Introduction
Prostate cancer (PCa) is the most commonly diagnosed
malignancy and the second leading cause of cancer-related deaths in
the western male population, while metastasis contributes to
majority of deaths (1,2). The five-year survival rate of patients
who present with localized disease is ~90%, which was significantly
higher than those with metastatic disease. Only ~33% of men
presented with metastatic tumors live beyond 5 years. Although
prostate cancer treatment has achieved numerous breakthroughs in
the last decade, effective therapies to prevent prostate cancer
metastasis to other parts of body are still lacking (3). Therefore, novel approaches are
urgently needed for the prevention and treatment of the metastasis
of prostate carcinoma.
Calcium (Ca2+), an intracellular
messenger, is indispensable for various cellular functions, such as
proliferation, apoptosis and metastasis (4,5). In
non-excitable cells, store-operated calcium entry (SOCE) is the
major influx of Ca2+ and is regulated by a
store-operated calcium channel (SOC), which consists of two
important components, stromal-interacting molecule 1 (STIM1) and
calcium release-activated calcium channel protein 1 (ORAI1). STIM1
is located in the endoplasmic reticulum (ER) and acts as
Ca2+ sensor. Once the Ca2+ store in ER is
depleted, STIM1 is activated and forms an oligomer with ORAI1, a
transmembrane protein, to form pores for Ca2+ influx
(6).
STIM1 participates in a variety of cellular
functions, including muscle contraction, the release of
neurotransmitters and hormones, gene transcription, cell
proliferation and metastasis (7).
Recently, STIM1 has attracted more and more attention due to its
oncogenic potential. STIM1 inhibition suppresses cell
proliferation, migration and invasion in a variety of cancer models
both in vitro and in vivo (8–11).
However, to date, its function on metastasis of prostate cancer is
unclear.
The phosphatidylinositol 3-kinase/protein kinase-B
(PI3K/Akt) signaling pathway plays a pivotal role in cell growth,
differentiation, proliferation and metastasis. PI3K phosphorylates
and activates Akt, affecting its downstream signaling molecules. It
has been demonstrated that activation of Akt promotes migration of
tumor cells (12–14). Numerous studies have revealed that
the PI3K/Akt signaling pathway is extensively activated in
migration and invasion of various types of cancers, including
liver, breast and pancreatic cancer (15–17).
PI3K/Akt inhibition by specific inhibitors suppresses cell
migration and invasion (18–21).
Whether the PI3K/Akt signaling pathway is involved in the
regulation of invasion and migration by STIM1 is unclear and needs
to be further explored.
In the present study, we determined whether STIM1
knockdown inhibited cell migration and invasion of prostate cancer
and further explored the potential underlying mechanism of STIM1 in
the process with a focus on the regulation of the PI3K/Akt
signaling pathway.
Materials and methods
Reagents
The PI3K inhibitor LY294002 was obtained from
Selleckchem (Houston, TX, USA). Matrigel was purchased from BD
Biosciences (San Jose, CA, USA), Transwell Minicells were obtained
from Millipore (Darmstadt, Germany). A BCA protein qualification
kit was purchased from Pierce (Rockford, IL, USA). The anti-STIM1
antibody for immunohistochemistry (IHC) staining was purchased from
Abgent (San Diego, CA, USA). Anti-STIM1 (4916), anti-p-Akt (Thr308)
(13038), anti-t-Akt (4691), anti-GAPDH (5174), anti-E-cadherin
(3195), anti-N-cadherin (13116), anti-vimentin (5741), anti-Snail
(3879) and secondary antibodies anti-rabbit (14708) and anti-mouse
(14709) antibodies were obtained from Cell Signaling Technology
(Boston, MA, USA).
Prostate tissue acquisition and IHC
staining
Prostate cancer specimens were obtained from
prostate cancer patients undergoing prostatectomy or prostate
biopsy. Benign prostatic hyperplasia (BPH) tissues were obtained
from BPH patients undergoing surgery at the Second Affiliated
Hospital of Soochow University and were used as a normal control.
The present study was approved by the Research Ethics Committee of
the Second Affiliated Hospital of Soochow University and was in
compliance with the Helsinki Declaration. Tissues were examined by
pathologists to confirm the diagnosis before IHC analyses. All
specimens were fixed in 10% formalin, embedded in paraffin, and cut
into 4-µm thick slides. The slides were dewaxed, and the endogenous
peroxidase activity was blocked by treatment with 3% hydrogen
peroxide solution in methanol for 20 min. Non-specific binding was
prevented by blocking with normal goat serum (1:100) for 30 min.
The staining procedure was carried out using the
avidin-biotin-peroxidase complex method. The expression of STIM1
was evaluated by staining with a mouse anti-STIM1 antibody. After
incubation with a primary antibody for 60 min, the slides were
washed using phosphate-buffered saline (PBS) 3 times, and then
incubated with a biotinylated goat anti-mouse IgG (H+L) at 37°C for
30 min, followed by incubation with a streptavidin-biotinylated HRP
complex (Sigma, St. Louis, MO, USA) for 30 min. Reactive products
were visualized with 3,3′-diaminobenzidene (DAB) as the chromogen,
and slides were counterstained with hematoxylin. Sections
previously known to express STIM1 were included in each run,
receiving either the primary antibody as the positive control or a
mouse IgG as the negative control. Stained slides were observed by
microscope.
All slides were evaluated twice at
different time-points by two independent pathologists
The expression level of STIM1 was assigned a score
based on the percentage of positive tumor cells over total tumor
cells and their staining intensities. The proportion of positive
tumor cells ≤10%, 11–25%, 26–50% and >50% were scored as 1, 2, 3
and 4. In addition, when the staining intensities were a
non-significant brown, a slight brown, a moderate brown and a deep
brown, they were scored as 1, 2, 3 and 4. Then, the two scores were
added. A score of 2–3 was graded as weak, and a score of 4–8 was
graded as strong.
Cell culture
Normal prostate cell line RWPE-1 and prostate cancer
cell lines LNCap, PC-3 and DU-145 were purchased from the American
Type Culture Collection (ATCC; Manassas, VA, USA). These were
cultured according to the instructions of ATCC. C4-2 was obtained
from UroCor Inc., (Oklahoma City, OK, USA) and grown in RPMI-1640
medium containing 10% fetal bovine serum (FBS) (Atlanta
Biologicals, Flowery Branch, GA, USA) and 1%
penicillin-streptomycin solution (Invitrogen, Carlsbad, CA, USA).
The cells were observed and images were obtained using an inverted
microscope (Olympus, Tokyo, Japan). When the cell density reached
90–100%, the cells were sub-cultured and seeded to plates according
to the experimental design. The culture medium was replaced by
fresh one every 2–3 days or according to the experimental
design.
Total RNA isolation, cDNA reversion
and polymerase chain reaction (PCR)
Total RNA was isolated using Trizol reagent
(Invitrogen) according to the manufacturer's instructions. The
concentration of total RNA was detected by UV spectrophotometry.
RT-PCR was performed by the two-step method. Synthesis of cDNA was
performed using a cDNA Synthesis kit (Thermo Fisher Scientific,
Franklin Lakes, NJ, USA). The PCR reaction conditions were: 95°C
for 5 min, 94°C for 30 sec, 56°C for 30 sec, 72°C for 30 sec for 40
cycles; the total volume was 20 µl. GAPDH was used as an internal
standard. The sequences of the primers used were: GAPDH forward,
TGTGG GCATCAATGGATTTGG and reverse, ACACCATGTATTC CGGGTCAAT; and
STIM1 forward, AGTCACAGTGAGAA GGCG AC and reverse,
ACACCATGTATTCCGGGTCAAT. All experiments were performed in
triplicate.
Western blot analysis
Total protein was extracted by radio
immunoprecipitation assay (RIPA) buffer (0.15 mM NaCl, 0.05 mM
Tris-HCl, pH 7.5, 1% Triton, 0.1% SDS, 0.1% sodium deoxycholate and
1% NP40). Sample extracts (30 µg) were loaded to 12%
SDS-polyacrylamide gels (PAGE) using a minigel apparatus and
transferred to polyvinylidene difluoride (PVDF) membranes (both
from Bio-Rad, Hercules, CA, USA). The membranes were blocked for 1
h with 5% skim milk, then incubated overnight with primary
antibody. Then the membranes were washed using Tris-buffered saline
with Tween-20 (TBST) 3 times and incubated with a secondary
antibody for 1 h at room temperature. Blots were visualized by
enhanced chemiluminescence (ECL) system.
Construction and infection of
lentivirus-mediated short hairpin vector
For short hairpin RNA (shRNA)-mediated knockdown of
STIM1, cells were transfected with lentiviral particles produced
using the STIM1-pGCSIL-GFP plasmid purchased from GeneChem
Corporation (Shanghai, China). The targeting senses of the shRNAs
were: shSTIM1-1, 5′-GCTCTCCACATTTGGATTCTT-3′; and shSTIM1-2,
5′-GGAGGATAATGGCTCTATT-3′. The negative control was a
double-stranded shRNA without sequence homology to any known human
genes. For gene silencing, purified lentiviruses (shSTIM1-1 and
shSTIM1-2) were added to cells at a multiplicity of infection of 20
for 8 h, and was washed twice with medium. Infection with a
multiplicity of infection of 20 resulted in a >90% infection of
cancer cells after 72 h, as monitored by GFP expression. Therefore,
we used a multiplicity of infection of 20 for the lentivirus in all
of the experiments, as it yielded optimal knockdown of the gene in
the required time. Control cells were infected with a negative
control shRNA, as a vector control according to the same
protocol.
Wound healing assay
Cells were seeded to 6-well plates at a
concentration of 5×105 cells/well and incubated
overnight. The cells were then infected with lentiviruses and
incubated for an additional 48 h until the cell monolayers formed.
The cells were starved by serum-free medium overnight. The wounds
were scratched by sterile 200 µl pipet tips. After scratching, the
cells were washed with PBS twice and cultured in serum-free medium.
Images of the wounds were captured at time-points of 0, 12 and 24 h
by an inverted microscope (magnification, ×40).
Transwell migration and invasion
assays
A Transwell migration assay was performed using
Transwell chambers consisting of 8 µm membrane filter inserts
(Corning, Corning NY, USA). Matrigel was diluted in serum-free
medium (1:5) and pre-paved to insert membranes 4 h before cell
seeding. The cells were infected with a lentivirus for 72 h, and
then re-suspended in serum-free medium. The cells in 500 µl
serum-free medium (3×104 cells for migration and
10×104 cells for invasion) were added to the upper
chamber, and the lower chamber was filled with 1 ml normal culture
medium containing 10% FBS. After incubation for 24 h, the cells
were fixed with 4% paraformaldehyde and stained with crystal
violet. Migrated and invaded cells were observed using a microscope
(magnification, ×100) and images were captured.
Statistical analysis
To carry out statistical analysis, we used the
software SPSS 23 (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism
6 (GraphPad Software, San Diego, CA, USA). The correlation between
STIM1 expression and clinicopathological parameters was analyzed by
Chi-square (χ2) test. The rest of the experimental data
were represented from at least 3 independent experiments.
Statistical significance was evaluated using the Students t-test. A
P-value <0.05 was considered to indicate a statistically
significant result.
Results
STIM1 is overexpressed in prostate
cancer tissues
To evaluate the expression of STIM1 in human
prostate cancer tissues and benign prostate tissues (BPH), we
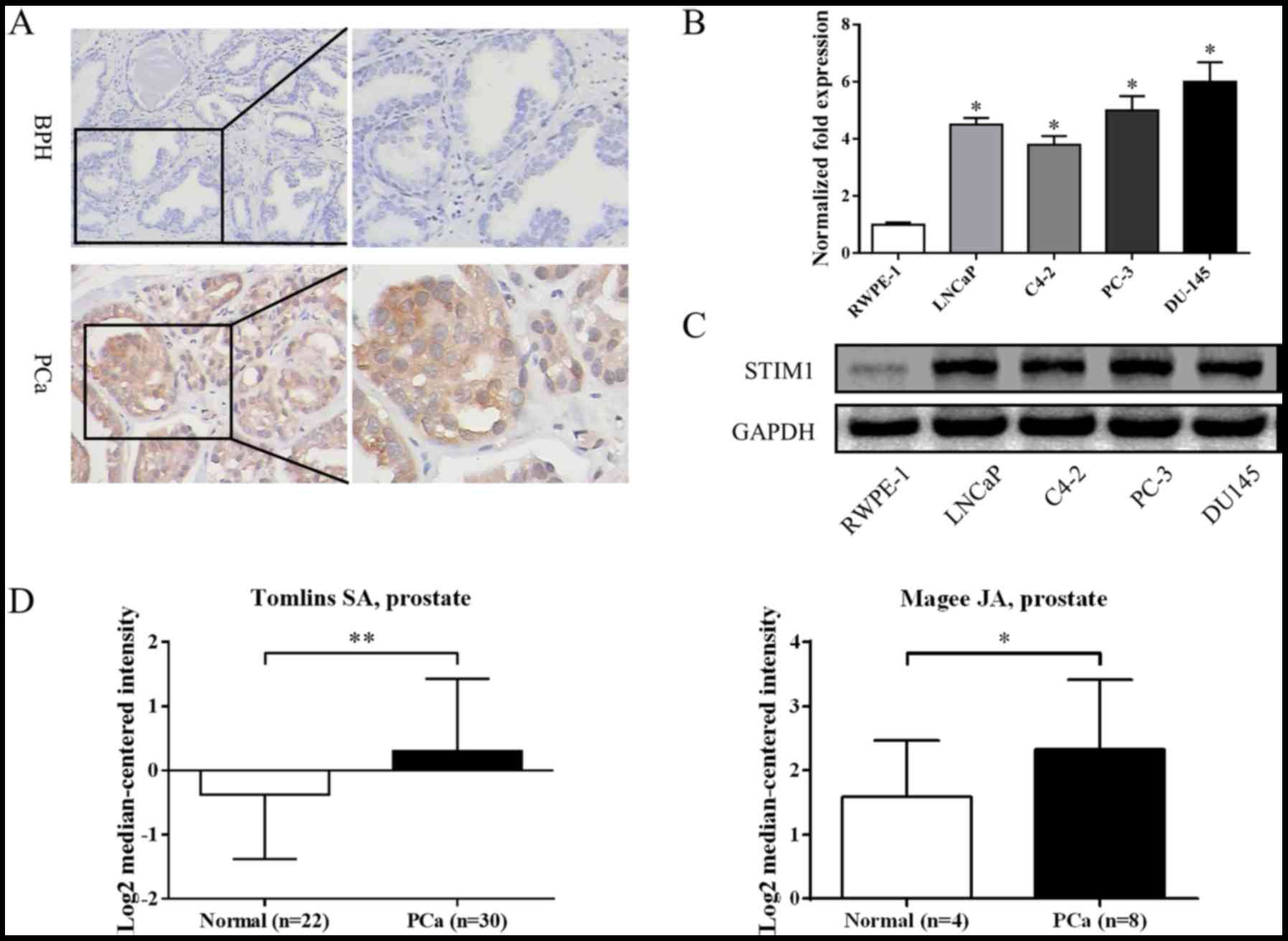
examined the specimens by IHC. As shown in Fig. 1A, the expression of STIM1 in
prostate cancer was higher than benign prostate hyperplasia (BPH)
tissues. Overall, STIM1 was expressed in 3 out of 10 (30.0%) BPH
specimens; whereas it was expressed in 37 out of 47 prostate cancer
specimens (78.7%). The difference was statistically significant
(P=0.007). We also searched in databases such as Oncomine
(www.oncomine.org) and found studies conducted by
Magee et al (22) and Tomlin
et al (23) which revealed
similar results (Fig. 1D).
To study whether STIM1 was correlated to prostate
cancer progression, the association between STIM1 expression and
clinicopathological characteristics was analyzed. As shown in
Table I, the expression of STIM1
was found to be significantly associated with stage grouping. The
STIM1 expression level of stage I+II was significantly lower than
that of stage III+IV (P=0.016). However, there was no significant
correlation between STIM1 expression and age or prostate cancer
Gleason score (P>0.05).
 | Table I.Association between STIM1 expression
and clinicopathological features in patients recruited. |
Table I.
Association between STIM1 expression
and clinicopathological features in patients recruited.
|
|
| STIM1
expression |
|
|---|
|
|
|
|
|
|---|
|
Characteristics | No. | Weak | Strong | P-values |
|---|
| Diagnosis |
|
|
| 0.007 |
|
BPH | 10 | 7 | 3 |
|
|
PCa | 47 | 10 | 37 |
|
| Age (PCa) |
|
|
| 0.487 |
|
≤65 | 19 | 5 | 14 |
|
|
>65 | 28 | 5 | 23 |
|
| PCa Gleason
score |
|
|
| 0.709 |
| ≤7 | 14 | 2 | 12 |
|
|
>7 | 33 | 8 | 25 |
|
| PCa stage
grouping |
|
|
| 0.016 |
|
I+II | 12 | 6 | 6 |
|
|
III+IV | 35 | 4 | 31 |
|
We also investigated the expression of STIM1 in cell
lines derived from normal prostate or prostate cancers. As shown in
Fig. 1B and C, the expression of
STIM1 in prostate cancer cell lines was much higher than that in
normal prostate epithelial cell line (RWPE-1) at both the mRNA and
protein levels. These results indicate that STIM1 may be an
oncogene in prostate tumorigenesis.
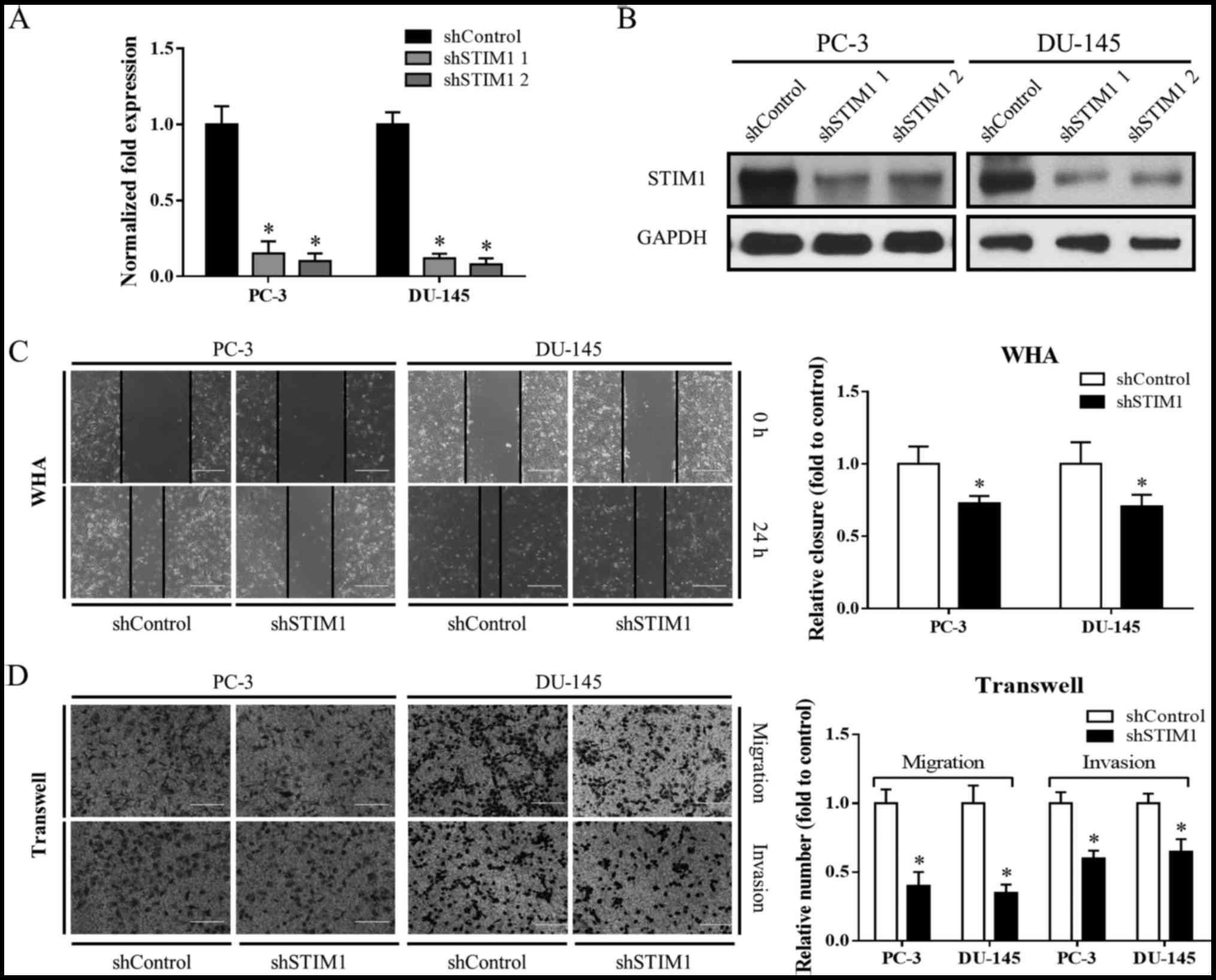
STIM1 knockdown inhibits migration and
invasion in prostate cancer cells
To study the role of STIM1 in prostate cancer, we
first designed two independent STIM1 shRNAs to knock down STIM1 in
PC-3 and DU-145 cell lines. A double-stranded shRNA without
sequence homology to any known human genes was used as control
(Fig. 2A and B).
Wound healing assay and Transwell migration were
then utilized to study the impact of STIM1 knockdown on cell
migration. As shown in Fig. 2C and
D, the STIM1 knockdown groups exhibited less wound closure and
migrated cells as compared to the shControl, indicating that cell
migration was inhibited when STIM1 was knocked down. Cell invasion
was also suppressed by STIM1 knockdown as determined by Transwell
invasion assay (Fig. 2D).
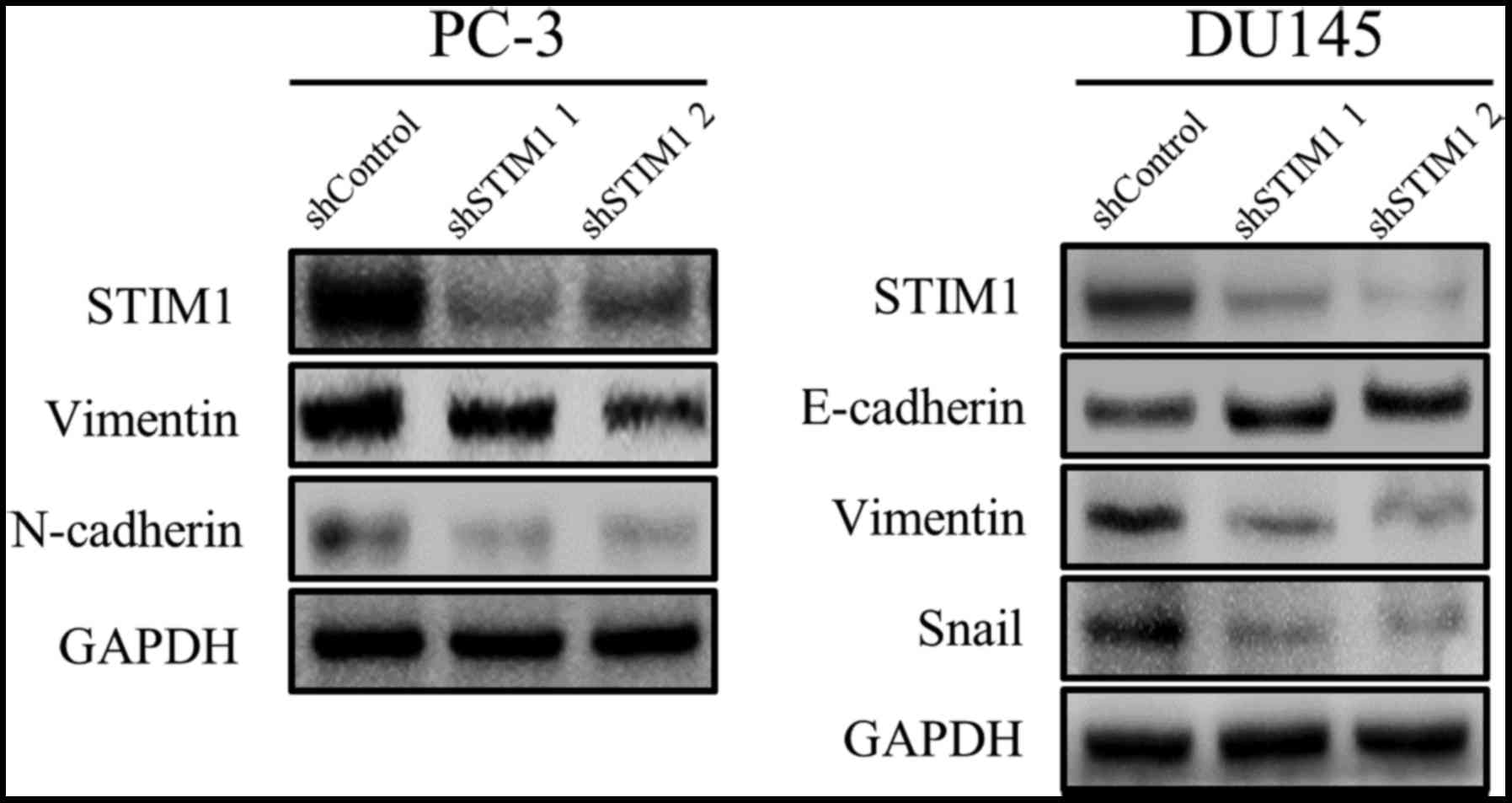
Motility inhibition by STIM1 knockdown
is associated with EMT suppression
To understand the potential mechanism involved in
the suppressed migration and invasion by STIM1 knockdown, we
examined EMT-related markers since STIM1 has been reported to
promote cancer cell metastasis through the induction of EMT. As
shown in Fig. 3, vimentin and
N-cadherin were downregulated by STIM1 knockdown in PC-3 cells. In
DU-145 cells, STIM1 knockdown decreased the expression of vimentin
and Snail but increased the expression of E-cadherin. These results
demonstrated that the migration and invasion inhibited by STIM1
knockdown were associated with EMT suppression.
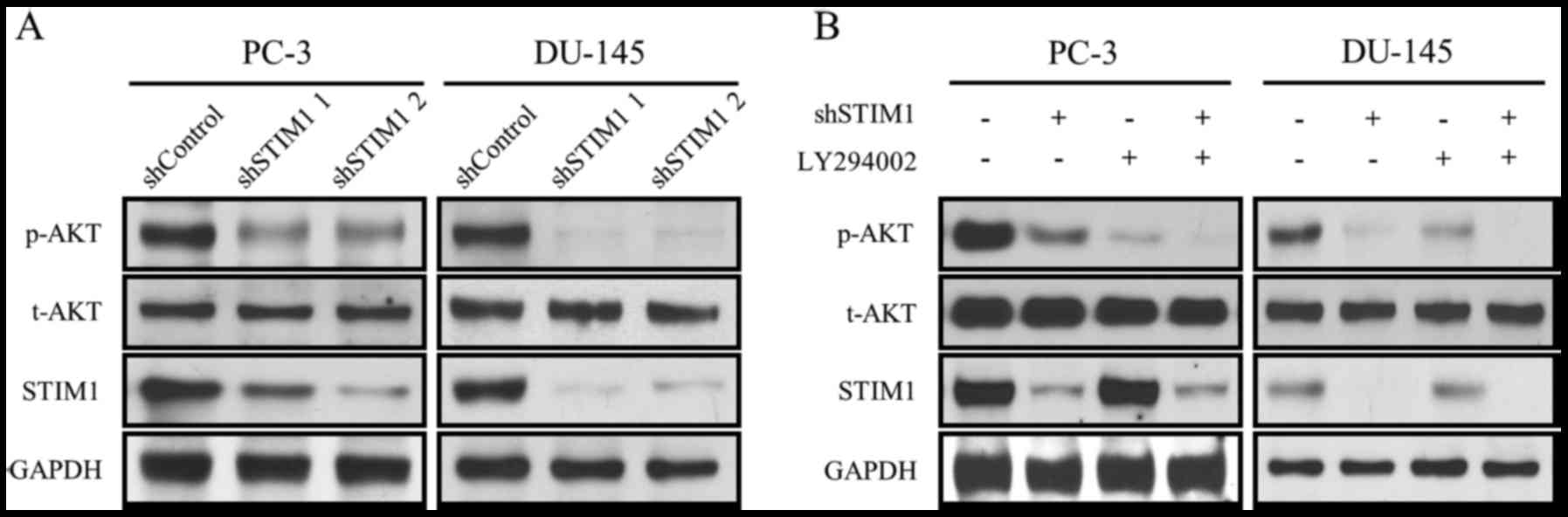
The PI3K/Akt signaling pathway is
inactivated by STIM1 knockdown
To study the influence of STIM1 knockdown on the
PI3K/Akt signaling pathway, we assessed the expression of p-Akt,
t-Akt and STIM1 in PC-3 and DU-145 cells after 72 h of infection
with shSTIM1. As shown in Fig. 4A,
p-Akt (Thr308) was suppressed by STIM1 knockdown with no change
observed in t-Akt. To further confirm the involvement of PI3K/Akt,
LY294002, a classic PI3K inhibitor, was used. As shown in Fig. 4B, both LY294002 and STIM1 decreased
the levels of p-Akt without affecting the expression of t-Akt. When
these two reagents were used in combination, the PI3K/Akt
inhibition effect was further enhanced (Fig. 4B).
These results revealed that the PI3K/Akt signaling
pathway was suppressed by STIM1 knockdown in prostate cancer
cells.
The PI3K/Akt signaling pathway is
involved in the inhibition of migration and invasion induced by
STIM1 knockdown
To determine whether PI3K/Akt inactivation mediated
the suppression of STIM1 knockdown on cell migration and invasion,
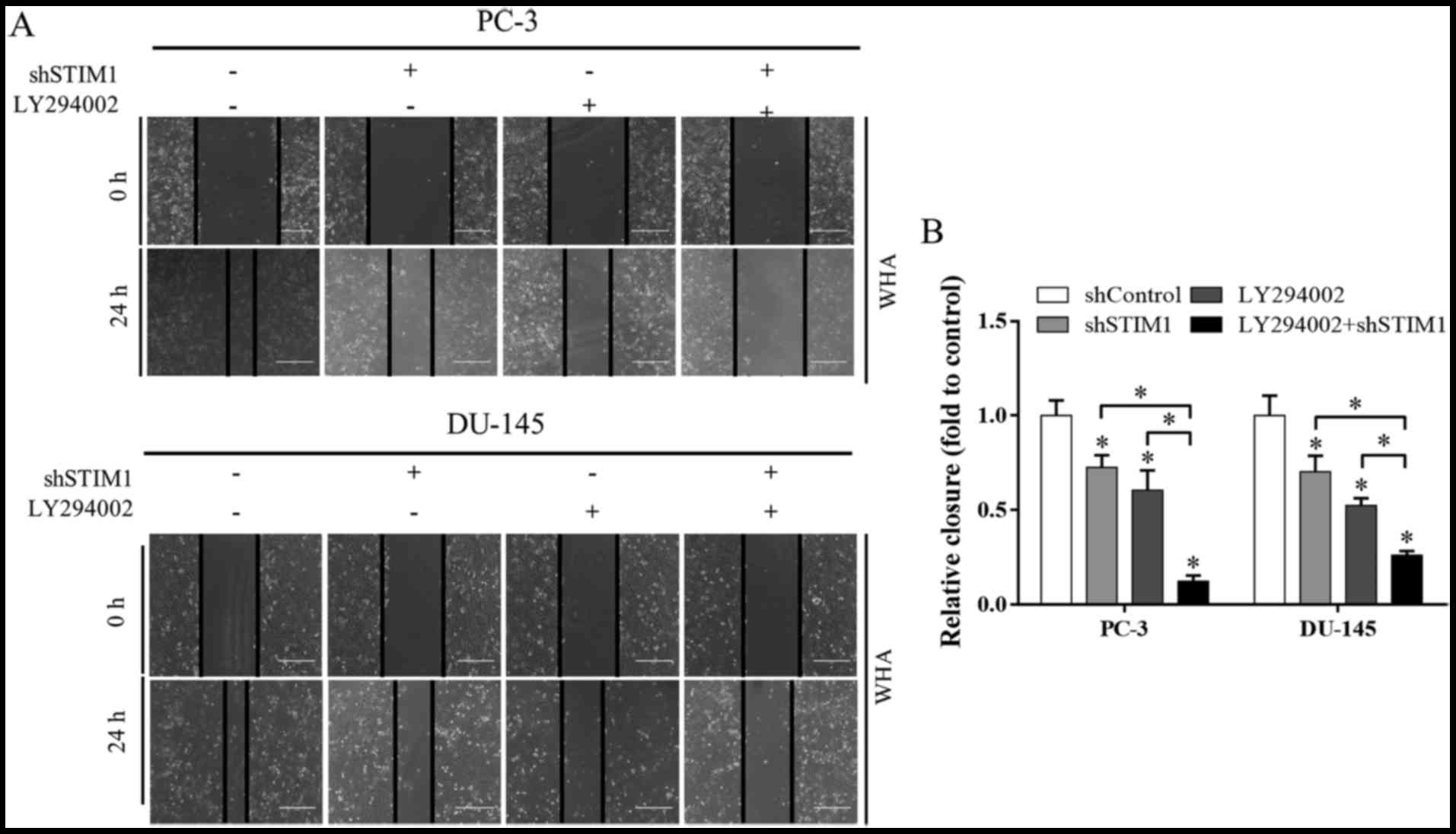
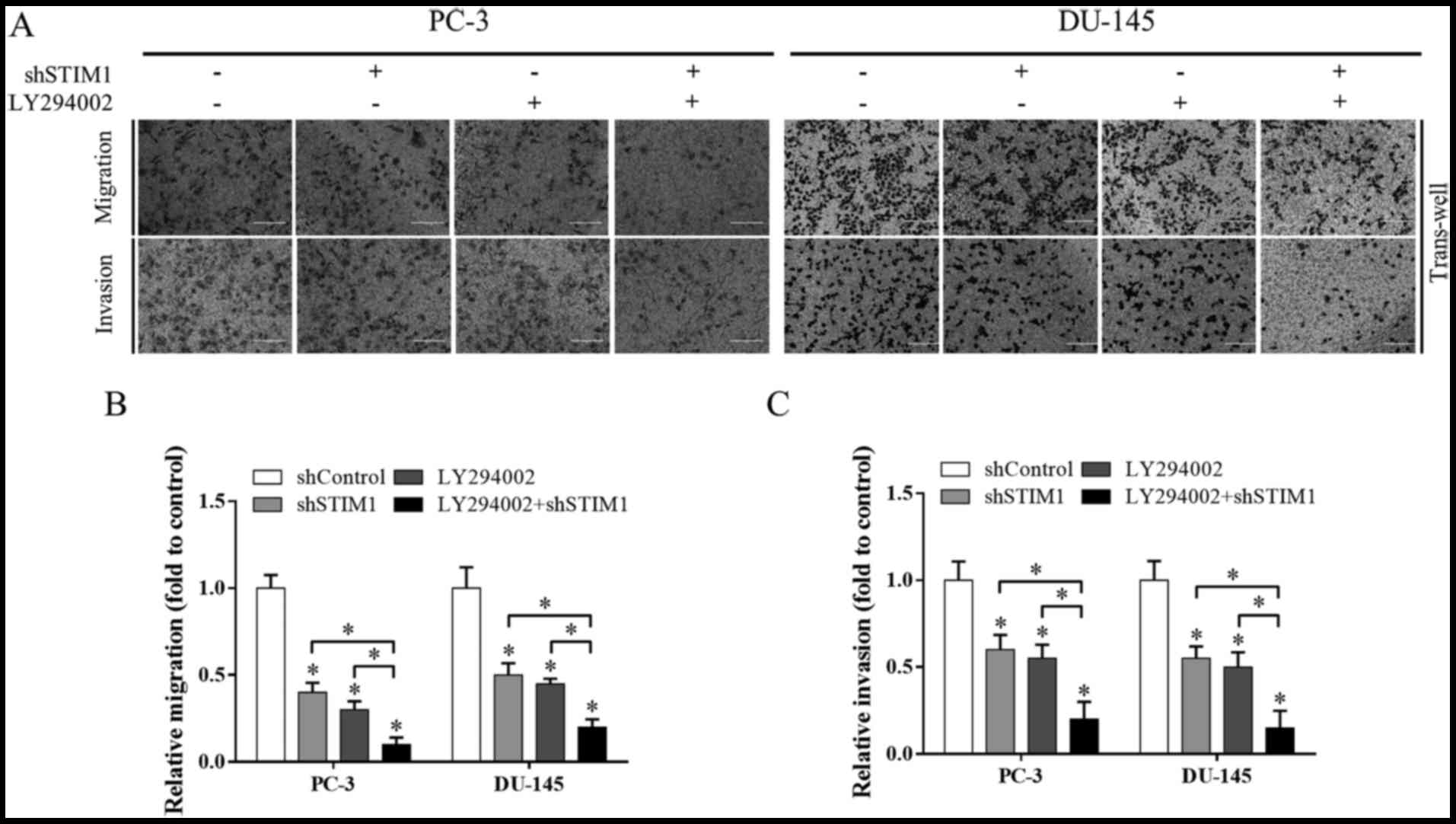
we detected cell migration under the treatment of LY294002 and/or
STIM1 knockdown. Both LY294002 and STIM1 inhibited cell migration,
and when these two treatments were used in combination, cell
migration inhibition was markedly enhanced, as determined by wound
healing assay (Fig. 5) and
Transwell migration assay (Fig. 6A and
B). A similar phenomenon was observed in the Transwell invasion
assay when studying cell invasion (Fig.
6A and C).
These data reveal that inactivation of PI3K/Akt is a
potential underlying mechanism involved in migration and invasion
inhibition induced by STIM1 knockdown.
Discussion
Calcium signaling regulates a variety of cellular
functions by activating or inhibiting cellular genes and signaling
pathways (24,25). A number of studies have documented
that tumor progression is generally associated with dysregulated
expression of Ca2+ channels and other molecules involved
in Ca2+ homeostasis (26–28).
STIM1 is one of the important components of major Ca2+
entry in non-excitable cells and has been reported to be aberrantly
expressed in various types of cancer. STIM1 plays an important role
in cell proliferation, migration and invasion in cervical cancer,
hepatocarcinoma, glioblastoma and gastric cancer. Thus, STIM1 is
considered to be an oncogene and a potential therapeutic target for
these types of cancer (29,30). A recent study revealed that STIM1
was downregulated in prostate cancer (31), which was contrary to other studies
and databases. Thus, there is a need to clarify the exact role of
STIM1 in prostate cancer and further explore its functions. In the
present study, we found that STIM1 was upregulated both in prostate
cancer tissues and prostate cancer cell lines. STIM1 played a
pivotal role in prostate cancer cell migration and invasion as
STIM1 knockdown decreased both cell migration and invasion. We then
examined EMT-related markers and found that mobility inhibited by
STIM1 knockdown was associated with EMT reversion.
Increasing studies indicate that Ca2+ is
an important regulator of the PI3K/Akt signaling pathway, and an
increase of intracellular Ca2+ activates the PI3K/Akt
pathway (32–37). PI3K/Akt signaling is one of the most
important intracellular pathways regulating cellular homeostasis.
PI3K/Akt is over-activated in a various types of cancer, and is
associated with progression of malignancy and, in particular,
metastasis. Inhibition of the PI3K/Akt pathway was revealed to
suppress invasion and migration in a variety of cancer models and
was considered to be a potential therapeutic approach to combat
cancer metastases (38–40).
However, in prostate cancer, whether migration and
invasion which were regulated by STIM1 involved the PI3K/Akt
pathway is still uncertain. Our mechanistic study revealed that the
PI3K/Akt signaling pathway was inactivated by STIM1 knockdown and
was involved in the inhibition of migration and invasion induced by
STIM1. In addition, the effect of STIM1 depletion was further
enhanced by the combination with LY294002, suggesting that STIM1
knockdown inhibits the migration and invasion of prostate cancer
cells involving the PI3K/Akt signaling pathway. However, besides
the PI3K/Akt pathway, some other signaling pathways are also
sensitive to calcium level changes within cells, and have been
reported to be regulated by STIM1, such as the MAPK and AMPK
pathways. Knockdown of STIM1 attenuates cytosolic calcium and
subsequently increases AMPK phosphorylation (41). Furthermore, AMPK is a regulator of
STIM1. STIM1 phosphorylation was reported to be mediated by the
AMPK-p38 MAPK signaling axis (42).
These signaling pathways differ from each other, but also have
cross-talks. They work together to form a complex network while
STIM1 is an important trigger. Our study just elucidated a part of
the picture and more light is needed to be shed on the detailed
mechanism involved in the future.
Collectively, our results indicated that STIM1
knockdown inhibited the migration and invasion of prostate cancer
cells involving the inactivation of PI3K/Akt signaling pathway.
These findings shed new light on our understanding of STIM1 and
suggest that STIM1 may be a potential target in the prevention of
prostate cancer metastasis.
Acknowledgements
The present study was supported by the Foundation of
Health Bureau of Suzhou (SYSD2012084), the Natural Science
Foundation of Jiangsu Province (BK20161222) and the Suzhou
Foundation for the Development of Science and Technology
(SYS201629), P.R. China.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lucas JM, Heinlein C, Kim T, Hernandez SA,
Malik MS, True LD, Morrissey C, Corey E, Montgomery B, Mostaghel E,
et al: The androgen-regulated protease TMPRSS2 activates a
proteolytic cascade involving components of the tumor
microenvironment and promotes prostate cancer metastasis. Cancer
Discov. 4:1310–1325. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shah ET, Upadhyaya A, Philp LK, Tang T,
Skalamera D, Gunter J, Nelson CC, Williams ED and Hollier BG:
Repositioning ‘old’ drugs for new causes: Identifying new
inhibitors of prostate cancer cell migration and invasion. Clin Exp
Metastasis. 33:385–399. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abdullaev IF, Bisaillon JM, Potier M,
Gonzalez JC, Motiani RK and Trebak M: Stim1 and Orai1 mediate CRAC
currents and store-operated calcium entry important for endothelial
cell proliferation. Circ Res. 103:1289–1299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berridge MJ, Lipp P and Bootman MD: The
versatility and universality of calcium signalling. Nat Rev Mol
Cell Biol. 1:11–21. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu J, Qin K, Zhang Y, Gong J, Li N, Lv D,
Xiang R and Tan X: Downregulation of transcription factor Oct4
induces an epithelial-to-mesenchymal transition via enhancement of
Ca2+ influx in breast cancer cells. Biochem Biophys Res
Commun. 411:786–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee KP, Yuan JP, Hong JH, So I, Worley PF
and Muallem S: An endoplasmic reticulum/plasma membrane junction:
STIM1/Orai1/TRPCs. FEBS Lett. 584:2022–2027. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang S, Zhang JJ and Huang XY: Orai1 and
STIM1 are critical for breast tumor cell migration and metastasis.
Cancer Cell. 15:124–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen YF, Chiu WT, Chen YT, Lin PY, Huang
HJ, Chou CY, Chang HC, Tang MJ and Shen MR: Calcium store sensor
stromal-interaction molecule 1-dependent signaling plays an
important role in cervical cancer growth, migration, and
angiogenesis. Proc Natl Acad Sci USA. 108:pp. 15225–15230. 2011;
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang N, Tang Y, Wang F, Zhang H, Xu D,
Shen Y, Sun S and Yang G: Blockade of store-operated
Ca2+ entry inhibits hepatocarcinoma cell migration and
invasion by regulating focal adhesion turnover. Cancer Lett.
330:163–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li G, Zhang Z, Wang R, Ma W, Yang Y, Wei J
and Wei Y: Suppression of STIM1 inhibits human glioblastoma cell
proliferation and induces G0/G1 phase arrest. J Exp Clin Cancer
Res. 32:202013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
King D, Yeomanson D and Bryant HE: PI3King
the lock: Targeting the PI3K/Akt/mTOR pathway as a novel
therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol.
37:245–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen X, Wang YW, Xing AY, Xiang S, Shi DB,
Liu L, Li YX and Gao P: Suppression of SPIN1-mediated PI3K-Akt
pathway by miR-489 increases chemosensitivity in breast cancer. J
Pathol. 239:459–472. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Broek R Vander, Mohan S, Eytan DF, Chen Z
and Van Waes C: The PI3K/Akt/mTOR axis in head and neck cancer:
Functions, aberrations, cross-talk, and therapies. Oral Dis.
21:815–825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim D, Kim S, Koh H, Yoon SO, Chung AS,
Cho KS and Chung J: Akt/PKB promotes cancer cell invasion via
increased motility and metalloproteinase production. FASEB J.
15:1953–1962. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoeli-Lerner M, Yiu GK, Rabinovitz I,
Erhardt P, Jauliac S and Toker A: Akt blocks breast cancer cell
motility and invasion through the transcription factor NFAT. Mol
Cell. 20:539–550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tanno S, Tanno S, Mitsuuchi Y, Altomare
DA, Xiao GH and Testa JR: AKT activation up-regulates insulin-like
growth factor I receptor expression and promotes invasiveness of
human pancreatic cancer cells. Cancer Res. 61:589–593.
2001.PubMed/NCBI
|
|
18
|
Mundi PS, Sachdev J, McCourt C and
Kalinsky K: AKT in cancer: New molecular insights and advances in
drug development. Br J Clin Pharmacol. 82:943–956. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang SX, Polley E and Lipkowitz S: New
insights on PI3K/AKT pathway alterations and clinical outcomes in
breast cancer. Cancer Treat Rev. 45:87–96. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen H, Zhou L, Wu X, Li R, Wen J, Sha J
and Wen X: The PI3K/AKT pathway in the pathogenesis of prostate
cancer. Front Biosci. 21:1084–1091. 2016. View Article : Google Scholar
|
|
21
|
Foster K, Wang Y, Zhou D and Wright C:
Dependence on PI3K/Akt signaling for malignant rhabdoid tumor cell
survival. Cancer Chemother Pharmacol. 63:783–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Magee JA, Araki T, Patil S, Ehrig T, True
L, Humphrey PA, Catalona WJ, Watson MA and Milbrandt J: Expression
profiling reveals hepsin overexpression in prostate cancer. Cancer
Res. 61:5692–5696. 2001.PubMed/NCBI
|
|
23
|
Tomlins SA, Mehra R, Rhodes DR, Cao X,
Wang L, Dhanasekaran SM, Kalyana-Sundaram S, Wei JT, Rubin MA,
Pienta KJ, et al: Integrative molecular concept modeling of
prostate cancer progression. Nat Genet. 39:41–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wiegert JS and Bading H:
Activity-dependent calcium signaling and ERK-MAP kinases in
neurons: A link to structural plasticity of the nucleus and gene
transcription regulation. Cell Calcium. 49:296–305. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Müller I, Lipp P and Thiel G:
Ca2+ signaling and gene transcription in
glucose-stimulated insulinoma cells. Cell Calcium. 52:137–151.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Monteith GR, McAndrew D, Faddy HM and
Roberts-Thomson SJ: Calcium and cancer: Targeting Ca2+
transport. Nat Rev Cancer. 7:519–530. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu M, Chen L, Zhao P, Zhou H, Zhang C, Yu
S, Lin Y and Yang X: Store-operated Ca2+ entry regulates
glioma cell migration and invasion via modulation of Pyk2
phosphorylation. J Exp Clin Cancer Res. 33:982014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Umemura M, Baljinnyam E, Feske S, De
Lorenzo MS, Xie LH, Feng X, Oda K, Makino A, Fujita T, Yokoyama U,
et al: Store-operated Ca2+ entry (SOCE) regulates
melanoma proliferation and cell migration. PLoS One. 9:e892922014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu Z, Qing J, Xia Y, Wang K and Zhang F:
Suppression of stromal interaction molecule 1 inhibits SMMC7721
hepatocellular carcinoma cell proliferation by inducing cell cycle
arrest. Biotechnol Appl Biochem. 62:107–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xia J, Wang H, Huang H, Sun L, Dong S,
Huang N, Shi M, Bin J, Liao Y and Liao W: Elevated Orai1 and STIM1
expressions upregulate MACC1 expression to promote tumor cell
proliferation, metabolism, migration, and invasion in human gastric
cancer. Cancer Lett. 381:31–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu Y, Zhang S, Niu H, Ye Y, Hu F, Chen S,
Li X, Luo X, Jiang S, Liu Y, et al: STIM1 accelerates cell
senescence in a remodeled microenvironment but enhances the
epithelial-to-mesenchymal transition in prostate cancer. Sci Rep.
5:117542015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Y, Zhang T, Wu C, Xia Q and Xu D:
ASIC1a mediates the drug resistance of human hepatocellular
carcinoma via the Ca2+/PI3-kinase/AKT signaling pathway.
Lab Invest. 97:53–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li GW, Xing WJ, Bai SZ, Hao JH, Guo J, Li
HZ, Li HX, Zhang WH, Yang BF, Wu LY, et al: The calcium-sensing
receptor mediates hypoxia-induced proliferation of rat pulmonary
artery smooth muscle cells through MEK1/ERK1,2 and PI3K pathways.
Basic Clin Pharmacol Toxicol. 108:185–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Divolis G, Mavroeidi P, Mavrofrydi O and
Papazafiri P: Differential effects of calcium on PI3K-Akt and
HIF-1α survival pathways. Cell Biol Toxicol. 32:437–449. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu ZM, Chen GG, Vlantis AC, Tse GM, Shum
CK and van Hasselt CA: Calcium-mediated activation of PI3K and p53
leads to apoptosis in thyroid carcinoma cells. Cell Mol Life Sci.
64:1428–1436. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang C, Chi Y, Li J, Miao Y, Li S, Su W,
Jia S, Chen Z, Du S, Zhang X, et al: FAM3A activates PI3K p110α/Akt
signaling to ameliorate hepatic gluconeogenesis and lipogenesis.
Hepatology. 59:1779–1790. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Danciu TE, Adam RM, Naruse K, Freeman MR
and Hauschka PV: Calcium regulates the PI3K-Akt pathway in
stretched osteoblasts. FEBS Lett. 536:193–197. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ke L, Xiang Y, Guo X, Lu J, Xia W, Yu Y,
Peng Y, Wang L, Wang G, Ye Y, et al: c-Src activation promotes
nasopharyngeal carcinoma metastasis by inducing the
epithelial-mesenchymal transition via PI3K/Akt signaling pathway: A
new and promising target for NPC. Oncotarget. 7:28340–28355. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Niessner H, Schmitz J, Tabatabai G, Schmid
AM, Calaminus C, Sinnberg T, Weide B, Eigentler TK, Garbe C,
Schittek B, et al: PI3K pathway inhibition achieves potent
antitumor activity in melanoma brain metastases in vitro and in
vivo. Clin Cancer Res. 22:5818–5828. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim MS, Lee WS, Jeong J, Kim SJ and Jin W:
Induction of metastatic potential by TrkB via activation of
IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget.
6:40158–40171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mungai PT, Waypa GB, Jairaman A, Prakriya
M, Dokic D, Ball MK and Schumacker PT: Hypoxia triggers AMPK
activation through reactive oxygen species-mediated activation of
calcium release-activated calcium channels. Mol Cell Biol.
31:3531–3545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sundivakkam PC, Natarajan V, Malik AB and
Tiruppathi C: Store-operated Ca2+ entry (SOCE) induced
by protease-activated receptor-1 mediates STIM1 protein
phosphorylation to inhibit SOCE in endothelial cells through
AMP-activated protein kinase and p38β mitogen-activated protein
kinase. J Biol Chem. 288:17030–17041. 2013. View Article : Google Scholar : PubMed/NCBI
|