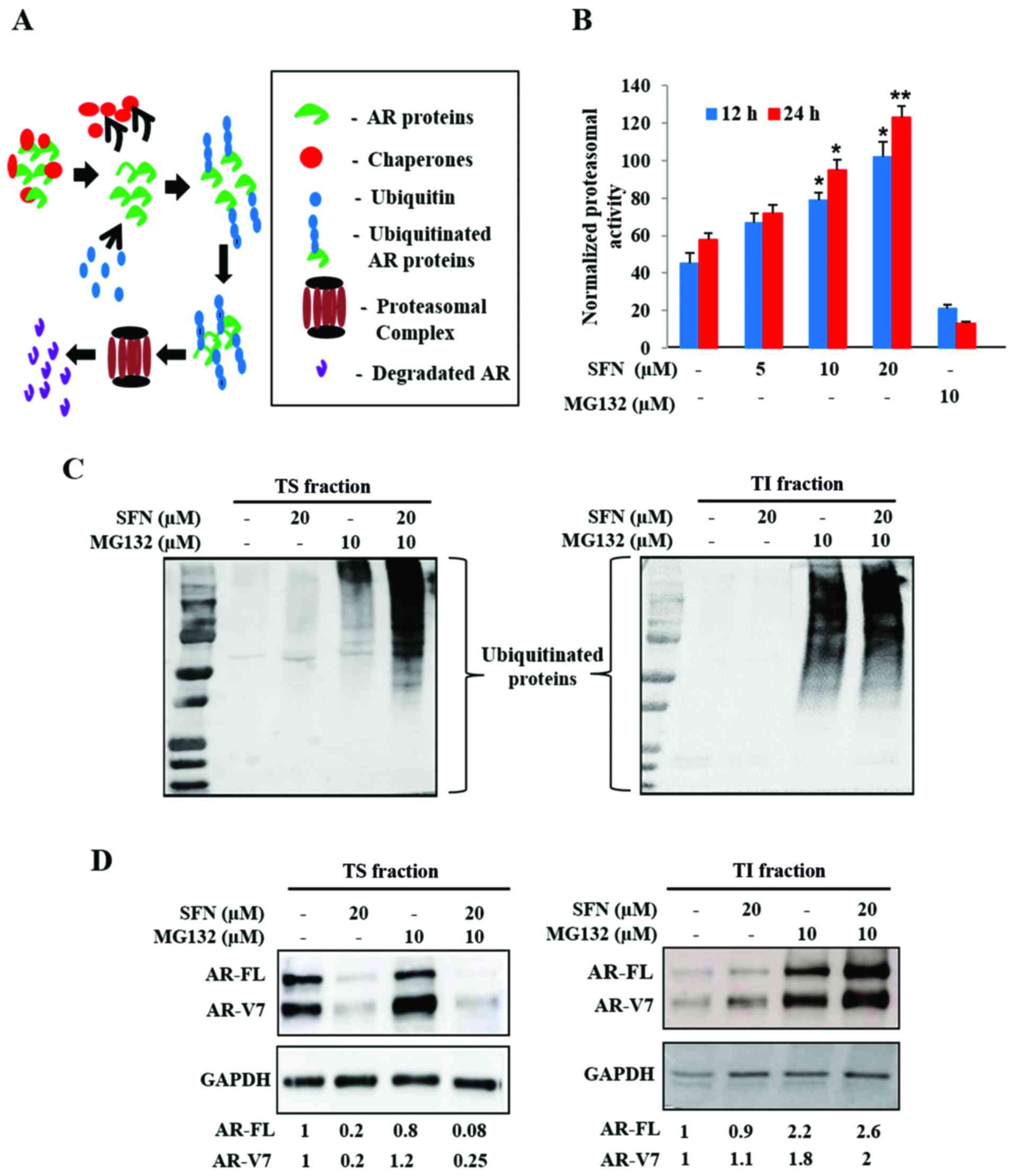
Intracellular proteins are continually degraded to
their constituent amino acids via the ubiquitin-proteasome system
(UPS) and are often protected from degradation via heat shock
protein (Hsp) chaperones. Stability of AR proteins is similarly
maintained via chaperone binding, ubiquitination and transit to the
26S proteasomal complex (Fig. 4A).
Since our studies implicated that SFN may decrease the stability of
both AR-FL and AR-V7, we tested if SFN treatment resulted in
increased proteasomal degradation of AR. Exposure to SFN
significantly increased proteasomal activity in 22Rv1 cells in both
dose (5–20 µM) and time (12, 24 h) dependent manner, an effect
validated by using MG132, a pharmacological proteasomal inhibitor
(Fig. 4B). These findings suggested
that SFN-mediated increase in proteasomal activity may be
responsible for degradation of both AR-FL and AR-V7.
To document AR-FL and AR-V7 levels in both the
soluble cytosolic fraction and in ubiquitinated aggresomal
fraction, we isolated both Triton-soluble (TS) and Triton-insoluble
(TI) fractions, respectively. Immunoblot analyses of TS (20 sec
exposure) and TI (60 sec exposure) indicated that proteasomal
blockade via MG132 increases ubiquitinated protein levels in both
TS and TI fractions (Fig. 4C).
Although exposure to SFN alone showed negligible increase in
ubiquitination, a striking enhancement of the ubiquitinated protein
bands was evident upon co-exposure to MG132, in both TS fraction
(Fig. 4C, left) and TI fraction
(Fig. 4C, right). This indicated
that SFN mediates ubiquitination and aggregation of cellular
proteins.
An augmented UPS may be responsible for rapid
suppression of both AR-FL and AR-V7 in SFN treated 22Rv1 cells.
Therefore, immunoblot analyses of AR-FL and AR-V7 levels were
carried out in both TS and TI fractions (Fig. 4D). Negligible quantities of AR
proteins were present in insoluble aggregates (TI fraction) under
basal conditions. Inhibition of proteasomal degradation by MG132
slightly increased AR levels in the TS fraction; however, it
significantly augmented both AR-FL and AR-V7 levels in the TI
aggregates. Although exposure to SFN (20 µM) reduced both AR-FL and
AR-V7 levels in the TS fraction (Fig.
4D, left), some of these AR proteins were consistently found in
the TI fraction (Fig. 4D, right).
Co-exposure to MG132 considerably increased the amount of
aggregated AR proteins in the TI fraction. These findings
implicated that the multimodal actions of SFN increase
ubiquitination, aggregation and proteasomal degradation to enable a
rapid and profound decrease in both AR-FL and AR-V7 protein levels
in 22Rv1 cells.
Inhibition of Hsp90 or activation of
Nrf2 reduces AR protein levels in 22Rv1 cells
Several past studies have reported that SFN
functions by inhibiting the chaperone activity of Hsp90 (43–45)
and by decreasing oxidative stress via increased Nrf2 function
(40,41). Therefore, we investigated whether
the AR suppressive effects of SFN can be replicated by targeting
the above two metabolic pathways. Treatment of 22Rv1 cells with the
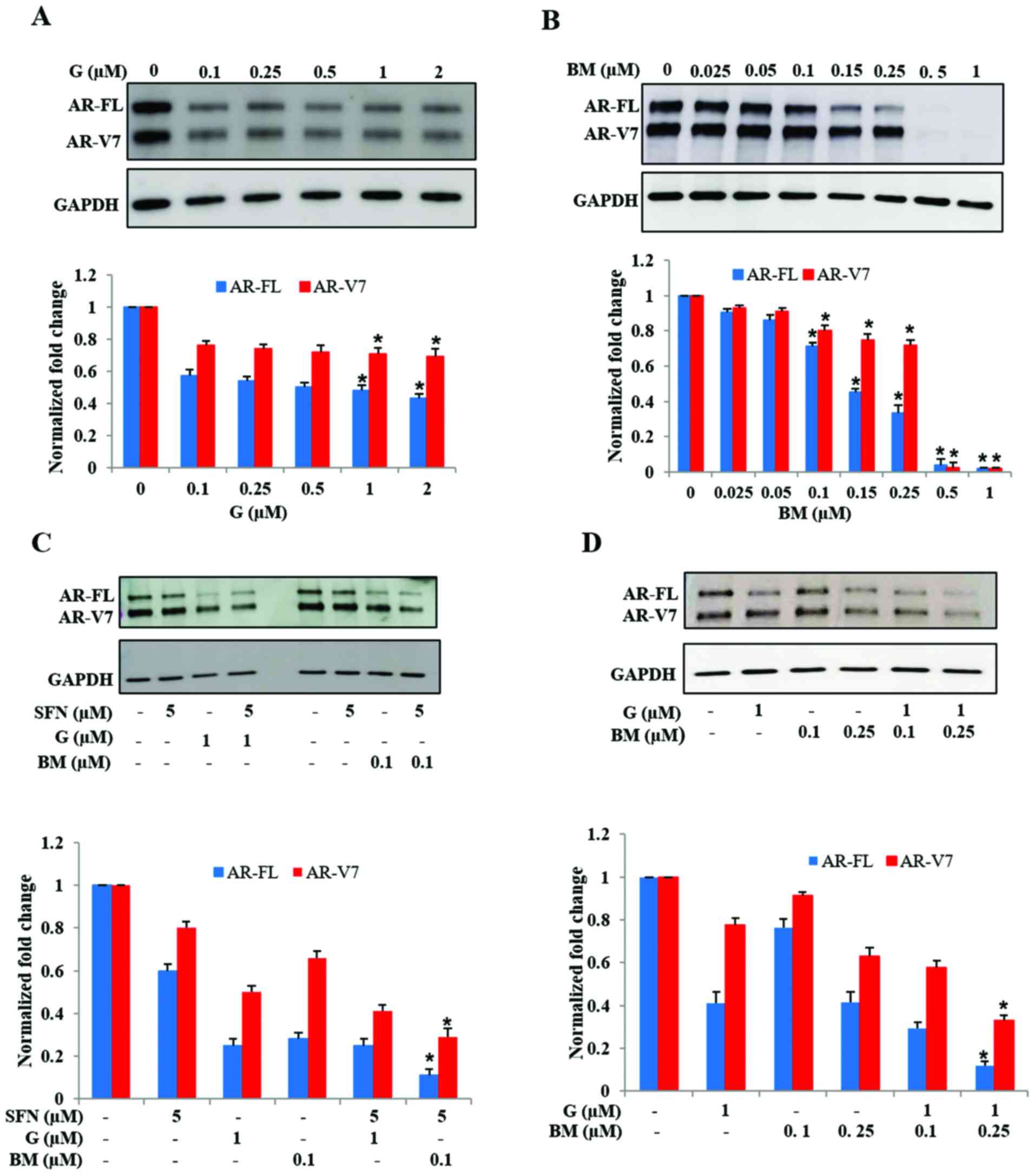
Hsp90 inhibitor, ganetespib (G) decreased both AR-FL and AR-V7
protein levels (Fig. 5A). Notably,
exposure to G did not show a dose-dependent effect in suppression
of AR-FL and AR-V7 levels. However, exposure to the Nrf2 activator,
bardoxolone methyl (BM) showed a pronounced effect in abrogating
both AR-FL and AR-V7 levels (Fig.
5B). Treatment with BM significantly reduced both AR-FL and
AR-V7 protein levels in a dose-dependent manner. We observed a
precipitous decrease in AR levels at the highest doses of BM used
(0.5 and 1.0 µM) which totally eliminated both AR-FL and AR-V7
proteins within 24 h. These findings indicated that both Hsp90
inhibition and Nrf2 induction, two pathways targeted by SFN, are
involved in suppressing AR protein levels in 22Rv1 cells, but may
have different potency.
We investigated whether augmentation of the Hsp90
inhibitory activity or the Nrf2 inductive effect of SFN will
enhance its efficacy, by using low dose SFN (5 µM) in presence of
either G (1 µM) or BM (0.1 µM) (Fig.
5C). Immunoblot studies indicated that increasing the Hsp90
inhibitory function of SFN, via the addition of G, did not
significantly enhance its potency in suppressing AR proteins.
However, co-exposure to SFN and low-dose BM showed significantly
increased suppression, as compared to SFN or BM alone. In addition,
we observed that, even in the absence of SFN, combined treatment
with low-dose G (1 µM) and BM (0.1 or 0.25 µM) was able to reduce
AR protein levels, which was much more than with either drug alone
(Fig. 5D). These findings indicated
that the knowledge obtained from the multimodal actions of SFN can
be utilized to use a pharmaceutical drug combination that
suppresses both AR-FL and AR-V7 in 22Rv1 cells.
Co-targeting of Hsp90 and Nrf2
sensitizes 22Rv1 cells to the anticancer effects of ENZ
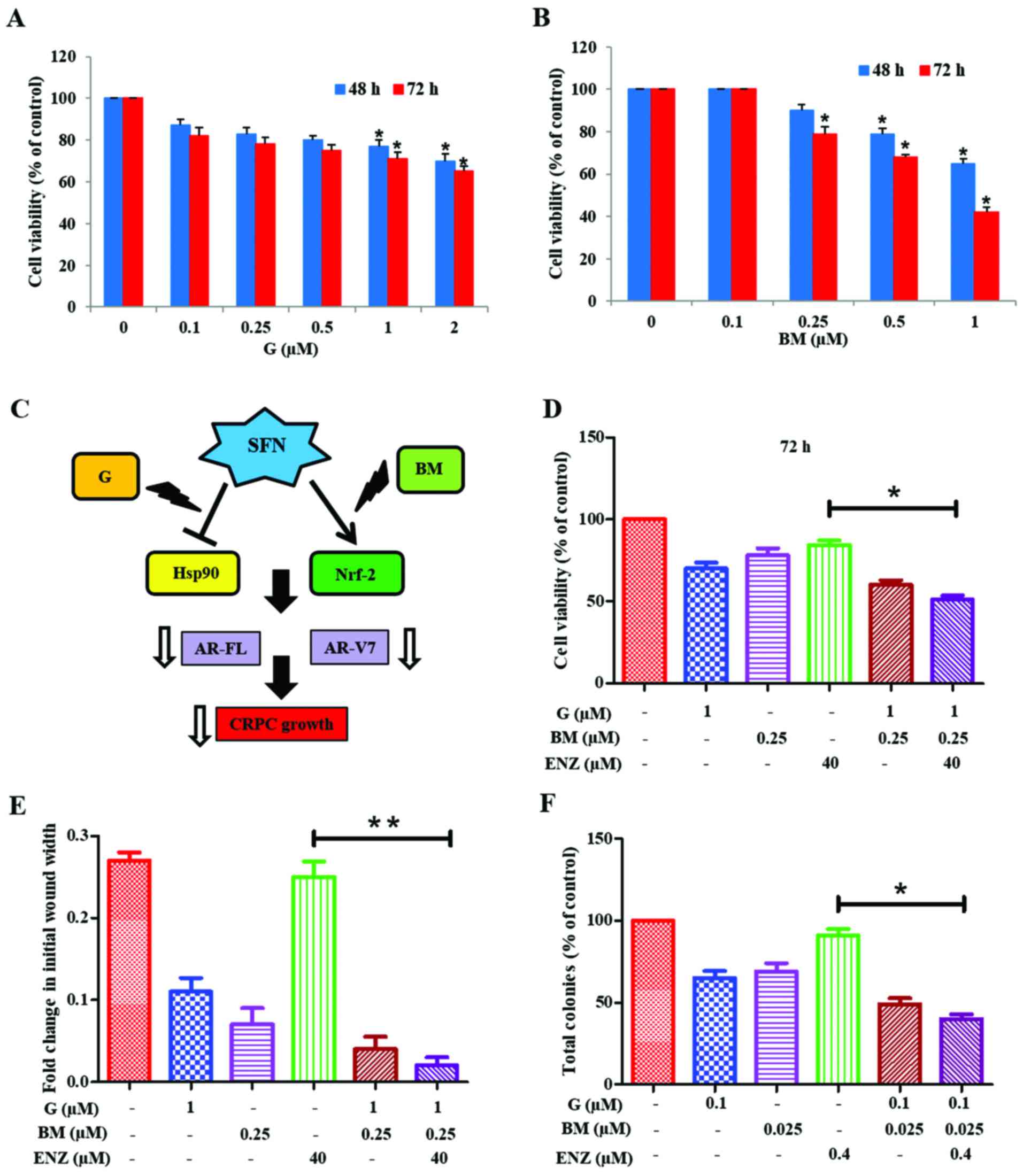
We investigated whether physiologically achievable
concentrations of G and BM can be used to sensitize 22Rv1 cells to
the anticancer effects of ENZ (Fig.
6). MTT cell viability assays were first carried out to
document the cytotoxic effects of increasing doses of G (0.1–2.0
µM) or BM (0.1–1.0 µM) alone (Fig. 6A
and B). Since the anticancer effects of SFN may involve
targeting of both Hsp90 and Nrf2 pathways, we investigated whether
co-exposure to G+BM would show potent anticancer and drug
sensitizing effects, as well (Fig.
6C). Our investigations clearly showed that combined treatment
with G (1 µM) and BM (0.25 µM) resulted in reduction of cell
viability and sensitized 22Rv1 cells to ENZ (40 µM) treatment. This
suppressive effect was evident at 48 h (not shown) and
significantly (P<0.05) more profound at 72 h (Fig. 6D). Furthermore, combination studies
using both wound-healing assays (Fig.
6E) and CFU assays (Fig. 6F)
demonstrated the efficacy of G+BM combination in suppressing both
the migratory behavior and clonogenic ability, respectively of
22Rv1 cells. Thus, our observations show that the knowledge of
multimodal effects of SFN may be exploited to formulate a potent
drug combination that works at nanomolar concentrations, in order
to sensitize the AR-V7 expressing CRPC cells to ENZ.
Discussion
The AR signaling axis is critical in both the
development and progression of PCa to CRPC (1). Nuclear translocation of AR and
induction of androgen response elements (ARE) regulated genes can
augment tumor proliferation, invasion and therapeutic resistance
(2–4). Therefore, strategies to suppress AR
function have been a standard of care in PCa patients (5). Efficacy of this approach is
corroborated by the survival benefits of newer and more potent
AR-targeted agents, such as enzalutamide (ENZ) and abiraterone
acetate (ABI) (51,52). However, despite the initial
favorable response, the CRPC tumors develop typically within 1–2
years in nearly all men (1).
Numerous clinical evidence suggests that AR variants may be the
functional drivers of PCa progression to CRPC (13–21).
The most clinically significant AR variant is reported to be AR-V7
(~80 kDa) (13–17). Hu et al showed an average of
20-fold higher expression of AR variant mRNA in CRPC tumors than in
hormone-naïve PCa samples. These investigators also showed that
AR-V7 mRNA, but not AR-V1 mRNA, was highly predictive of
biochemical recurrence and CRPC progression (15).
There is an urgent need to suppress both AR-FL and
AR-V7 for better therapeutic efficacy in patients with CRPC.
Although recent studies using the anti-helminthic drug, niclosamide
have documented its potent ability to suppress AR-V7 levels; no
significant suppression of AR-FL levels was documented (53,54).
Sarwar et al, used the phosphatidylinositol-4-phosphate
5-kinase α (PIP5Kα) inhibitor, ISA-2011B to similarly disrupt
stabilization of AR-V7 protein, which circumvented resistance to
the anti-androgen enzalutamide (21). However, although ISA-2011B was very
potent and targeted both AR-FL and AR-V7 levels at nanomolar doses,
this experimental drug is not currently in any clinical trials and
translation to CRPC patients will involve significant time. Our
in vitro studies using 22Rv1 cells showed that the
phytochemical SFN, or the pharmaceutical agents G and BM, that are
both in several clinical trials (55–61),
may provide a more promising approach and can be rapidly
implemented as an adjunct agent in PCa patients, especially those
with therapeutic resistance.
The 22Rv1 cell line is a CRPC line that expresses
AR-FL and multiple AR splice variants, out of which AR-V7 is the
most abundant (62). Similar to
multiple previous studies (13–17,20,21),
our investigations, comparing the anticancer efficacy of
hormone-deprivation and ENZ confirmed that 22Rv1 cells are more
resistant than C4-2B and LNCaP cells. Overexpression of AR-V7 was
found to be sufficient in driving the growth of LNCaP cells even
under hormone-deprived conditions (16). In addition, studies demonstrated
that the knockdown of AR-V7 inhibits growth of 22Rv1 cells under
castrate conditions both in vitro and in vivo
(16,63). We observed that SFN can rapidly
decrease AR-FL and AR-V7 protein levels in 22Rv1 cells. The
precipitous decrease in AR proteins may enable the potent reduction
in cell proliferation, migration and clonogenic ability, observed
with SFN in 22Rv1 cells.
Intracellular proteins are continually degraded to
their constituent amino acids via the UPS pathway and cancer cells
can protect important proteins from degradation via several Hsp
chaperones (64). The stability of
AR in CRPC cells is also maintained via chaperone binding,
ubiquitination and transit to the 26S proteasomal complex (65). Thus, rapid degradation of AR-FL and
AR-V7 via SFN is most likely associated with the increased
proteasomal activity and ubiquitination rates observed following
exposure to SFN. This is in line with earlier reports showing
SFN-mediated enhancement of proteasomal activity (66). Of note, this was shown to be via the
upregulation of Hsp27, another chaperone protein known to support
cancer cell survival under stressful conditions by regulating both
ubiquitination and proteasomal activity. Our studies using both
Triton soluble and insoluble fractions (TS and TI) clearly showed
that the SFN-mediated reduction in AR protein levels in the TS
fraction was not reversed by MG132 co-exposure. However, AR levels
increased in the TI fraction in SFN and SFN+MG132 treated groups.
Indeed, significant increases in insoluble protein aggregates have
been reported following exposure to proteasomal inhibitors
(67–69). Protein aggregation is enhanced when
the accumulation of ubiquitinated proteins exceed beyond the
capacity of proteasomes to degrade them (70).
The Hsp90 protein is necessary for the stabilization
and correct folding of AR in both normal and malignant prostate
cells (71). However, as compared
to normal prostate epithelial cells, the expression of Hsp90 is
reported to significantly increase in the malignant PCa (72). Studies have shown that SFN
hyper-acetylates and inactivates Hsp90 (43) by targeting the function of histone
deacetylase 6 (HDAC6) (44,45) which may be associated with its
ability to degrade both AR-FL and AR-V7. However, we also observed
that potent inhibition of Hsp90 via high doses of G (2 µM) was
unable to fully abrogate AR protein levels. Recent studies have
shown that Hsp90 inhibitors alone are not very effective against
CRPC tumors (73). This clearly
underscores the importance of targeting parallel pathways such as
oxidative stress or Nrf2 to increase the efficacy of Hsp90
inhibitors against CRPC tumors.
Cancer cells are characterized by increased reactive
oxygen species (ROS) levels and oxidative stress (74). Nrf2 is a transcription factor which
is known to upregulate many cellular antioxidant proteins. In its
inactive state, Nrf2 is found in the cytoplasm bound by its
negative regulator Kelch-like ECH-associated protein 1 (Keap-1)
which prevents Nrf2 nuclear translocation and directs it for
proteasomal degradation. SFN has been shown to interact with Keap-1
thereby eliminating its inhibitory effect on Nrf2 (31,40,41).
In line with these earlier studies, we observed that treatment of
22Rv1 cells with a pharmacological activator of Nrf2, BM
drastically reduced both AR-FL and AR-V7 protein levels.
Noteworthy, co-exposure to low-dose BM enhanced the suppressive
effect of G. Thus, knowledge of the multimodal actions of SFN,
i.e., Hsp90 inhibition and Nrf2 activation, can be used to
formulate a safe pharmaceutical combination to potently decrease
AR-FL and AR-V7 levels in CRPC cells. The efficacy of this
combination is further supported by our observations that similar
to SFN, combined treatment with physiologic doses (nanomolar) of G
and BM suppressed proliferation, migration and clonogenic ability,
and most importantly, resensitized these AR-V7 expressing cells to
the anticancer effects of ENZ.
Although we did not carry out in depth mechanistic
studies using the G+BM combination, numerous investigators have
shown the potent Hsp90 inhibitory effect of G (72,75)
and the potent Nrf2 inductive effect of BM (76,77).
In addition, our previous published finding documented that
overexpression of Nrf2 can suppress the expression and function of
AR-FL in both LNCaP and C4-2B cells (42). Since SFN functions via the above two
mechanisms (31,40,41,43–45),
it is likely that the G+BM combination can simultaneously target
them in order to decrease both AR-FL and AR-V7 levels.
Although we have not investigated the in vivo
efficacies of either SFN or our pharmaceutical drug combination,
i.e. G+BM, our findings clearly implicate in vivo potential
of these agents as evident from numerous past studies (55–61).
Our in vitro findings on the potent effects of SFN alone
(micromolar doses) or the G+BM combination (nanomolar doses) in
suppressing proliferation, migration and clonogenic ability of
22Rv1 cells, parameters that dictate aggressive tumor growth in
vivo (49,78,79),
clearly suggest that these agents may be effective against in
vivo tumors. In summary, our findings suggest that either SFN,
or the combination of G+BM, may provide an effective adjunct to
current treatment in CRPC patients.
Acknowledgements
This study was supported by funds from the Louisiana
Cancer Research Consortium (LCRC) to D.M. and from the Laboratory
Training funds to S.C.S.
References
|
1
|
Yap TA, Zivi A, Omlin A and de Bono JS:
The changing therapeutic landscape of castration-resistant prostate
cancer. Nat Rev Clin Oncol. 8:597–610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hodgson MC, Bowden WA and Agoulnik IU:
Androgen receptor footprint on the way to prostate cancer
progression. World J Urol. 30:279–285. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang KH, Ercole CE and Sharifi N:
Androgen metabolism in prostate cancer: From molecular mechanisms
to clinical consequences. Br J Cancer. 111:1249–1254. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scher HI, Buchanan G, Gerald W, Butler LM
and Tilley WD: Targeting the androgen receptor: Improving outcomes
for castration resistant prostate cancer. Endocr Relat Cancer.
11:459–476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Godbole AM and Njar VC: New insights into
the androgen-targeted therapies and epigenetic therapies in
prostate cancer. Prostate Cancer. 2011:9187072011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim W and Ryan CJ: Androgen receptor
directed therapies in castration-resistant metastatic prostate
cancer. Curr Treat Options Oncol. 13:189–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harris WP, Mostaghel EA, Nelson PS and
Montgomery B: Androgen deprivation therapy: Progress in
understanding mechanisms of resistance and optimizing androgen
depletion. Nat Clin Pract Urol. 6:76–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lamont KR and Tindall DJ: Minireview:
Alternative activation pathways for the androgen receptor in
prostate cancer. Mol Endocrinol. 25:897–907. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brooke GN and Bevan CL: The role of
androgen receptor mutations in prostate cancer progression. Curr
Genomics. 10:18–25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Armstrong CM and Gao AC: Drug resistance
in castration resistant prostate cancer: Resistance mechanisms and
emerging treatment strategies. Am J Clin Exp Urol. 3:64–76.
2015.PubMed/NCBI
|
|
11
|
Antonarakis ES, Armstrong AJ, Dehm SM and
Luo J: Androgen receptor variant-driven prostate cancer: Clinical
implications and therapeutic targeting. Prostate Cancer Prostatic
Dis. 19:231–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lokhandwala PM, Riel SL, Haley L, Lu C,
Chen Y, Silberstein J, Zhu Y, Zheng G, Lin MT, Gocke CD, et al:
Analytical validation of androgen receptor splice variant 7
detection in a clinical laboratory improvement amendments (CLIA)
laboratory setting. J Mol Diagn. 19:115–125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Del Re M, Biasco E, Crucitta S, Derosa L,
Rofi E, Orlandini C, Miccoli M, Galli L, Falcone A, Jenster GW, et
al: The detection of androgen receptor splice variant 7 in
plasma-derived exosomal RNA strongly predicts resistance to
hormonal therapy in metastatic prostate cancer patients. Eur Urol.
71:680–687. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dehm SM, Schmidt LJ, Heemers HV, Vessella
RL and Tindall DJ: Splicing of a novel androgen receptor exon
generates a constitutively active androgen receptor that mediates
prostate cancer therapy resistance. Cancer Res. 68:5469–5477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu R, Dunn TA, Wei S, Isharwal S, Veltri
RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al:
Ligand-independent androgen receptor variants derived from splicing
of cryptic exons signify hormone-refractory prostate cancer. Cancer
Res. 69:16–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo Z, Yang X, Sun F, Jiang R, Linn DE,
Chen H, Chen H, Kong X, Melamed J, Tepper CG, et al: A novel
androgen receptor splice variant is up-regulated during prostate
cancer progression and promotes androgen depletion-resistant
growth. Cancer Res. 69:2305–2313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun S, Sprenger CC, Vessella RL, Haugk K,
Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et
al: Castration resistance in human prostate cancer is conferred by
a frequently occurring androgen receptor splice variant. J Clin
Invest. 120:2715–2730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Morrissey C, Sun S, Ketchandji M,
Nelson PS, True LD, Vakar-Lopez F, Vessella RL and Plymate SR:
Androgen receptor variants occur frequently in castration resistant
prostate cancer metastases. PLoS One. 6:e279702011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dehm SM and Tindall DJ: Alternatively
spliced androgen receptor variants. Endocr Relat Cancer.
18:R183–R196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Antonarakis ES, Lu C, Wang H, Luber B,
Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et
al: AR-V7 and resistance to enzalutamide and abiraterone in
prostate cancer. N Engl J Med. 371:1028–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sarwar M, Semenas J, Miftakhova R,
Simoulis A, Robinson B, Gjörloff Wingren A, Mongan NP, Heery DM,
Johnsson H, Abrahamsson PA, et al: Targeted suppression of AR-V7
using PIP5K1α inhibitor overcomes enzalutamide resistance in
prostate cancer cells. Oncotarget. 7:63065–63081. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Watson PA, Chen YF, Balbas MD, Wongvipat
J, Socci ND, Viale A, Kim K and Sawyers CL: Constitutively active
androgen receptor splice variants expressed in castration-resistant
prostate cancer require full-length androgen receptor. Proc Natl
Acad Sci USA. 107:pp. 16759–16765. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cao B, Qi Y, Zhang G, Xu D, Zhan Y,
Alvarez X, Guo Z, Fu X, Plymate SR, Sartor O, et al: Androgen
receptor splice variants activating the full-length receptor in
mediating resistance to androgen-directed therapy. Oncotarget.
5:1646–1656. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W,
Gambhir SS, Lee P, Sartor O, Flemington EK, et al: Androgen
receptor splice variants dimerize to transactivate target genes.
Cancer Res. 75:3663–3671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y and Tang L: Discovery and
development of sulforaphane as a cancer chemopreventive
phytochemical. Acta Pharmacol Sin. 28:1343–1354. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clarke JD, Dashwood RH and Ho E:
Multi-targeted prevention of cancer by sulforaphane. Cancer Lett.
269:291–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheung KL and Kong AN: Molecular targets
of dietary phenethyl isothiocyanate and sulforaphane for cancer
chemoprevention. AAPS J. 12:87–97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fawzy Elbarbry and Nehad Elrody: Potential
health benefits of sulforaphane: A review of the experimental,
clinical and epidemiological evidences and underlying mechanisms. J
Med Plants Res. 5:473–484. 2011.
|
|
29
|
Shapiro TAI, Fahey JW, Dinkova-Kostova AT,
Holtzclaw WD, Stephenson KK, Wade KL, Ye L and Talalay P: Safety,
tolerance, and metabolism of broccoli sprout glucosinolates and
isothiocyanates: a clinical phase I study. Nutr Cancer. 55:53–62.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Petri N, Tannergren C, Holst B, Mellon FA,
Bao Y, Plumb GW, Bacon J, O'Leary KA, Kroon PA, Knutson L, et al:
Absorption/metabolism of sulforaphane and quercetin, and regulation
of phase II enzymes, in human jejunum in vivo. Drug Metab Dispos.
31:805–813. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Keum YS, Khor TO, Lin W, Shen G, Kwon KH,
Barve A, Li W and Kong AN: Pharmacokinetics and pharmacodynamics of
broccoli sprouts on the suppression of prostate cancer in
transgenic adenocarcinoma of mouse prostate (TRAMP) mice:
Implication of induction of Nrf2, HO-1 and apoptosis and the
suppression of Akt-dependent kinase pathway. Pharm Res.
26:2324–2331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Traka MH, Melchini A and Mithen RF:
Sulforaphane and prostate cancer interception. Drug Discov Today.
19:1488–1492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SH and Singh SV: D,L-Sulforaphane
causes transcriptional repression of androgen receptor in human
prostate cancer cells. Mol Cancer Ther. 8:1946–1954. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wiczk A, Hofman D, Konopa G and
Herman-Antosiewicz A: Sulforaphane, a cruciferous vegetable-derived
isothiocyanate, inhibits protein synthesis in human prostate cancer
cells. Biochim Biophys Acta. 1823:1295–1305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Burnett JP, Lim G, Li Y, Shah RB, Lim R,
Paholak HJ, McDermott SP, Sun L, Tsume Y, Bai S, et al:
Sulforaphane enhances the anticancer activity of taxanes against
triple negative breast cancer by killing cancer stem cells. Cancer
Lett. 394:52–64. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li QQ, Xie YK, Wu Y, Li LL, Liu Y, Miao
XB, Liu QZ, Yao KT and Xiao GH: Sulforaphane inhibits cancer
stem-like cell properties and cisplatin resistance through
miR-214-mediated downregulation of c-MYC in non-small cell lung
cancer. Oncotarget. 8:12067–12080. 2017.PubMed/NCBI
|
|
37
|
Singh SV, Srivastava SK, Choi S, Lew KL,
Antosiewicz J, Xiao D, Zeng Y, Watkins SC, Johnson CS, Trump DL, et
al: Sulforaphane-induced cell death in human prostate cancer cells
is initiated by reactive oxygen species. J Biol Chem.
280:19911–19924. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xiao D, Powolny AA, Antosiewicz J, Hahm
ER, Bommareddy A, Zeng Y, Desai D, Amin S, Herman-Antosiewicz A and
Singh SV: Cellular responses to cancer chemopreventive agent
D,L-sulforaphane in human prostate cancer cells are initiated by
mitochondrial reactive oxygen species. Pharm Res. 26:1729–1738.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pei Y, Wu B, Cao Q, Wu L and Yang G:
Hydrogen sulfide mediates the anti-survival effect of sulforaphane
on human prostate cancer cells. Toxicol Appl Pharmacol.
257:420–428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang C, Su Z-Y, Khor TO, Shu L and Kong
AN: Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP
C1 cells through epigenetic regulation. Biochem Pharmacol.
85:1398–1404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kensler TW, Egner PA, Agyeman AS,
Visvanathan K, Groopman JD, Chen JG, Chen TY, Fahey JW and Talalay
P: Keap1-nrf2 signaling: A target for cancer prevention by
sulforaphane. Top Curr Chem. 329:163–177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schultz MA, Hagan SS, Datta A, Zhang Y,
Freeman ML, Sikka SC, Abdel-Mageed AB and Mondal D: Nrf1 and Nrf2
transcription factors regulate androgen receptor transactivation in
prostate cancer cells. PLoS One. 9:e872042014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gibbs A, Schwartzman J, Deng V and Alumkal
J: Sulforaphane destabilizes the androgen receptor in prostate
cancer cells by inactivating histone deacetylase 6. Proc Natl Acad
Sci USA. 106:pp. 16663–16668. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Myzak MC, Hardin K, Wang R, Dashwood RH
and Ho E: Sulforaphane inhibits histone deacetylase activity in
BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis.
27:811–819. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tortorella SM, Royce SG, Licciardi PV and
Karagiannis TC: Dietary sulforaphane in cancer chemoprevention: The
role of epigenetic regulation and HDAC inhibition. Antioxid Redox
Signal. 22:1382–1424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Khurana N, Talwar S, Chandra PK, Sharma P,
Abdel-Mageed AB, Mondal D and Sikka SC: Sulforaphane increases the
efficacy of anti-androgens by rapidly decreasing androgen receptor
levels in prostate cancer cells. Int J Oncol. 49:1609–1619. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu HC, Hsieh JT, Gleave ME, Brown NM,
Pathak S and Chung LW: Derivation of androgen-independent human
LNCaP prostatic cancer cell sublines: Role of bone stromal cells.
Int J Cancer. 57:406–412. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li Y, Karagöz GE, Seo YH, Zhang T, Jiang
Y, Yu Y, Duarte AM, Schwartz SJ, Boelens R, Carroll K, et al:
Sulforaphane inhibits pancreatic cancer through disrupting
Hsp90-p50(Cdc37) complex and direct interactions with amino acids
residues of Hsp90. J Nutr Biochem. 23:1617–1626. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Uygur B and Wu W-S: SLUG promotes prostate
cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol
Cancer. 10:1392011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Scher HI, Fizazi K, Saad F, Taplin ME,
Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et
al: AFFIRM Investigators: Increased survival with enzalutamide in
prostate cancer after chemotherapy. N Engl J Med. 367:1187–1197.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ryan CJ and Cheng ML: Abiraterone acetate
for the treatment of prostate cancer. Expert Opin Pharmacother.
14:91–96. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz
CT, Evans CP and Gao AC: Niclosamide inhibits androgen receptor
variants expression and overcomes enzalutamide resistance in
castration-resistant prostate cancer. Clin Cancer Res. 20:3198–210.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liu C, Armstrong C, Zhu Y, Lou W and Gao
AC: Niclosamide enhances abiraterone treatment via inhibition of
androgen receptor variants in castration resistant prostate cancer.
Oncotarget. 7:32210–32220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Goldman JW, Raju RN, Gordon GA, El-Hariry
I, Teofilivici F, Vukovic VM, Bradley R, Karol MD, Chen Y, Guo W,
et al: A first in human, safety, pharmacokinetics, and clinical
activity phase I study of once weekly administration of the Hsp90
inhibitor ganetespib (STA-9090) in patients with solid
malignancies. BMC Cancer. 13:1522013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Thakur MK, Heilbrun LK, Sheng S, Stein M,
Liu G, Antonarakis ES, Vaishampayan U, Dzinic SH, Li X, Freeman S,
et al: A phase II trial of ganetespib, a heat shock protein 90
Hsp90) inhibitor, in patients with docetaxel-pretreated metastatic
castrate-resistant prostate cancer (CRPC)-a prostate cancer
clinical trials consortium (PCCTC) study. Invest New Drugs.
34:112–118. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Goyal L, Wadlow RC, Blaszkowsky LS, Wolpin
BM, Abrams TA, McCleary NJ, Sheehan S, Sundaram E, Karol MD, Chen
J, et al: A phase I and pharmacokinetic study of ganetespib
(STA-9090) in advanced hepatocellular carcinoma. Invest New Drugs.
33:128–137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hong DS, Kurzrock R, Supko JG, He X, Naing
A, Wheler J, Lawrence D, Eder JP, Meyer CJ, Ferguson DA, et al: A
phase I first-in-human trial of bardoxolone methyl in patients with
advanced solid tumors and lymphomas. Clin Cancer Res. 18:3396–3406.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gao X, Deeb D, Liu Y, Arbab AS, Divine GW,
Dulchavsky SA and Gautam SC: Prevention of prostate cancer with
oleanane synthetic triterpenoid CDDO-Me in the TRAMP mouse model of
prostate cancer. Cancers (Basel). 3:3353–3369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Alumkal JJ, Slottke R, Schwartzman J,
Cherala G, Munar M, Graff JN, Beer TM, Ryan CW, Koop DR, Gibbs A,
et al: A phase II study of sulforaphane-rich broccoli sprout
extracts in men with recurrent prostate cancer. Invest New Drugs.
33:480–489. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shapiro TA, Fahey JW, Dinkova-Kostova AT,
Holtzclaw WD, Stephenson KK, Wade KL, Ye L and Talalay P: Safety,
tolerance, and metabolism of broccoli sprout glucosinolates and
isothiocyanates: a clinical phase I study. Nutr Cancer. 55:53–62.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cunningham D and You Z: In vitro and in
vivo model systems used in prostate cancer research. J Biol
Methods. 2:e172015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li Y, Hwang TH, Oseth LA, Hauge A,
Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA and
Dehm SM: AR intragenic deletions linked to androgen receptor splice
variant expression and activity in models of prostate cancer
progression. Oncogene. 31:4759–4767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lilienbaum A: Relationship between the
proteasomal system and autophagy. Int J Biochem Mol Biol. 4:1–26.
2013.PubMed/NCBI
|
|
65
|
Jaworski T: Degradation and beyond:
control of androgen receptor activity by the proteasome system.
Cell Mol Biol Lett. 11:109–131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gan N, Wu YC, Brunet M, Garrido C, Chung
FL, Dai C and Mi L: Sulforaphane activates heat shock response and
enhances proteasome activity through up-regulation of Hsp27. J Biol
Chem. 285:35528–35536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Miyahara K, Kazama H, Kokuba H, Komatsu S,
Hirota A, Takemura J, Hirasawa K, Moriya S, Abe A, Hiramoto M, et
al: Targeting bortezomib-induced aggresome formation using
vinorelbine enhances the cytotoxic effect along with ER stress
loading in breast cancer cell lines. Int J Oncol. 49:1848–1858.
2016.PubMed/NCBI
|
|
68
|
Guthrie CR and Kraemer BC: Proteasome
inhibition drives HDAC6-dependent recruitment of tau to aggresomes.
J Mol Neurosci. 45:32–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zaarur N, Meriin AB, Bejarano E, Xu X,
Gabai VL, Cuervo AM and Sherman MY: Proteasome failure promotes
positioning of lysosomes around the aggresome via local block of
microtubule-dependent transport. Mol Cell Biol. 34:1336–1348. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Moriya S, Komatsu S, Yamasaki K, Kawai Y,
Kokuba H, Hirota A, Che XF, Inazu M, Gotoh A, Hiramoto M, et al:
Targeting the integrated networks of aggresome formation,
proteasome, and autophagy potentiates ER stress-mediated cell death
in multiple myeloma cells. Int J Oncol. 46:474–486. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Azad AA, Zoubeidi A, Gleave ME and Chi KN:
Targeting heat shock proteins in metastatic castration-resistant
prostate cancer. Nat Rev Urol. 12:26–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
He S, Zhang C, Shafi AA, Sequeira M,
Acquaviva J, Friedland JC, Sang J, Smith DL, Weigel NL, Wada Y, et
al: Potent activity of the Hsp90 inhibitor ganetespib in prostate
cancer cells irrespective of androgen receptor status or variant
receptor expression. Int J Oncol. 42:35–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Shafi AA, Cox MB and Weigel NL: Androgen
receptor splice variants are resistant to inhibitors of Hsp90 and
FKBP52, which alter androgen receptor activity and expression.
Steroids. 78:548–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shiota M, Yokomizo A and Naito S:
Oxidative stress and androgen receptor signaling in the development
and progression of castration-resistant prostate cancer. Free Radic
Biol Med. 51:1320–1328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gillis JL, Selth LA, Centenera MM, Townley
SL, Sun S, Plymate SR, Tilley WD and Butler LM:
Constitutively-active androgen receptor variants function
independently of the HSP90 chaperone but do not confer resistance
to HSP90 inhibitors. Oncotarget. 4:691–704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang YY, Yang YX, Zhe H, He ZX and Zhou
SF: Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update
on its pharmacokinetic and pharmacodynamic properties. Drug Des
Devel Ther. 8:2075–2088. 2014.PubMed/NCBI
|
|
77
|
Probst BL, McCauley L, Trevino I, Wigley
WC and Ferguson DA: Cancer cell growth is differentially affected
by constitutive activation of NRF2 by KEAP1 deletion and
pharmacological activation of NRF2 by the synthetic triterpenoid,
RTA 405. PLoS One. 10:e01352572015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Huo C, Kao Y-H and Chuu C-P: Androgen
receptor inhibits epithelial-mesenchymal transition, migration, and
invasion of PC-3 prostate cancer cells. Cancer Lett. 369:103–111.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Aapro MS, Eliason JF, Krauer F and Alberto
P: Colony formation in vitro as a prognostic indicator for primary
breast cancer. J Clin Oncol. 5:890–896. 1987. View Article : Google Scholar : PubMed/NCBI
|