Introduction
Pancreatic cancer is one of the lethal malignancies,
and the 5-year survival rate for pancreatic cancer is only 8%
(1). Although surgical resection is
regarded as the effective treatment for cure, gemcitabine (GEM) is
often used as a standard anticancer drug following surgical
resection (2). The treatment of
GEM, a nucleoside analogue, induces cell cycle arrest at S phase
(3) and triggers the
phosphorylation of checkpoint kinase 1 (Chk1), a cell cycle
checkpoint protein (4,5). Since it has been expected that GEM
treatment and Chk1 inhibition are likely to increase cell death
owing to lethal chromosomal instability caused by cell cycle
progression without repairing damaged DNA at the arrested S phase,
this combined effect has been examined. As expected, this combined
treatment inhibits colony formation (4), increases clonogenic cell death
(6) and inhibits tumor growth
compared with GEM treatment alone (7–10).
Phase I clinical trials have been initiated, and the recommended
phase II dose has been shown (11–15).
However, concerns about unpredicted effects of Chk1 inhibitors on
normal cells have been raised (12,16).
Therefore, to increase the inhibitory effect of GEM without causing
side effects on normal cells, the identification of a molecular
target specific for cancer cells is necessary.
Doublecortin-like kinase 1 (Dclk1) encodes a
Ca2+/calmodulin-dependent kinase (CaM kinase)-like
domain and regulates microtubule polymerization (17,18).
Dclk1 is highly expressed in human pancreatic cancer compared with
human normal pancreas (19) and
positively regulates tumor growth, invasion, metastasis,
pluripotency factors, angiogenic factors, and
epithelial-mesenchymal transition (EMT)-related genes in pancreatic
cancer cells (20–22). Two small molecule kinase inhibitors,
XMD8-92 and LRRK2-IN-1 (LRRK), inhibit tumor growth and the
expression levels of EMT-, pluripotency-related genes, and
oncogenes such as c-MYC and KRAS through Dclk1
inhibition (23,24). However, the Dclk1-signaling pathway,
including its substrate proteins, remains to be elucidated.
In this study, we identified Chk1 as a candidate
substrate protein phosphorylated by Dclk1, using a cancer-related
phosphorylated protein microarray. We examined the effects of GEM,
LRRK, and combined treatment with GEM and LRRK on p-Chk1
expression, DNA damage, apoptosis, and cell survival rate in
pancreatic cancer cells.
Materials and methods
Cell lines and culture
The human pancreatic cancer-derived cell lines MIA
Paca2 and PANC-1 were purchased from Riken BioResource Center
(Tsukuba, Japan) and maintained in DMEM and RPMI-1640,
respectively, with 10% fetal bovine serum (FBS) at 37°C in a
humidified atmosphere containing 5% CO2.
Reagents
LRRK2-IN-1 was purchased from Merck Millipore
(Darmstadt, Germany) and dissolved in dimethyl sulfoxide (DMSO).
Gemcitabine was purchased from Sigma-Aldrich (Tokyo, Japan) and
dissolved in PBS. LRRK2-IN-1 and gemcitabine were stored at
4°C.
Transfection of siRNAs
siRNAs were synthesized by GE Dharmacon (Chalfont,
UK). Transfection of On-Target plus Human DCLK1 (9201) siRNA-SMART
pool (L-004884-00-0005) or On-Target plus Non-targeting Pool
(D-001810-10-05) control siRNA was performed with Lipofectamine
RNAiMAX (Invitrogen, Carlsbad, CA, USA) following the
manufacturer's protocol. Following incubation for 72 h, PANC-1
cells transfected with Non-target or Dclk1 siRNA were harvested to
prepare cell lysates.
Protein microarray
To identify proteins phosphorylated by Dclk1, we
used a protein microarray, Cancer Signaling Phospho Antibody Array
(Full Moon BioSystems Inc., Sunnyvale, CA, USA). This array
features 269 highly specific antibodies that are important in
cancer signaling pathways. Cell lysates prepared from MIA Paca2
cells treated with DMSO or LRRK2-IN-1 were used, as well as PANC-1
cells transfected with non-target or Dclk1 siRNA. The arrays were
scanned using GenePix 4000B microarray scanner (Molecular Devices,
Sunnyvale, CA, USA), and array images were analyzed with GenePix
Pro7.
Western blot analysis
MIA Paca2 cells were treated with DMSO, GEM (20 or
40 nM), LRRK (10 µM) treatment, or the combined treatment with GEM
(20 or 40 nM) and LRRK (10 µM) for 48 h. PANC-1 cells were
transfected with Non-target or Dclk1 siRNA and incubated for 72 h.
After harvesting, the cells were lysed with RIPA buffer [50 mM
Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate,
0.1% SDS, and 1X protease inhibitor] and incubated on ice for 30
min. After centrifugation at 20000 × g for 20 min at 4°C, and the
supernatants were collected as cell lysates. Protein concentrations
were determined by BCA protein assay (Thermo Fisher Scientific,
Waltham, MA, USA). Equivalent amounts of cell lysates (30 µg) were
mixed with sample buffer containing reducing reagent (Nacalai
Tesque, Kyoto, Japan) and incubated at room temperature for 20 min.
Cell lysates mixed with sample buffer were separated by SDS-PAGE
using polyacrylamide gel, SuperSep Ace 10 or 15% (Wako, Osaka,
Japan) and transferred to an Immobilon-P PVDF transfer membrane
(Merck Millipore). After blocking with TBST (Tris-buffered saline
containing Tween-20) including 5% w/v bovine serum albumin for 1 h
at room temperature, the membrane was immunoblotted with the
appropriate primary antibodies diluted at 1:1000 and incubated
overnight at 4°C. After washing, the membrane was incubated with
the appropriate secondary antibodies conjugated with horseradish
peroxidase diluted at 1:5000 for 45 min at room temperature. After
washing, the immune complexes reacted with Amersham ECL Prime
Western blotting detection reagent (GE Healthcare Life Sciences,
Chicago, IL, USA) were detected using an Amersham Imager 600 (GE
Healthcare Life Sciences). Band intensity was measured using ImageJ
software.
The primary antibodies used in this study were
anti-DCAMKL1 antibody (Abgent, San Diego, CA, USA), anti-Chk1
antibody (Abcam, Cambridge, UK), anti-Phospho-Chk1 (S345) antibody
(Cell Signaling Technology, Danvers, MA, USA), anti-PARP-1 (F-2)
antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
anti-Caspase-3 (31A1067) antibody (Santa Cruz Biotechnology), and
anti-β-actin antibody (Sigma-Aldrich, St. Louis, MO, USA). The
secondary antibodies used in this study were goat polyclonal
anti-mouse immunoglobulins and anti-rabbit immunoglobulins
conjugated with horseradish peroxidase (HRP) (Dako, Glostrup,
Denmark).
Flow cytometry and cell cycle
analysis
MIA Paca2 cells were seeded at 2×105
cells/2 ml media in each well of a 6-well plate. To synchronize
cells at G0 phase of cell cycle, DMEM containing 10% FBS was
substituted with serum-free DMEM on the following day. After serum
starvation for 24 h, serum-free DMEM was substituted with DMEM
containing 10% FBS, and cells were treated with DMSO, GEM (40 nM),
LRRK (10 µM) treatment, or the combined treatment with GEM (40 nM)
and LRRK (10 µM) for 24 h. After harvesting, cells were fixed with
70% ethanol and incubated for 30 min at −20°C, followed by washing
three times with Cell Staining Buffer (BioLegend, San Diego, CA,
USA). Cells were incubated with propidium iodide (PI)/RNase
(Immunostep, Salamanca, Spain) and FITC anti-H2A.X Phospho (Ser139)
antibody (BioLegend, San Diego, CA, USA). After incubation, the
cells were applied to a BD FACSAria III flow cytometer (BD
Biosciences, San Jose, CA, USA).
Cell survival assay
MIA Paca2 cells were seeded at 1×104
cells/well on 96-well plates. After incubation for 24 h, cells were
treated with DMSO, GEM (40 nM), LRRK (10 µM) treatment, or the
combined treatment with GEM (40 nM) and LRRK (10 µM) for 72 h and
analyzed by Cell Count Reagent SF (Nacalai Tesque). To evaluate the
number of viable cells, absorbance at 450 nm was measured using an
iMark microplate reader (Bio-Rad, Hercules, CA, USA). Absorbance
value was normalized to that in control cells.
Statistical analysis
Statistical analyses were performed using JMP Pro
11.2.0 software (SAS Institute Inc.). Tukey's test was performed to
analyze the differences between multiple groups. P-values <0.05
were considered statistically significant.
Results
Identification of candidate substrate
proteins phosphorylated by Dclk1, using protein microarray
To identify proteins phosphorylated by Dclk1, we
used a protein microarray that features highly specific antibodies
against 269 phosphorylated proteins that play important roles in
cancer signaling pathways. Cell lysates were prepared from MIA
Paca2 cells treated with either DMSO or LRRK, as well as PANC-1
cells transfected with either Non-target or Dclk1 siRNA. Tables I and II show cancer-related phosphorylated
proteins with expression levels decreased by >50% in
Dclk1-inhibited pancreatic cancer cells. The expression levels of
p-Chk1 and p-cdc25A, a downstream protein of p-Chk1, were reduced
in both Dclk1-inhibited PANC-1 and MIA Paca2 pancreatic cancer
cells. These proteins belong to the ATR pathway and regulate the
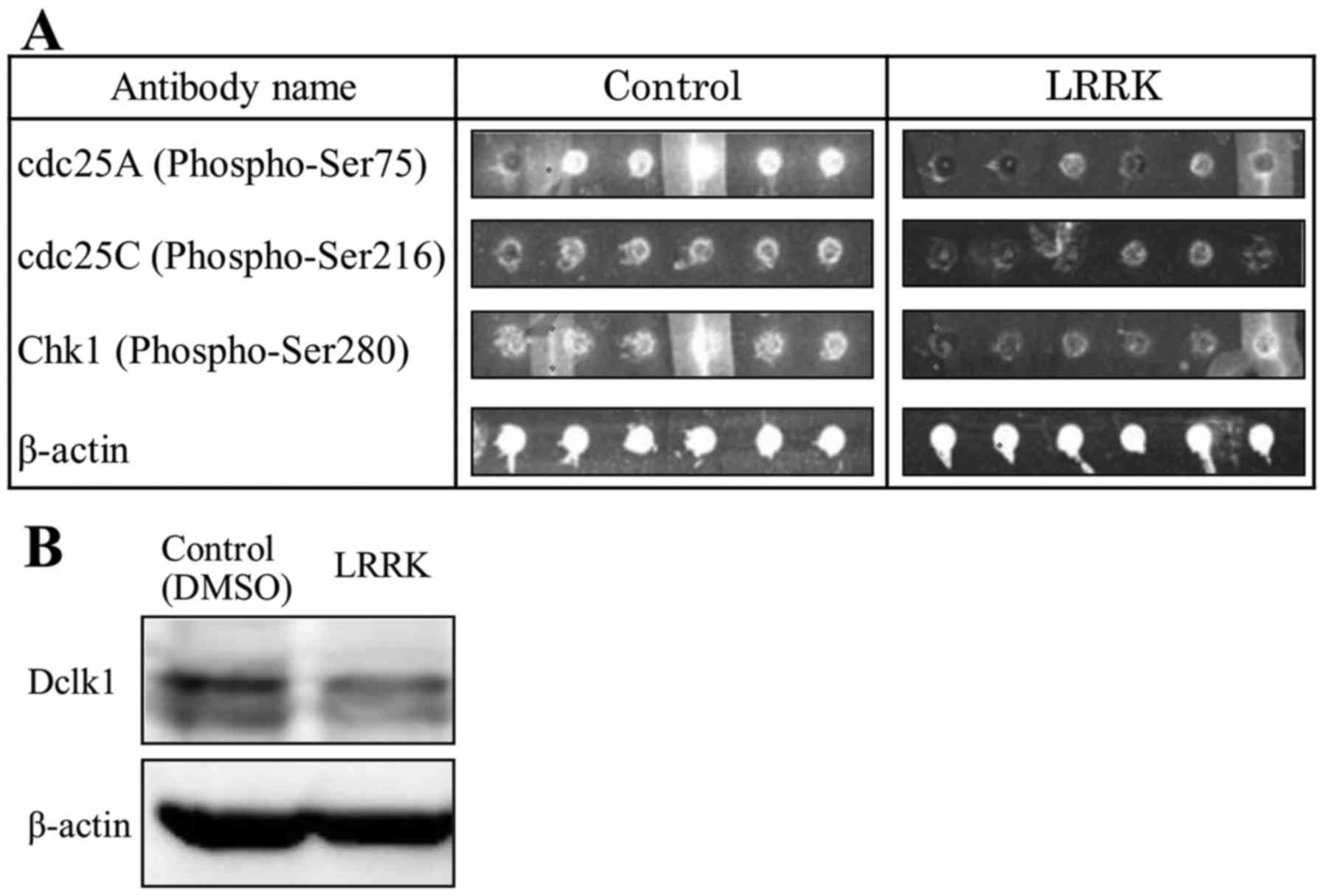
cell cycle checkpoint. The array images of p-cdc25A, p-Chk1, and
β-actin in MIA Paca2 cells treated with either DMSO or LRRK are
shown (Fig. 1A). LRRK treatment
tended to decrease Dclk1 expression compared with DMSO treatment
(Fig. 1B). Thus, these results
indicate that Dclk1 is involved in the regulation of the cell cycle
checkpoint through phosphorylation of cdc25A and Chk1.
 | Table I.The expression of phosphorylated
proteins which decreased by >50% in Dclk1-inhibited MIA Paca2
cells. |
Table I.
The expression of phosphorylated
proteins which decreased by >50% in Dclk1-inhibited MIA Paca2
cells.
|
| Intensity |
|
|---|
|
|
|
|
|---|
| Antibody name | Control (DMSO) | LRRK (50 µM) | Ratio (%)
(LRRK/Control) |
|---|
| Akt
(Phospho-Ser473) | 15278.5 | 500.5 | 3.28 |
| cdc25A
(Phospho-Ser75) | 26021.0 | 1914.0 | 7.36 |
| STAT1
(Phospho-Ser727) | 27948.0 | 2471.0 | 8.84 |
| JAK2
(Phospho-Tyr221) | 16696.0 | 1557.0 | 9.33 |
| FAK
(Phospho-Tyr925) | 18429.0 | 3475.0 | 18.86 |
| c-Jun
(Phospho-Ser243) | 18280.5 | 4807.0 | 26.30 |
| cdc25C
(Phospho-Ser216) | 19121.5 | 5211.0 | 27.25 |
| BAD
(Phospho-Ser112) | 23047.5 | 7167.0 | 31.10 |
| NFκB-p105/p50
(Phospho-Ser893) | 12876.5 | 4189.0 | 32.53 |
| eEF2K
(Phospho-Ser366) | 17454.5 | 5932.0 | 33.99 |
| JAK2
(Phospho-Tyr1007) | 12732.0 | 5130.0 | 40.29 |
| CDK2
(Phospho-Thr160) | 17259.0 | 6985.0 | 40.47 |
| BCL-2
(Phospho-Ser70) | 16820.0 | 6861.0 | 40.79 |
| Raf1
(Phospho-Ser259) | 15261.5 | 6241.5 | 40.90 |
| TYK2
(Phospho-Tyr1054) | 16798.0 | 7063.0 | 42.05 |
| BCL-2
(Phospho-Thr56) | 14397.5 | 6312.5 | 43.84 |
| c-Jun
(Phospho-Thr239) | 11045.5 | 5136.0 | 46.50 |
| NFκB-p105/p50
(Phospho-Ser907) | 23004.5 | 11145.5 | 48.45 |
| Chk1
(Phospho-Ser280) | 17482.0 | 8578.5 | 49.07 |
| Table II.Expression of the phosphorylated
proteins which decreased by >50% in Dclk1-silenced PANC-1
cells. |
Table II.
Expression of the phosphorylated
proteins which decreased by >50% in Dclk1-silenced PANC-1
cells.
|
| Intensity |
|
|---|
|
|
|
|
|---|
| Antibody name | Control
(non-target) | si-Dclk1 | Ratio (%)
(si-Dclk1/Control) |
|---|
| cdc25A
(Phospho-Ser75) | 14935.2 | 3961.8 | 26.53 |
| Fak
(Phospho-Tyr397) | 13761.0 | 4523.5 | 32.87 |
| STAT4
(Phospho-Tyr693) | 10683.7 | 4292.0 | 40.17 |
| Chk1
(Phospho-Ser317) | 19384.0 | 8719.8 | 44.98 |
| PTEN
(Phospho-Ser380/Thr382/Thr383) | 13016.7 | 6084.8 | 46.75 |
| Src
(Phospho-Tyr529) | 25736.5 | 12031.7 | 46.75 |
GEM induces phosphorylation of Chk1,
and combined treatment with GEM and LRRK significantly decreases
p-Chk1 expresssion
To evaluate the individual and combined effects of
GEM and LRRK treatment on phosphorylation of Chk1, MIA Paca2 cells
were treated with DMSO, GEM, LRRK treatment, or the combined
treatment with GEM and LRRK for 48 h and analyzed by western
blotting. Dclk1 expression tended to be decreased following LRRK
treatment and remain unchanged following GEM treatment compared
with DMSO treatment (Fig. 2A and
B). As expected, GEM treatment significantly induced Chk1
phosphorylation in MIA Paca2 cells (Fig. 2C and D). Notably, combined treatment
with GEM and LRRK significantly reduced the expression of p-Chk1
compared with GEM treatment alone (Fig.
2C and D).
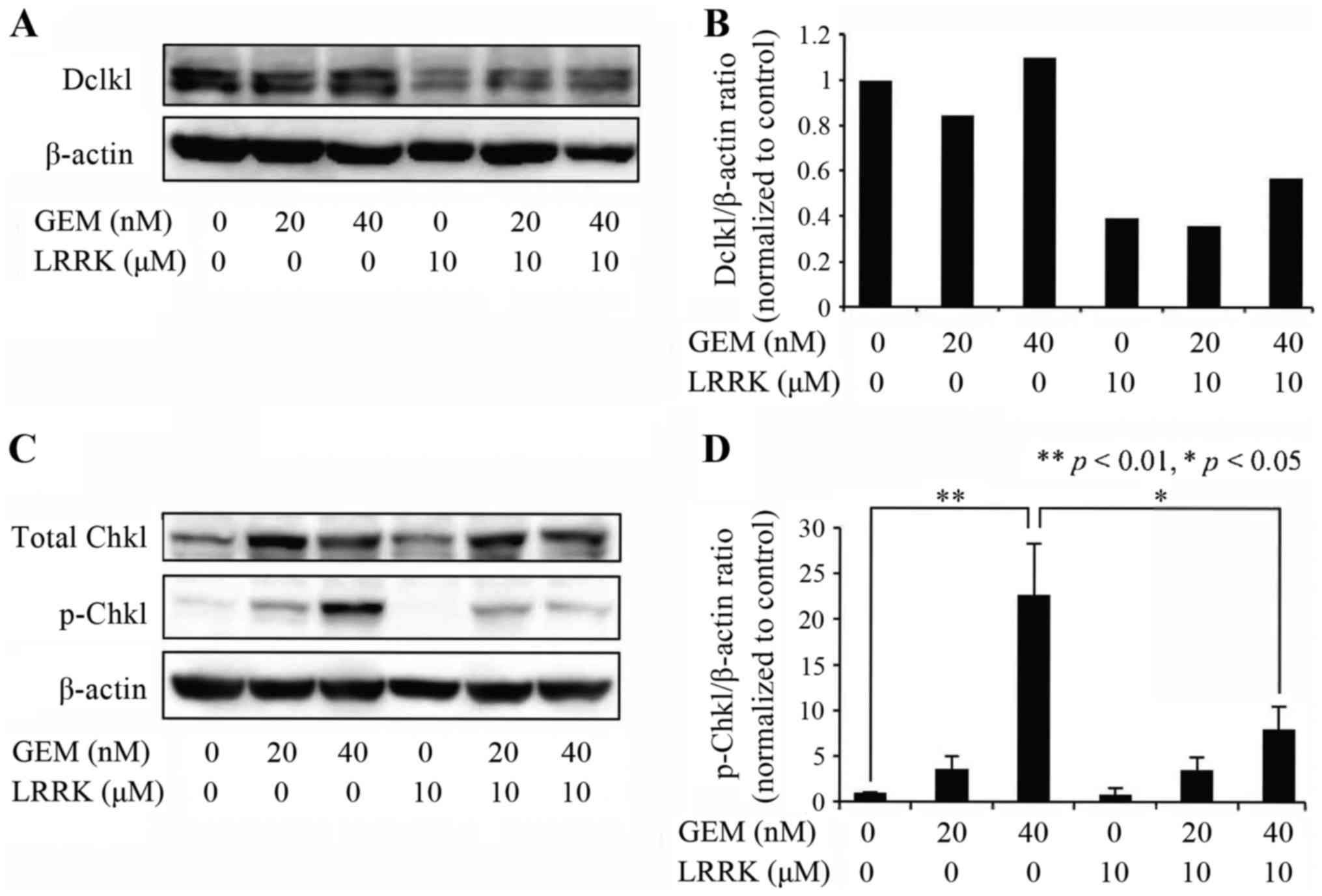 | Figure 2.Effect of DMSO, GEM, LRRK treatment,
or the combined treatment with GEM and LRRK on the expression of
p-Chk1. (A) Cell lysates were prepared from MIA Paca2 cells treated
with DMSO, GEM (20 or 40 nM), LRRK (10 µM) treatment, or the
combined treatment with GEM (20 or 40 nM) and LRRK (10 µM) for 48
h, and the expression levels of Dclk1 and β-actin were detected by
western blotting. β-actin was used to assess the total amount of
proteins loaded on the gel. (B) The intensity of each band was
quantified using ImageJ software. The ratio of Dclk1 to β-actin was
normalized to that in control cells. (C) Cell lysates were prepared
from MIA Paca2 cells treated with DMSO, GEM (20 or 40 nM), LRRK (10
µM) treatment, or the combined treatment with GEM (20 or 40 nM) and
LRRK (10 µM) for 48 h, and the expression levels of Chk1, p-Chk1,
and β-actin were detected by western blotting. β-actin was used to
assess the total amount of proteins loaded on the gel. (D) The
intensity of each band was quantified using ImageJ software. The
ratio of p-Chk1 to β-actin was normalized to that in control cells.
Each bar represents the mean ± SE of three experiments. *P<0.05;
**P<0.01, significantly different. |
Combined treatment with GEM and LRRK
abolished GEM-induced cell cycle arrest and increased DNA damage
compared with GEM or LRRK treatment alone
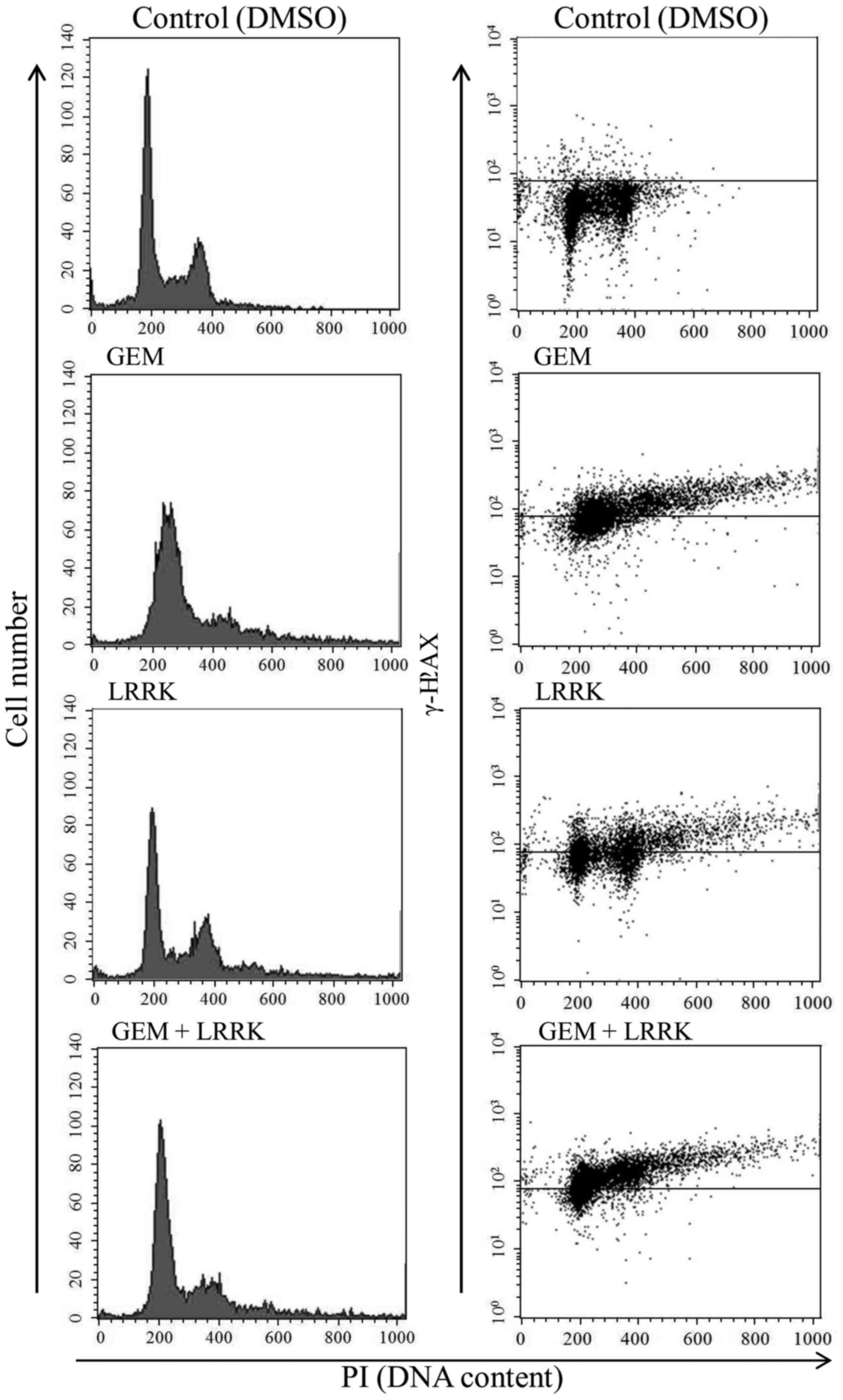
To evaluate the individual and combined effects of
GEM and LRRK treatment on cell cycle progression and DNA damage, we
performed flow cytometry analyses of cells stained with PI and
γ-H2AX, respectively. After serum starvation for 24 h, most MIA
Paca2 cells were observed at G0/G1 phase of the cell cycle (data
not shown). As previously reported, individual treatment with GEM
induced cell cycle arrest at S phase (3) and increased the number of
γ-H2AX-positive cells (Fig. 3 and
Table III). Individual treatment
with LRRK proceeded cell cycle and increased the number of
γ-H2AX-positive cells (Fig. 3 and
Table III). Notably, combined
treatment with GEM and LRRK allowed cell cycle progression without
arresting at S phase and increased the number of γ-H2AX-positive
cells compared with individual treatments with GEM or LRRK
(Fig. 3 and Table III).
| Table III.The proportion of γ-H2AX-positive
cells in each phase of the cell cycle. |
Table III.
The proportion of γ-H2AX-positive
cells in each phase of the cell cycle.
| Treatment | sub-G1 | G1 | S | G2/M | Total |
|---|
| Control |
|
|
|
|
|
| γ-H2AX
(+) | 0.81 | 1.37 | 0.98 | 2.49 | 5.65 |
| γ-H2AX
(−) | 4.01 | 53.81 | 12.49 | 24.04 | 94.35 |
|
Total | 4.82 | 55.18 | 13.47 | 26.53 | 100.00 |
| GEM |
|
|
|
|
|
| γ-H2AX
(+) | 0.66 | 11.19 | 27.37 | 29.14 | 68.36 |
| γ-H2AX
(−) | 1.32 | 12.00 | 15.81 | 2.51 | 31.64 |
|
Total | 1.98 | 23.19 | 43.18 | 31.65 | 100.00 |
| LRRK |
|
|
|
|
|
| γ-H2AX
(+) | 0.95 | 16.42 | 5.85 | 29.79 | 53.01 |
| γ-H2AX
(−) | 1.65 | 28.53 | 4.41 | 12.40 | 46.99 |
|
Total | 2.60 | 44.95 | 10.26 | 42.19 | 100.00 |
| GEM + LRRK |
|
|
|
|
|
| γ-H2AX
(+) | 0.61 | 38.10 | 13.35 | 28.76 | 80.82 |
| γ-H2AX
(−) | 0.48 | 16.02 | 1.56 | 1.12 | 19.18 |
|
Total | 1.09 | 54.12 | 14.91 | 29.88 | 100.00 |
LRRK treatment alone, and combined
treatment with GEM and LRRK induces caspase-3 activation and PARP1
cleavage
To evaluate the individual and combined effects of
GEM and LRRK treatment on apoptosis, MIA Paca2 cells were treated
with DMSO, GEM, LRRK treatment, or the combined treatment with GEM
and LRRK for 48 h and analyzed by western blotting. Notably,
individual treatment with LRRK, and combined treatment with GEM and
LRRK induced caspase-3 activation and PARP1 cleavage, although
individual treatment with GEM almost did not (Fig. 4A). PARP1 cleavage upon combined
treatment with GEM and LRRK significantly increased compared with
that upon GEM treatment alone and tended to increase compared with
that upon LRRK treatment alone (Fig.
4B). Notably, GEM treatment alone tended to increase the
expression of intact PARP1, a substrate protein of activated
caspase-3, and combined treatment with GEM and LRRK tended to
increase cleaved caspase-3 levels. These are reasons why combined
treatment with GEM and LRRK tended to increase cleaved PARP1
levels.
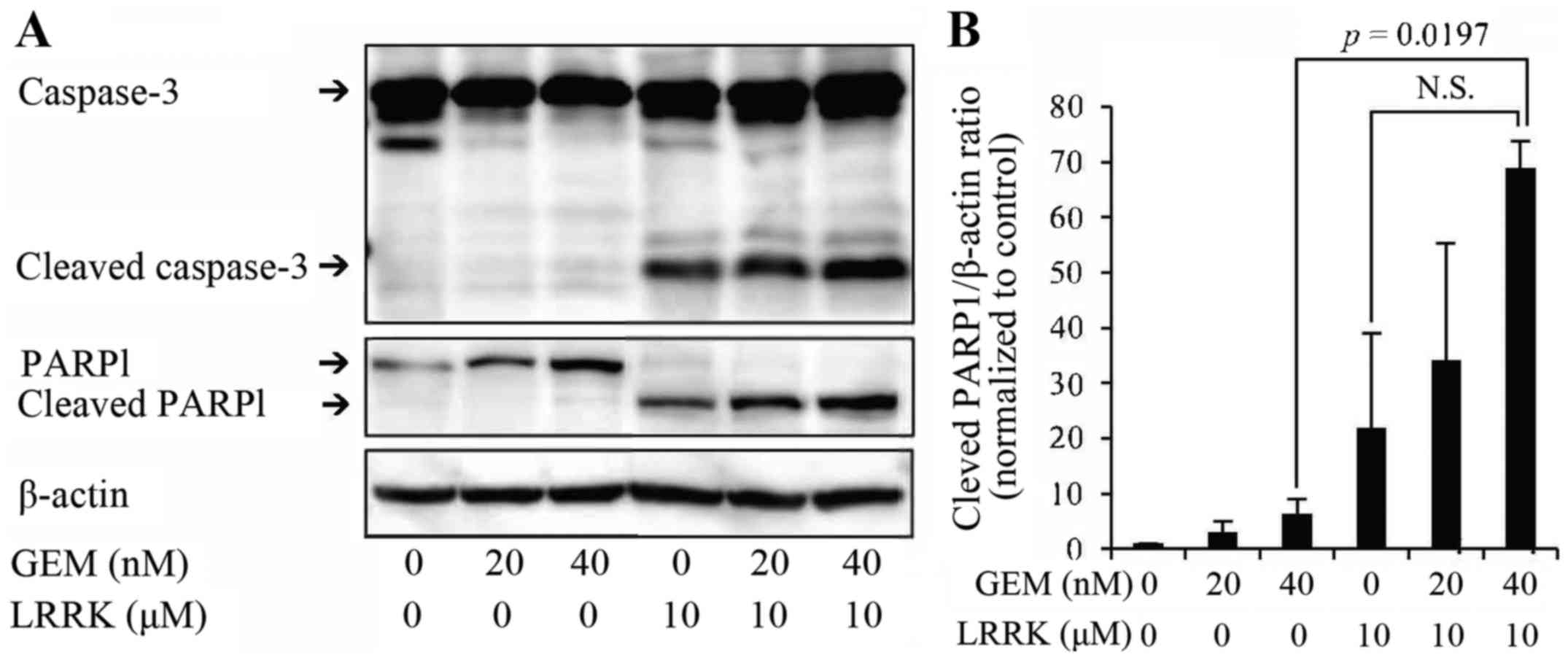 | Figure 4.Effect of DMSO, GEM, LRRK treatment,
or the combined treatment with GEM and LRRK on caspase-3 activation
and PARP1 cleavage. (A) Cell lysates were prepared from MIA Paca2
cells treated with DMSO, GEM (20 or 40 nM), LRRK (10 µM) treatment,
or the combined treatment with GEM (20 or 40 nM) and LRRK (10 µM)
for 48 h, and the expression levels of caspase-3, cleaved
caspase-3, PARP1, cleaved PARP1, and β-actin were detected by
western blotting. β-actin was used to assess the total amount of
proteins loaded on the gel. (B) The intensity of each band was
quantified using ImageJ software. The ratio of cleaved PARP1 to
β-actin was normalized to that in control cells. Each bar
represents the mean ± SE of three experiments. N.S., no significant
difference. |
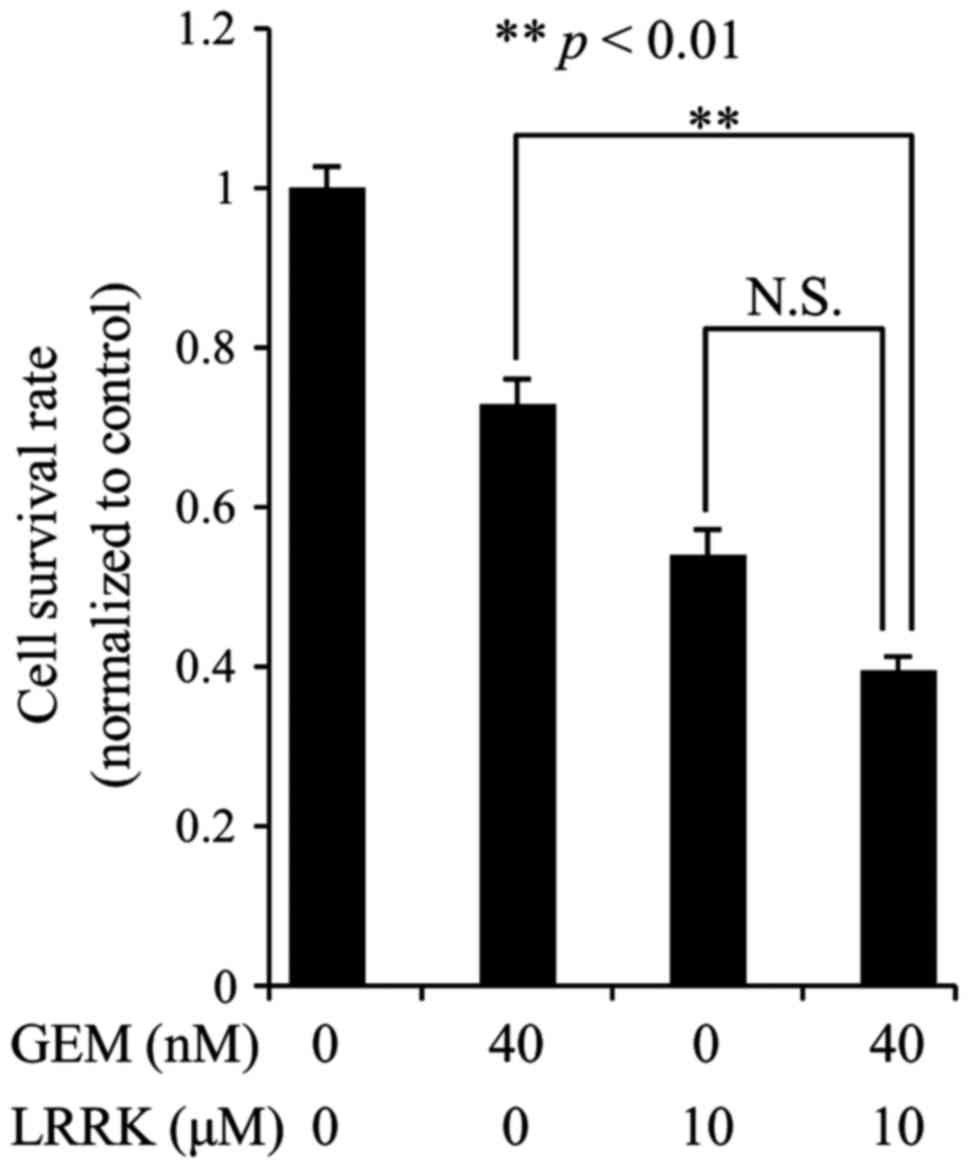
Combined treatment with GEM and LRRK
significantly decrease survival rate of MIA Paca2 cells compared to
individual treatment with GEM
To evaluate the individual and combined effects of
GEM and LRRK treatment on MIA Paca2 cell survival, we performed
cell survival assay following DMSO, GEM, LRRK treatment, or the
combined treatment with GEM and LRRK. Individual treatment with GEM
or LRRK decreased cell survival to a modest extent (Fig. 5). Combined treatment with GEM and
LRRK significantly decreased cell survival compared to individual
treatment with GEM and tended to decrease cell survival compared to
individual treatment with LRRK (Fig.
5).
Discussion
In this study, we identified candidate substrate
proteins phosphorylated by Dclk1, using a cancer-related
phosphorylated protein microarray of Dclk1-inhibited MIA Paca2
cells. P-cdc25A and p-Chk1 proteins were included among the
phosphorylated proteins whose expression levels were decreased by
>50% through functional inhibition of Dclk1. These proteins
belong to the ATR pathway, which regulates the cell cycle
checkpoint. Chk1 is phosphorylated by activated ATR, and p-Chk1
phosphorylates cdc25A, leading to the induction of cell cycle
arrest. That is, p-Chk1 is located upstream of cdc25A, and cdc25A
is, therefore, a substrate protein of p-Chk1. Thus, we focused on
Chk1 as a candidate substrate protein phosphorylated by Dclk1. As
expected, GEM-induced p-Chk1 expression level is significantly
decreased by Dclk1 inhibition, and combined treatment with GEM and
LRRK allowed cell cycle progression without arresting at S phase.
Furthermore, combined treatment increased DNA damage, apoptosis,
and cell death compared with those upon individual treatment with
GEM. Based on these results, we propose that the mechanism by which
the combined treatment increased cell death is as follows. Dclk1
inhibition decreased GEM-induced p-Chk1 expression, and the cell
cycle checkpoint was impaired. Consequently, cell cycle proceeded
without repairing damaged DNA at the arrested S phase, leading to
cell death due to lethal chromosome instability. Furthermore,
O'Connell et al recently reported that the short form of
Dclk1 is mainly expressed in human colon cancer cells, whereas the
long form is mainly expressed in normal colon cells (25). In this study, we detected the
expression of the short form of Dclk1 in MIA Paca2 cells. To
determine whether the expression of the short form of Dclk1 in
human pancreatic cancer cells is universal, further studies will be
needed to investigate which form of Dclk1 is expressed in many
other human pancreatic cancer cell lines, pancreatic cancer
tissues, and normal human pancreatic cells.
In conclusion, combined treatment with GEM and a
Dclk1 inhibitor, LRRK, significantly reduced the cell survival rate
compared to individual treatment with GEM, by impairing the cell
cycle checkpoint. Targeting Dclk1, in combination with GEM
treatment, might offer an excellent opportunity for future
pancreatic cancer treatments.
Acknowledgements
We would like to thank Professor Yoichi Mizukami
(Chairman of the Center for Gene Research at Yamaguchi University)
for useful suggestions on cell cycle analysis. We also thank Ms.
Yukari Hironaka for technical assistance. This study was supported
by Grant-in-Aids for Young Scientific Research (B) (16K19932 to
Y.T.) from Japan Society for the Promotion of Science (JSPS) and
the Onkochishin Project Grant (to A.N.) from Yamaguchi
University.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi Z, Azuma A, Sampath D, Li YX, Huang P
and Plunkett W: S-Phase arrest by nucleoside analogues and
abrogation of survival without cell cycle progression by
7-hydroxystaurosporine. Cancer Res. 61:1065–1072. 2001.PubMed/NCBI
|
|
4
|
Karnitz LM, Flatten KS, Wagner JM,
Loegering D, Hackbarth JS, Arlander SJH, Vroman BT, Thomas MB, Baek
YU, Hopkins KM, et al: Gemcitabine-induced activation of checkpoint
signaling pathways that affect tumor cell survival. Mol Pharmacol.
68:1636–1644. 2005.PubMed/NCBI
|
|
5
|
Morgan MA, Parsels LA, Parsels JD,
Mesiwala AK, Maybaum J and Lawrence TS: Role of checkpoint kinase 1
in preventing premature mitosis in response to gemcitabine. Cancer
Res. 65:6835–6842. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parsels LA, Morgan MA, Tanska DM, Parsels
JD, Palmer BD, Booth RJ, Denny WA, Canman CE, Kraker AJ, Lawrence
TS, et al: Gemcitabine sensitization by checkpoint kinase 1
inhibition correlates with inhibition of a Rad51 DNA damage
response in pancreatic cancer cells. Mol Cancer Ther. 8:45–54.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Venkatesha VA, Parsels LA, Parsels JD,
Zhao L, Zabludoff SD, Simeone DM, Maybaum J, Lawrence TS and Morgan
MA: Sensitization of pancreatic cancer stem cells to gemcitabine by
Chk1 inhibition. Neoplasia. 14:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Montano R, Thompson R, Chung I, Hou H,
Khan N and Eastman A: Sensitization of human cancer cells to
gemcitabine by the Chk1 inhibitor MK-8776: Cell cycle perturbation
and impact of administration schedule in vitro and in vivo. BMC
Cancer. 13:6042013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koh SB, Courtin A, Boyce RJ, Boyle RG,
Richards FM and Jodrell DI: CHK1 inhibition synergizes with
gemcitabine initially by destabilizing the DNA replication
apparatus. Cancer Res. 75:3583–3595. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barnard D, Diaz HB, Burke T, Donoho G,
Beckmann R, Jones B, Barda D, King C and Marshall M: LY2603618, a
selective CHK1 inhibitor, enhances the anti-tumor effect of
gemcitabine in xenograft tumor models. Invest New Drugs. 34:49–60.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seto T, Esaki T, Hirai F, Arita S, Nosaki
K, Makiyama A, Kometani T, Fujimoto C, Hamatake M, Takeoka H, et
al: Phase I, dose-escalation study of AZD7762 alone and in
combination with gemcitabine in Japanese patients with advanced
solid tumours. Cancer Chemother Pharmacol. 72:619–627. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sausville E, Lorusso P, Carducci M, Carter
J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, et
al: Phase I dose-escalation study of AZD7762, a checkpoint kinase
inhibitor, in combination with gemcitabine in US patients with
advanced solid tumors. Cancer Chemother Pharmacol. 73:539–549.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Daud AI, Ashworth MT, Strosberg J, Goldman
JW, Mendelson D, Springett G, Venook AP, Loechner S, Rosen LS,
Shanahan F, et al: Phase I dose-escalation trial of checkpoint
kinase 1 inhibitor MK-8776 as monotherapy and in combination with
gemcitabine in patients with advanced solid tumors. J Clin Oncol.
33:1060–1066. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Doi T, Yoshino T, Shitara K, Matsubara N,
Fuse N, Naito Y, Uenaka K, Nakamura T, Hynes SM and Lin AB: Phase I
study of LY2603618, a CHK1 inhibitor, in combination with
gemcitabine in Japanese patients with solid tumors. Anticancer
Drugs. 26:1043–1053. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calvo E, Braiteh F, Von Hoff D, McWilliams
R, Becerra C, Galsky MD, Jameson G, Lin J, McKane S, Wickremsinhe
ER, et al: Phase I study of CHK1 inhibitor LY2603618 in combination
with gemcitabine in patients with solid tumors. Oncology.
91:251–260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goto H, Izawa I, Li Ping and Inagaki M:
Novel regulation of checkpoint kinase 1: Is checkpoint kinase 1 a
good candidate for anti-cancer therapy? Cancer Sci. 103:1195–1200.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin PT, Gleeson JG, Corbo JC, Flanagan L
and Walsh CA: DCAMKL1 encodes a protein kinase with homology to
doublecortin that regulates microtubule polymerization. J Neurosci.
20:9152–9161. 2000.PubMed/NCBI
|
|
18
|
Omori Y, Suzuki M, Ozaki K, Harada Y,
Nakamura Y, Takahashi E and Fujiwara T: Expression and chromosomal
localization of KIAA0369, a putative kinase structurally related to
Doublecortin. J Hum Genet. 43:169–177. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mohammed A, Janakiram NB, Madka V, Brewer
M, Ritchie RL, Lightfoot S, Kumar G, Sadeghi M, Patlolla JMR,
Yamada HY, et al: Targeting pancreatitis blocks tumor-initiating
stem cells and pancreatic cancer progression. Oncotarget.
6:15524–15539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sureban SM, May R, Qu D, Weygant N,
Chandrakesan P, Ali N, Lightfoot SA, Pantazis P, Rao CV, Postier
RG, et al: DCLK1 regulates pluripotency and angiogenic factors via
microRNA-dependent mechanisms in pancreatic cancer. PLoS One.
8:e739402013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sureban SM, May R, Lightfoot SA, Hoskins
AB, Lerner M, Brackett DJ, Postier RG, Ramanujam R, Mohammed A, Rao
CV, et al: DCAMKL-1 regulates epithelial-mesenchymal transition in
human pancreatic cells through a miR-200a-dependent mechanism.
Cancer Res. 71:2328–2338. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito H, Tanaka S, Akiyama Y, Shimada S,
Adikrisna R, Matsumura S, Aihara A, Mitsunori Y, Ban D, Ochiai T,
et al: Dominant expression of DCLK1 in human pancreatic cancer stem
cells accelerates tumor invasion and metastasis. PLoS One.
11:e01465642016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sureban SM, May R, Weygant N, Qu D,
Chandrakesan P, Bannerman-Menson E, Ali N, Pantazis P, Westphalen
CB, Wang TC, et al: XMD8-92 inhibits pancreatic tumor xenograft
growth via a DCLK1-dependent mechanism. Cancer Lett. 351:151–161.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weygant N, Qu D, Berry WL, May R,
Chandrakesan P, Owen DB, Sureban SM, Ali N, Janknecht R and Houchen
CW: Small molecule kinase inhibitor LRRK2-IN-1 demonstrates potent
activity against colorectal and pancreatic cancer through
inhibition of doublecortin-like kinase 1. Mol Cancer. 13:1032014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
O'Connell MR, Sarkar S, Luthra GK, Okugawa
Y, Toiyama Y, Gajjar AH, Qiu S, Goel A and Singh P: Epigenetic
changes and alternate promoter usage by human colon cancers for
expressing DCLK1-isoforms: Clinical implications. Sci Rep.
5:149832015. View Article : Google Scholar : PubMed/NCBI
|