Introduction
Chemotherapy is one of the major strategies for
cancer treatment, and functions by targeting the physiological
characteristics of cancer cells, including proliferation,
angiogenesis, apoptosis, invasion and migration (1). However, drug resistance (2) and severe side-effects (3) still hinder the effects of
chemotherapy. The use of drug combinations has numerous benefits
for cancer therapy. It enhances the therapeutic effects or
decreases the required dosages of each drug thereby reducing the
severity of adverse effects (4).
Thus, the search for combinations of agents that can achieve
synergistic antitumor effects remains an important strategy with
which to improve the effects of chemotherapy.
Acetylation is a major form of protein
post-translational modification and is responsible for regulating
various cellular processes, including cell proliferation and cell
survival (5). Acetylation is
catalyzed by histone acetyltransferases (HATs) and histone
deacetylases (HDACs). HATs transfer acetyl groups to lysine
residues, whereas HDACs remove acetyl groups from lysine residues
(6). In total, 18 human HDACs have
been identified, which are categorized into four classes: Class I
(HDAC1, HDAC2, HDAC3 and HDAC8), IIa (HDAC4, HDAC5, HDAC7 and
HDAC9), IIb (HDAC6 and 10) and IV (HDAC11) are classical HDACs,
whose activities are inhibited by pan-HDAC inhibitors, such as
trichostatin A; whereas class III HDACs, also known as sirtuins,
are not affected by trichostatin A (7).
HDACs are overexpressed in many types of tumors, and
inhibition of HDACs can result in the inhibition of cell
proliferation and induction of apoptosis (8). HDAC inhibitors can achieve antitumor
effects through the phosphatase and tensin homolog (PTEN)/protein
kinase B (AKT) signaling pathway (9,10). To
date, three pan-HDAC inhibitors, namely vorinostat (SAHA),
belinostat (PXD-101) and panobinostat (LBH-589), have been approved
by the US Food and Drug Administration (FDA) for the treatment of
cancer (11). Clinical application
of pan-HDAC inhibitors can result in several adverse side-effects,
such as fatigue, diarrhea, bone marrow toxicity and
thrombocytopenia (12). This is
likely due to the fact that most HDACs are necessary for important
biological processes; knockout of HDAC1, or most other HDACs
(except HDAC6), leads to death or severe defects during embryonic
development or shortly after birth (13). However, HDAC6-deficient mice
typically survive (14), and the
inhibition of HDAC6 has antitumor effects (15). Aberrant expression of HDAC6 is an
independent prognostic indicator in human breast cancer (16). Moreover, our previous study
demonstrated that the activation of PTEN through K163 acetylation,
in response to the inhibition of HDAC6, is an important mechanism
underlying the antitumor effects of pan-HDAC inhibitors (17). Therefore, HDAC6-specific inhibitors
may have potential clinical advantages as antitumor agents as they
do not affect other critical HDACs.
PTEN is a tumor-suppressor gene that plays a crucial
role in cell growth, development and apoptosis (18). PTEN function is commonly lost by
deletion or mutation in many types of human cancer, including
melanoma, pancreatic, colorectal and lung cancer (19). PTEN inhibits cell growth and
proliferation by removing a phosphate group from PIP3, thus
preventing the activation of AKT (20). Membrane-bound PTEN is the activated
form of the protein (21,22).
Cyclooxygenase-2 (COX-2) is an important
rate-limiting enzyme in the process of prostaglandin E2 (PGE2)
synthesis. PGE2 induces proliferation, invasion and migration of
cancer cells via several signaling pathways, including the
β-catenin and AKT pathways (23,24).
COX-2 is overexpressed in many different types of cancer (25). Inhibition of COX-2 induces the
inhibition of proliferation and apoptosis in several cancer cell
lines, including colorectal, prostate and breast cancer cell lines
(26). These studies suggest that
COX-2 may be a potential novel therapeutic target for chemotherapy.
The COX-2 selective inhibitor celecoxib, alone or in combination
with other agents, is currently being clinically tested in lung
cancer patients (27). Although
COX-2 inhibitors enhance the anti-angiogenic effects of pan-HDAC
inhibitors (28), it is not known
which HDAC is involved in the crosstalk with the COX-2 signaling
pathway. We previously demonstrated that COX-2 inhibitors
upregulate PTEN (22), and HDAC6
inhibitors can also activate PTEN (17). This prompted the question of whether
COX-2 inhibitors can enhance the antitumor effects of HDAC6
inhibitors through the synergistic activation of PTEN.
In the present study, celecoxib, a COX-2 inhibitor,
was shown to enhance the antitumor effects of an HDAC6 inhibitor
through synergistic activation of PTEN and inactivation of AKT in
CAL 27 and SACC-83 cells.
Materials and methods
Cell culture and treatments
CAL 27 cells derived from human tongue squamous cell
carcinoma (29) were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY,
USA) with 10% fetal bovine serum (FBS) at 37°C in an atmosphere
with 5% CO2. SACC-83 cells derived from human salivary
adenoid cystic cancer (30), were
cultured in RPMI-1640 medium (Gibco) with 10% FBS at 37°C with 5%
CO2. U-87 MG cells that had been stably transfected with
wild-type PTEN and PTEN-K163R were incubated in DMEM with 10% FBS
at 37°C with 5% CO2. Cells were treated with various
doses of tubastatin A or celecoxib alone for 24 h, or with a
combination of an HDAC6 inhibitor (tubastatin A or tubacin) and
celecoxib for 24 h.
Reagents and antibodies
Tubastatin A, tubacin and celecoxib were purchased
from Selleck Chemicals (Houston, TX, USA). The anti-PTEN (#9552)
and anti-phospho-AKT (S473; #4058) antibodies were purchased from
the Cell Signaling Technology, Inc. (Danvers, MA, USA), and the
anti-β-actin (I-19) antibody (TA-09) was purchased from ZSGB-BIO
Co. (Beijing, China).
Protein extraction and western
blotting
Whole cell lysates were extracted with RIPA lysis
buffer (Applygen Technologies Inc., Beijing, China). Membrane
proteins were extracted using a Nuclear-Cytosol Extraction kit
(Applygen Technologies Inc.). Protein concentrations were
determined via BCA protein assay (Thermo Fisher Scientific, Inc.,
Waltham, MA USA). Equal amounts of samples were subjected to 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then
transferred to a polyvinylidene fluoride membrane (EMD Millipore,
Billerica, MA, USA). The membrane was blocked with fat-free milk
(5%) in TBS-T (50 mmol/l Tris, pH 7.5; 150 mmol/l NaCl; 0.05%
Tween-20) for 1 h. Following incubation with the primary antibodies
(diluted 1:1,000 in TBS-T) overnight at 4°C, the membrane was
washed extensively with TBS-T three times (5 min/wash) at room
temperature and then incubated with a secondary antibody conjugated
with a fluorophore for 1 h at room temperature. After extensive
washing with TBS-T, the protein bands on the membrane were
visualized with an Odyssey infrared imaging system (Odyssey;
LI-COR, Lincoln, NE, USA).
Cell proliferation assay
The cell proliferation assay was performed using
Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) according to
the manufacturer's instructions. In brief, the cells were seeded
into 96-well plates (1.5×103 cells/well) and treated
with 20 µM celecoxib or 20 µM HDAC6 inhibitor (either tubastatin A
or tubacin), or a combination of 20 µM COX-2 inhibitor and 20 µM
HDAC6 inhibitor for 24 h. Subsequently, 10 µl CCK-8 reagent was
added to each well. Cells were further incubated at 37°C for 3 h,
then the absorbance at 450 nm (OD450) of each well was
measured. Data are presented as the mean ± standard deviation (SD)
of at least three independent experiments.
Analysis of drug interaction in
vitro
The coefficient of drug interaction (CDI) was used
to analyze the synergistic inhibitory effect of the drug
combination. CDI was calculated as follows: CDI = AB/(A × B). AB is
the OD450 ratio of the two-drug combination group to the
control group; A and B are the OD450 ratios of each of
the single-drug groups to the control group. CDI <1 indicates a
synergistic effect, CDI <0.7 indicates a significant synergistic
effect, CDI =1 indicates additivity, and CDI >1 indicates
antagonism (31).
Assessment of cell apoptosis
Cells were washed in phosphate-buffered saline (PBS)
three times, fixed with 10% formaldehyde for 5 min, and then
incubated with 5 mg/ml 4,6-diamidino-2-phenylindole dihydrochloride
(DAPI) in the dark for 5 min at room temperature. Following three
washes in PBS, the cells were examined under a fluorescence
microscope (Nikon Corporation, Tokyo, Japan). Apoptotic cells were
considered to be those that presented features of nuclear
condensation and fragmentation under fluorescence microscopy.
Apoptotic cells were counted within five randomly selected fields,
and the rate of apoptotic cells is presented as the mean ± SD of at
least three independent experiments.
Caspase-3/−7 activities were measured using
Caspase-Glo® 3/7 assay (Promega Corporation, Madison,
WI, USA) according to the manufacturer's instructions. The cells
were seeded into 96-well plates (1.5×103 cells/well) and
treated with 20 µM celecoxib or 20 µM HDAC6 inhibitor (either
tubastatin A or tubacin), or the combination of COX-2 and HDAC6
inhibitors for 24 h. Subsequently, the cells were separately
transferred into a white-walled 96-well plate (100 µl/well).
Caspase-Glo® 3/7 reagent (100 µl; prepared prior to the
start of the assay) was added to each well, which contained 100 µl
blank (culture medium without cells) or treated cells in culture
medium. The plate was gently shaken using a plate shaker at 300 rpm
for 30 sec, incubated at room temperature for up to 3 h, and then
measured for the luminescence of each well on an EnSpire Multimode
Plate Reader with Epic® Label-Free technology
(PerkinElmer, Bridgeville, PA, USA).
Transwell migration and invasion
assays
Cell migration and invasion assays were performed in
Transwell chambers (Corning Costar, Corning, NY, USA) with
polycarbonate membranes. For the migration assay, cells were seeded
into the upper chambers of each well (1×105 cells/well)
in serum-free culture medium, while culture medium containing 10%
FBS was added to the lower chambers. The cells were incubated for
12 h, after which the cells on the top surface of the membrane were
wiped off, and the cells on the bottom surface were fixed with 4%
paraformaldehyde and stained with 0.01% crystal violet. Cells on
the bottom surface of the membrane were examined under a light
microscope, counted and averaged from six randomly selected
fields.
The Transwell invasion assay was performed in the
same manner as the migration assay, except that the upper chambers
were coated with 20 µg extracellular matrix gel (Sigma-Aldrich, St.
Louis, MO, USA) prior to seeding the cells.
Xenograft tumor inoculation
BALB/c nude mice (6 weeks of age) were purchased
from the Beijing Vital River Laboratory Animal Technology Co. Ltd.
(Beijing, China). The care and treatment of experimental animals
followed institutional guidelines. Mice were randomly allocated to
four groups (n=4/group). CAL 27 cells were subcutaneously
inoculated (5×106 cells/mouse) into the left axilla of
each mouse. After 10 days, each group received daily celecoxib (40
mg/kg dissolved in methylcellulose) alone via gavage, or tubastatin
A (0.5 mg/kg dissolved in dimethyl sulfoxide) alone via
intraperitoneal injection, or a combination of celecoxib (40 mg/kg)
and tubastatin A (0.5 mg/kg), or their dissolvents, via gavage or
intraperitoneal injection, for three weeks. The mice were then
euthanized, and the weights of the xenograft tumors were
measured.
Statistical analysis
All statistical examinations were performed using
SPSS 21 for Windows (SPSS, Inc., Chicago, IL, USA). All data are
presented as the mean ± SD. Differences between multiple groups
were analyzed by one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
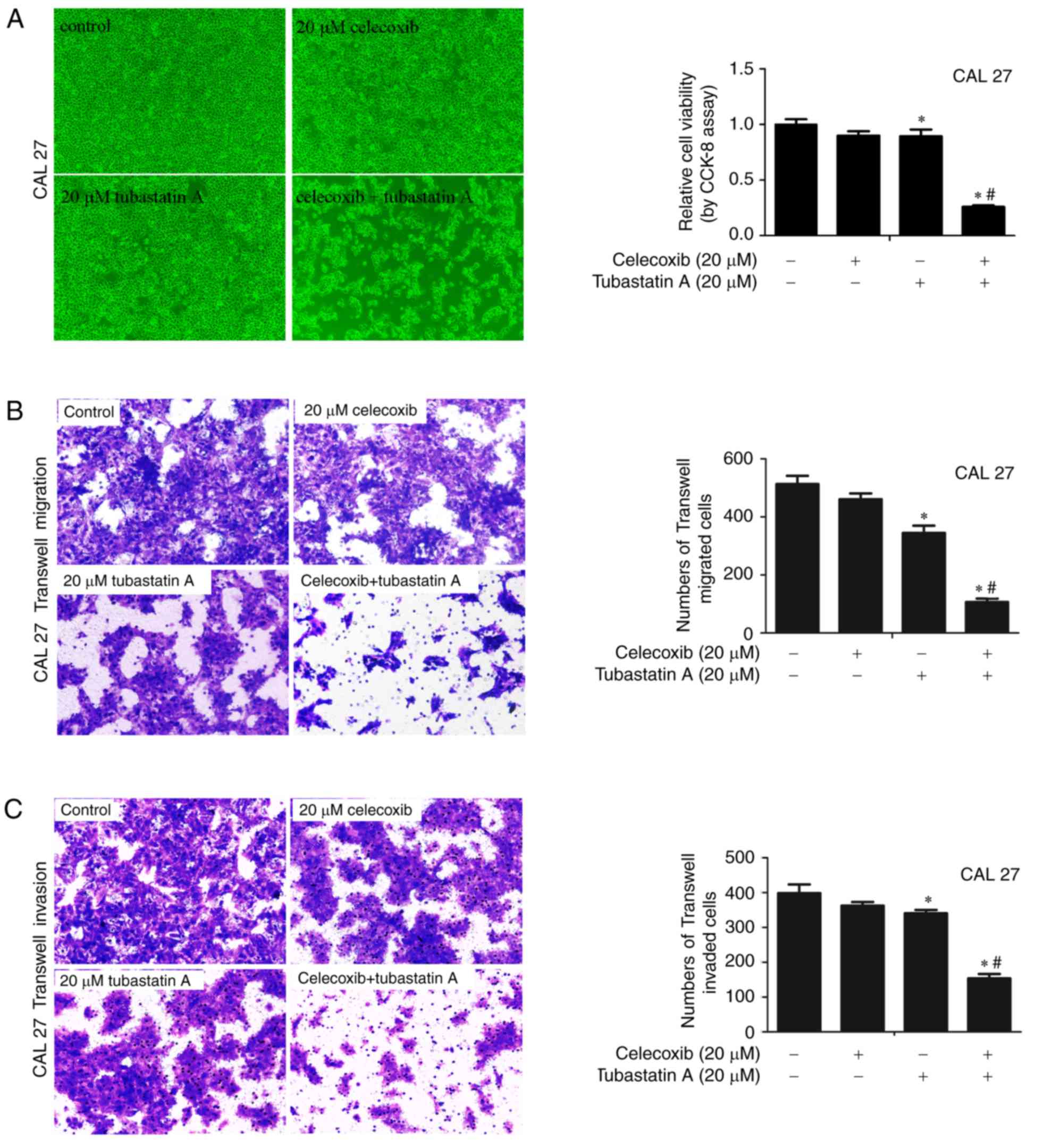
Combined treatment with an HDAC6
inhibitor and a COX-2 inhibitor exerts synergistic effects on the
proliferation, migration and invasion of CAL 27 cells
To examine whether the combination of HDAC6 and
COX-2 inhibitors achieves synergistic effects on cell
proliferation, migration and invasion, CAL 27 cells were treated
with 20 µM celecoxib alone, 20 µM tubastatin A alone, or a
combination of celecoxib and tubastatin A for 24 h. As shown in
Fig. 1A-C, following treatment of
the cells with celecoxib or tubastatin A alone, cell proliferation,
migration and invasion were slightly inhibited. The
combined-treatment group showed synergistic inhibition of
proliferation, and inhibition of cell migration and invasion. The
CDI of the HDAC6 inhibitor tubastatin A combined with the COX-2
inhibitor celecoxib was calculated as 0.32 in CAL 27 cells, which
indicated a significant synergistic effect.
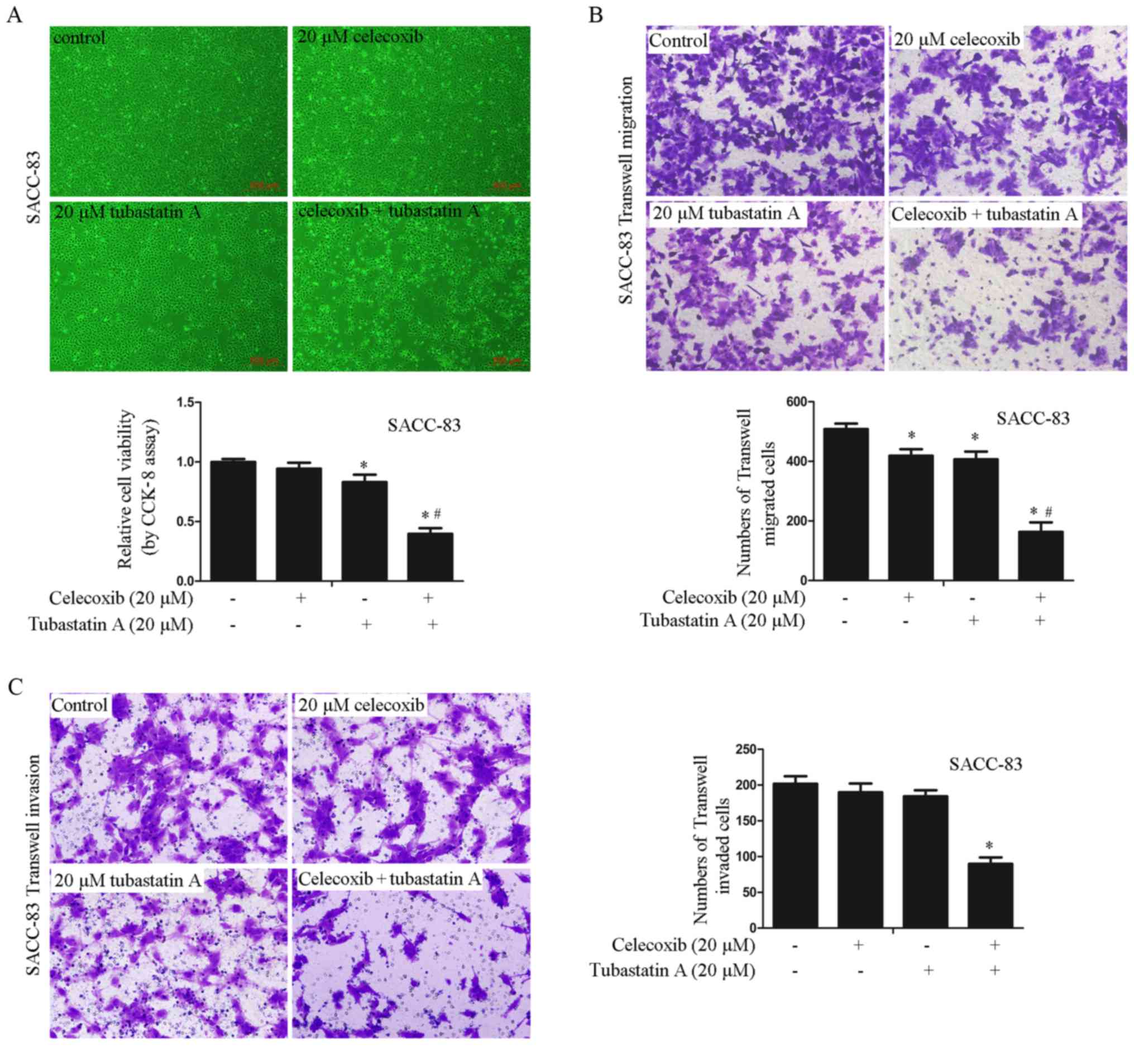
Combined treatment with HDAC6 and
COX-2 inhibitors exerts synergistic effects on the proliferation,
migration and invasion of SACC-83 cells
To examine whether the synergistic effects on cell
proliferation, migration and invasion are achieved by combining an
HDAC6 inhibitor with a COX-2 inhibitor in SACC-83 cells, the cells
were treated with 20 µM celecoxib alone, 20 µM tubastatin A alone,
or a combination of celecoxib and tubastatin A for 24 h. As shown
in Fig. 2A-C, proliferation, cell
migration and invasion were slightly inhibited in the cells treated
with celecoxib or tubastatin A alone whereas the combined-treatment
group showed synergistic inhibition of proliferation, and
inhibition of cell migration and invasion. The CDI of the combined
treatment was calculated as 0.51 in SACC-83 cells, which indicated
a significant synergistic effect.
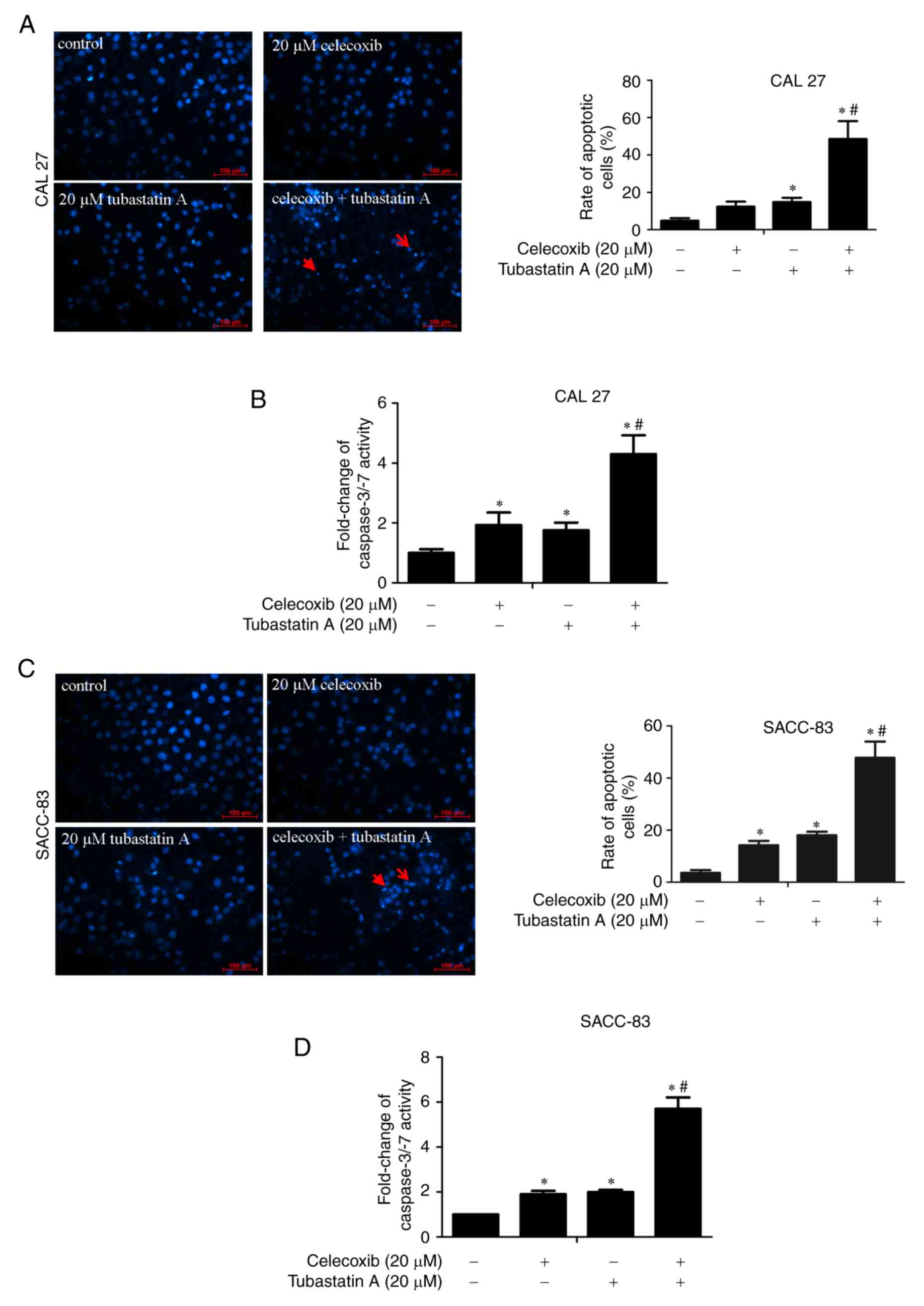
Apoptosis is induced following
treatment with tubastatin A and/or celecoxib
DAPI staining and caspase-3/−7 activity were
measured to detect the induction of apoptosis by tubastatin A
alone, celecoxib alone and the combination of tubastatin A and
celecoxib in CAL 27 and SACC-83 cells. The cells were treated with
20 µM celecoxib alone, 20 µM tubastatin A alone, or a combination
of celecoxib and tubastatin A for 24 h. As shown in Fig. 3A-D, treatment with celecoxib or
tubastatin A alone led to a slight induction of cell apoptosis and
marginal upregulation of caspase-3/−7 activity, whereas the
combined-treatment group showed significant induction of cell
apoptosis and significant upregulation of caspase-3/−7
activity.
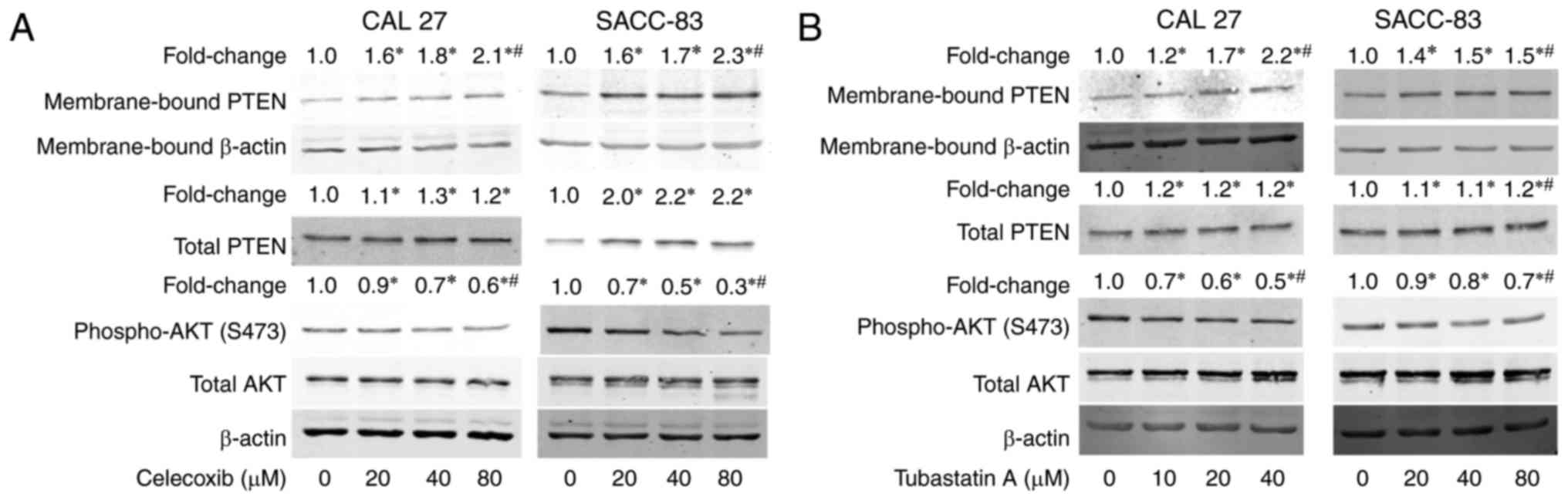
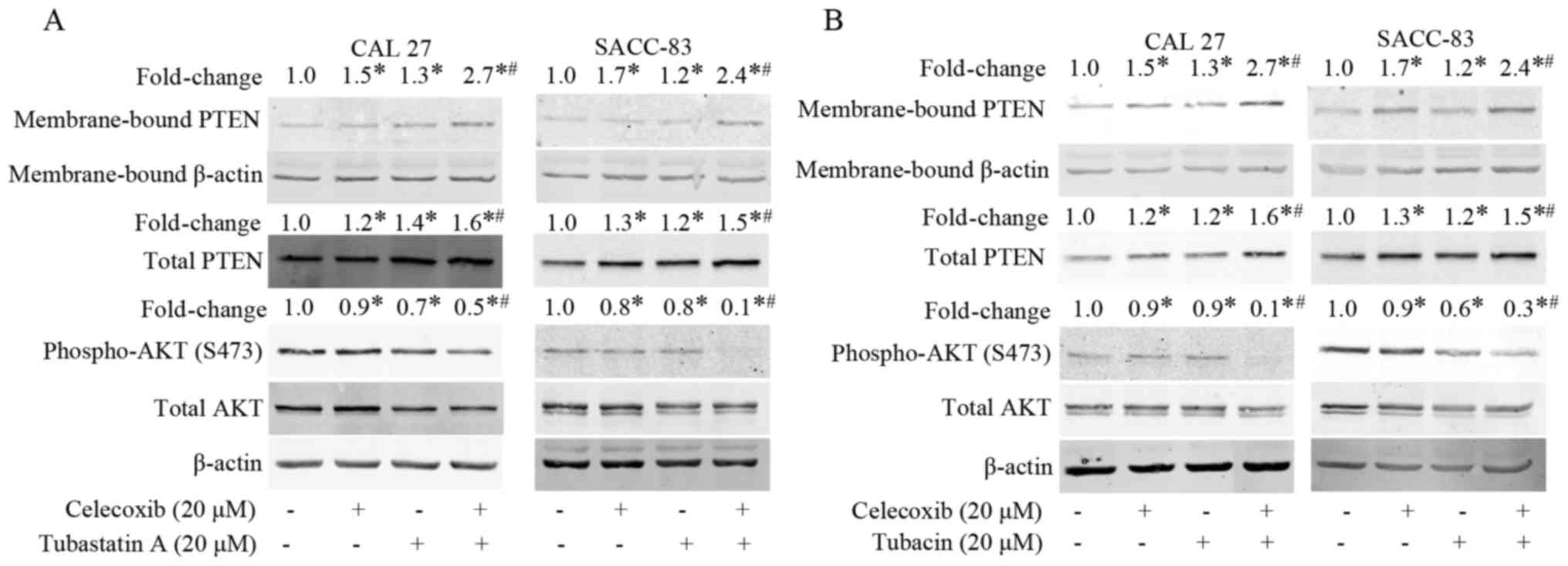
Treatment with celecoxib or tubastatin
A activates PTEN and inactivates AKT
To understand the mechanism underlying the
synergistic antitumor effects of the combination of celecoxib and
tubastatin A, we evaluated the levels of membrane-bound PTEN and
phospho-AKT in the cells after treatment with celecoxib for 24 h.
As shown in Fig. 4A, membrane-bound
PTEN and total PTEN were dose-dependently upregulated, whereas
phosphorylation of AKT was dose-dependently downregulated in the
CAL 27 and SACC-83 cells. Subsequently, the levels of
membrane-bound PTEN and phospho-AKT following treatment with
tubastatin A for 24 h were evaluated. As shown in Fig. 4B, tubastatin A induced the
dose-dependent upregulation of membrane-bound PTEN, but did not
affect total PTEN expression in the CAL 27 and SACC-83 cells.
Correspondingly, phospho-AKT was dose-dependently downregulated by
tubastatin A.
Combined treatment with an HDAC6
inhibitor and COX-2 inhibitor exerts synergistic effects on PTEN
and AKT
Finally, we examined whether the combination of
celecoxib and the HDAC6 inhibitor tubastatin A affects the
activation of PTEN and inactivation of AKT in the CAL 27 and
SACC-83 cells. As shown in Fig. 5A and
B, celecoxib or tubastatin A treatments alone slightly
upregulated membrane-bound PTEN and downregulated phospho-AKT,
whereas the combination of celecoxib and tubastatin A
synergistically upregulated membrane-bound PTEN and correspondingly
downregulated phospho-AKT. Similar results were observed when the
cells were treated with combined celecoxib and tubacin (another
HDAC6 inhibitor). These results suggest that the synergistic
antitumor effects achieved by the combination of an HDAC6 inhibitor
and a COX-2 inhibitor may be due, at least partially, to the
activation of PTEN and inactivation AKT.
Combined treatment with celecoxib and
tubastatin A does not induce synergistic inhibition of
proliferation in PTEN-deficient U-87 MG cells
To further examine whether the PTEN/AKT signaling
pathway is required for the synergistic antitumor effects of
celecoxib and tubastatin A, PTEN-deficient U-87 MG cells were
treated with 20 µM celecoxib alone, or 20 µM tubastatin A alone or
the two combined. As shown in Fig. 6A
and B, the combined treatment did not significantly affect
proliferation (CDI=1.0). Similarly, the level of phospho-AKT in the
combined-treatment group was not different from the level noted in
the celecoxib alone, tubastatin A alone, or control groups
(Fig. 6C).
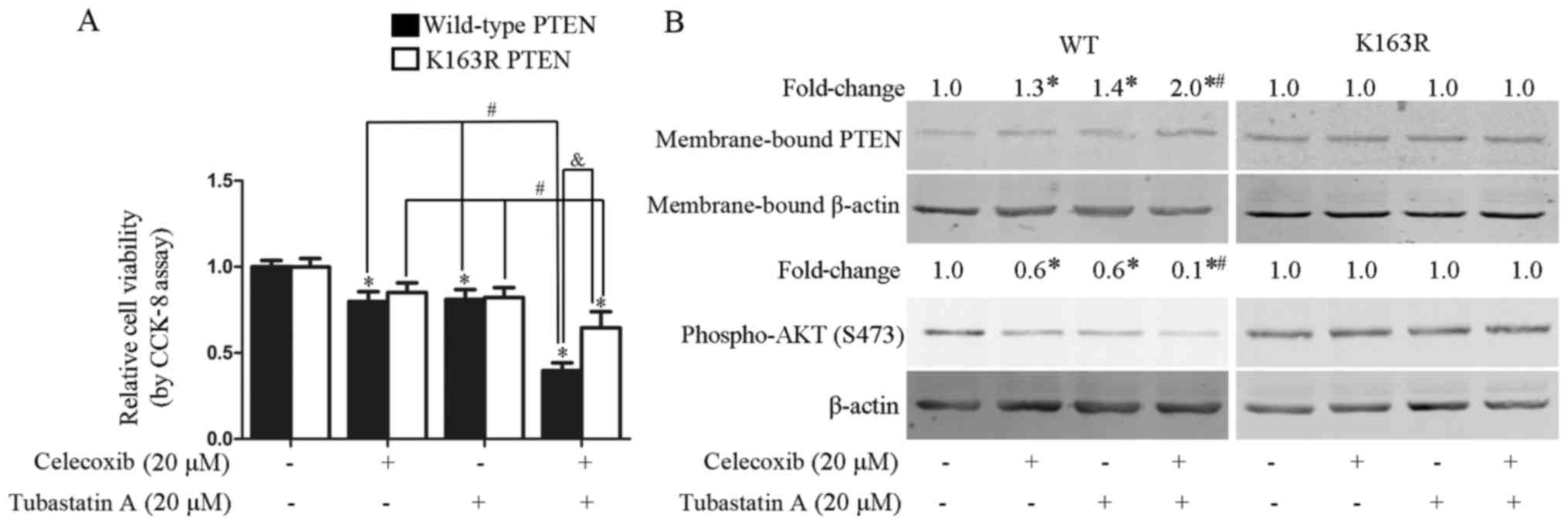
Combined treatment with celecoxib and
tubastatin A synergistically inhibits the proliferation of U-87 MG
cells stably transfected with wild-type PTEN, but not with mutant
PTEN-K163R
In our previous study, inhibition of HDAC6 was shown
to activate PTEN via PTEN acetylation at K163 (17). To examine whether the synergistic
antitumor effects of the combined treatment with tubastatin A and
celecoxib were dependent on PTEN acetylation at K163, U-87 MG cells
were stably transfected with wild-type PTEN and mutant PTEN-K163R
(with lysine replaced by arginine), and subsequently treated for 24
h with celecoxib alone, tubastatin A alone, or the two agents
combined. As shown in Fig. 7A,
synergistic inhibition of proliferation was achieved by the
combination of tubastatin A and celecoxib in the U-87 MG cells
stably transfected with wild-type PTEN (CDI=0.6), but not in the
cells stably transfected with the K163R mutant form of PTEN
(CDI=1.0). Correspondingly, the synergistic upregulation of
membrane-bound PTEN and downregulation of phospho-AKT was observed
in the U-87 MG cells stably transfected with wild-type PTEN, but
not the K163R mutant (Fig. 7B).
Similar findings were observed with individual celecoxib or
tubastatin A treatments, which slightly upregulated membrane-bound
PTEN and downregulated phospho-AKT in the cells transfected with
wild-type PTEN, but not in those transfected with mutant
PTEN-K163R.
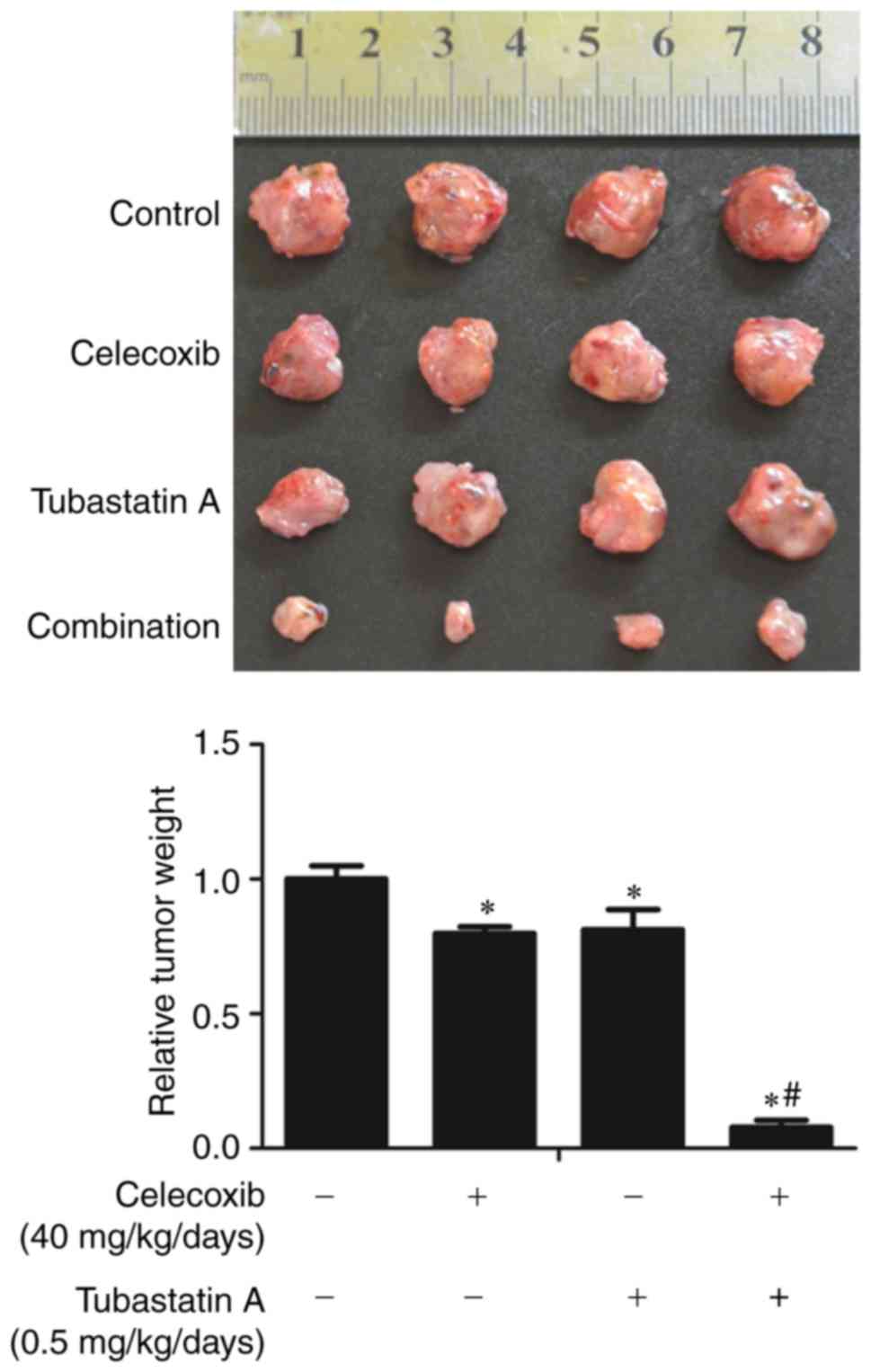
Combined treatment with inhibitor
tubastatin A and celecoxib synergistically inhibits xenograft tumor
growth
To further confirm the synergistic antitumor effects
of the HDAC6 inhibitor tubastatin A and the COX-2 inhibitor
celecoxib in vivo, nude mice were inoculated with CAL 27
cells for 10 days, and the mice were then treated for three weeks
with celecoxib and/or tubastatin A, or with the carriers as a
control. As shown in Fig. 8, the
weights of the xenograft tumors were significantly lower in the
combined-treatment group than that noted in the other groups.
Discussion
In the present study, we demonstrated that combining
an HDAC6 inhibitor with a COX-2 inhibitor produced synergistic
antitumor effects. Our observations suggest a potential novel
clinical strategy using HDAC6 inhibitors in combination with COX-2
inhibitors for antitumor therapy.
The combined treatment resulted in synergistic
antitumor effects by activation of PTEN and inactivation of AKT.
The combination of the HDAC6 inhibitor tubastatin A and the COX-2
inhibitor celecoxib induced apoptosis and inhibited proliferation,
migration and invasion to a greater extent than either of the
treatments alone in CAL 27 and SACC-83 cells. The synergistic
antitumor effects achieved by the combined treatment were further
confirmed in nude mice with CAL 27 tumor xenografts. The
combined-treatment group showed significantly decreased xenograft
tumor weights than each of the single-treatment groups. The
combination of tubastatin A and celecoxib achieved these affects by
activating PTEN and inactivating AKT, which was demonstrated by the
significant upregulation of membrane-bound PTEN (the activated
form), and the corresponding significant downregulation of
phospho-AKT, in the combined-treatment group compared with either
of the single-treatment groups. Moreover, the combination of
tubastatin A and celecoxib failed to produce synergistic inhibition
of proliferation in PTEN-deficient U-87 MG cells, whereas the
synergistic inhibition of proliferation was rescued in U-87 MG
cells that had been stably transfected with wild-type PTEN, but not
those transfected with mutant PTEN-K163R. In our previous study, it
was demonstrated that wild-type PTEN could be acetylated and
activated by a HDAC6 inhibitor, whereas the acetylation and
activation of the mutant PTEN-K163R could not be induced by an
HDAC6 inhibitor (17).
Consistently, the levels of membrane-bound PTEN and phospho-AKT in
the combined-treatment group were significantly upregulated and
downregulated, respectively, in the U-87 MG cells stably
transfected with wild-type PTEN, but not the mutant PTEN-K163R.
Therefore, these results suggest that PTEN is responsible for the
synergistic antitumor effects achieved by combination treatment
with HDAC6 and COX-2 inhibitors.
To the best of our knowledge, these results
demonstrated for the first time that COX-2 inhibitors are important
agents for enhancing the antitumor effects of HDAC6 inhibitors.
Therefore, these results provide the rationale for the use of
combined HDAC6 inhibitors and COX-2 inhibitors in cancer
chemotherapy. However, it should be noted that the combination of
HDAC6 inhibitors and COX-2 inhibitors may be better applied in
tumors that do not harbor aberrant or deleted PTEN. Otherwise, the
use of this combination may fail to generate synergistic antitumor
effects.
It was previously demonstrated that the upregulation
of PTEN protein expression by COX-2 inhibitors was partially
achieved through the downregulation of Sp1, whereas the
upregulation of membrane-bound PTEN by COX-2 inhibitors is due to
inhibition of phosphorylation (S380/T382/T383) in the PTEN
C-terminal tail, as well as the upregulation of PTEN protein
expression (22). Sp1 is an
important negative regulator of PTEN that acts by recruiting HDAC1
to the core promoter of PTEN. Therefore, knockdown of Sp1
upregulates PTEN mRNA and protein expression (32). Moreover, COX-2 inhibitors
downregulate Sp1 protein expression by promoting the degradation of
Sp1 (33). These previous studies
suggest that the underlying mechanism of COX-2 inhibitor-induced
upregulation of membrane-bound PTEN relies partially on the
downregulation of Sp1, and partially on reduced PTEN
phosphorylation. Correspondingly, the present study found that
celecoxib upregulated total PTEN and slightly upregulated the
membrane-bound PTEN in CAL 27 and SACC-83 cells. The upregulation
of membrane-bound PTEN in U-87 MG cells stably transfected with
wild-type PTEN may be due to the downregulation of PTEN
phosphorylation, as the total exogenous PTEN was not affected by
the COX-2 inhibitor. However, surprisingly, celecoxib did not
affect membrane-bound PTEN in U-87 MG cells stably transfected with
mutant PTEN-K163R. Currently, we do not have an explanation for
this phenomenon. We speculate that abolishing acetylation at K163
may somehow interfere with the regulation of PTEN membrane
translocation by phosphorylation at the C-terminal tail. Further
study is required to test this hypothesis.
In conclusion, the present study demonstrated that
the combination of an HDAC6 inhibitor and a COX-2 inhibitor
produced a synergistic antitumor effects through activation of PTEN
and inactivation of AKT. Application of this combination may be
suitable for the treatment of tumors without PTEN mutations or
deletions.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81472764).
References
|
1
|
Liu T, Liu X and Li W: Tetrandrine, a
Chinese plant-derived alkaloid, is a potential candidate for cancer
chemotherapy. Oncotarget. 7:40800–40815. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Volm M and Efferth T: Prediction of Cancer
Drug Resistance and Implications for Personalized Medicine. Front
Oncol. 5:2822015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Damaskos C, Karatzas T, Nikolidakis L,
Kostakis ID, Karamaroudis S, Boutsikos G, Damaskou Z, Kostakis A
and Kouraklis G: Histone deacetylase (HDAC) inhibitors: Current
evidence for therapeutic activities in pancreatic cancer.
Anticancer Res. 35:3129–3135. 2015.PubMed/NCBI
|
|
4
|
Bukowska B, Gajek A and Marczak A: Two
drugs are better than one. A short history of combined therapy of
ovarian cancer. Contemp Oncol. 19:350–353. 2015.
|
|
5
|
Drazic A, Myklebust LM, Ree R and Arnesen
T: The world of protein acetylation. Biochim Biophys Acta.
1864:1372–1401. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang C, Zhong JF, Stucky A, Chen XL,
Press MF and Zhang X: Histone acetylation: novel target for the
treatment of acute lymphoblastic leukemia. Clin Epigenetics.
7:1172015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mathias RA, Guise AJ and Cristea IM:
Post-translational modifications regulate class IIa histone
deacetylase (HDAC) function in health and disease. Mol Cell
Proteomics. 14:456–470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang H, Shang YP, Chen HY and Li J:
Histone deacetylases function as novel potential therapeutic
targets for cancer. Hepatol Res. Jul 26–2016.(Epub ahead of print).
doi: 10.1111/hepr.12757.
|
|
9
|
Gan YH and Zhang S: PTEN/AKT pathway
involved in histone deacetylases inhibitor induced cell growth
inhibition and apoptosis of oral squamous cell carcinoma cells.
Oral Oncol. 45:e150–e154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang WJ, Liang YC, Chuang SE, Chi LL, Lee
CY, Lin CW, Chen AL, Huang JS, Chiu CJ, Lee CF, et al: NBM-HD-1: A
novel histone deacetylase inhibitor with anticancer activity. Evid
Based Complement Alternat Med. 2012:7814172012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Manal M, Chandrasekar MJ, Priya J Gomathi
and Nanjan MJ: Inhibitors of histone deacetylase as antitumor
agents: A critical review. Bioorg Chem. 67:18–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ossenkoppele GJ, Lowenberg B, Zachee P,
Vey N, Breems D, Van de Loosdrecht AA, Davidson AH, Wells G,
Needham L, Bawden L, et al: A phase I first-in-human study with
tefinostat - a monocyte/macrophage targeted histone deacetylase
inhibitor - in patients with advanced haematological malignancies.
Br J Haematol. 162:191–201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Witt O, Deubzer HE, Milde T and Oehme I:
HDAC family: What are the cancer relevant targets? Cancer Lett.
277:8–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Kwon S, Yamaguchi T, Cubizolles
F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, et al:
Mice lacking histone deacetylase 6 have hyperacetylated tubulin but
are viable and develop normally. Mol Cell Biol. 28:1688–1701. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McConkey DJ, White M and Yan W: HDAC
inhibitor modulation of proteotoxicity as a therapeutic approach in
cancer. Adv Cancer Res. 116:131–163. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saji S, Kawakami M, Hayashi S, Yoshida N,
Hirose M, Horiguchi S, Itoh A, Funata N, Schreiber SL, Yoshida M,
et al: Significance of HDAC6 regulation via estrogen signaling for
cell motility and prognosis in estrogen receptor-positive breast
cancer. Oncogene. 24:4531–4539. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng Z, Jia LF and Gan YH: PTEN activation
through K163 acetylation by inhibiting HDAC6 contributes to tumour
inhibition. Oncogene. 35:2333–2344. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gu J, Wang D, Zhang J, Zhu Y, Li Y, Chen
H, Shi M, Wang X, Shen B, Deng X, et al: GFRα2 prompts cell growth
and chemoresistance through down-regulating tumor suppressor gene
PTEN via Mir-17-5p in pancreatic cancer. Cancer Lett. 380:434–441.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Milella M, Falcone I, Conciatori F, Incani
U Cesta, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S,
Cognetti F, et al: PTEN: Multiple functions in human malignant
tumors. Front Oncol. 5:242015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tesio M, Trinquand A, Macintyre E and
Asnafi V: Oncogenic PTEN functions and models in T-cell
malignancies. Oncogene. 35:3887–3896. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vazquez F, Matsuoka S, Sellers WR,
Yanagida T, Ueda M and Devreotes PN: Tumor suppressor PTEN acts
through dynamic interaction with the plasma membrane. Proc Natl
Acad Sci USA. 103:pp. 3633–3638. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meng Z and Gan YH: Activating PTEN by
COX-2 inhibitors antagonizes radiation-induced AKT activation
contributing to radiosensitization. Biochem Biophys Res Commun.
460:198–204. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ke J, Yang Y, Che Q, Jiang F, Wang H, Chen
Z, Zhu M, Tong H, Zhang H, Yan X, et al: Prostaglandin E2 (PGE2)
promotes proliferation and invasion by enhancing SUMO-1 activity
via EP4 receptor in endometrial cancer. Tumour Biol.
37:12203–12211. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gan L, Qiu Z, Huang J, Li Y, Huang H,
Xiang T, Wan J, Hui T, Lin Y, Li H, et al: Cyclooxygenase-2 in
tumor-associated macrophages promotes metastatic potential of
breast cancer cells through Akt pathway. Int J Biol Sci.
12:1533–1543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Salehifar E and Hosseinimehr SJ: The use
of cyclooxygenase-2 inhibitors for improvement of efficacy of
radiotherapy in cancers. Drug Discov Today. 21:654–662. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vosooghi M and Amini M: The discovery and
development of cyclooxygenase-2 inhibitors as potential anticancer
therapies. Expert Opin Drug Discov. 9:255–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu R, Xu KP and Tan GS: Cyclooxygenase-2
inhibitors in lung cancer treatment: Bench to bed. Eur J Pharmacol.
769:127–133. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang X, Li G, Wang A, Zhang Z, Merchan JR
and Halmos B: Combined histone deacetylase and cyclooxygenase
inhibition achieves enhanced antiangiogenic effects in lung cancer
cells. Mol Carcinog. 52:218–228. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gioanni J, Fischel JL, Lambert JC, Demard
F, Mazeau C, Zanghellini E, Ettore F, Formento P, Chauvel P,
Lalanne CM, et al: Two new human tumor cell lines derived from
squamous cell carcinomas of the tongue: Establishment,
characterization and response to cytotoxic treatment. Eur J Cancer
Clin Oncol. 24:1445–1455. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li SL: Establishment of a human cancer
cell line from adenoid cystic carcinoma of the minor salivary
gland. Zhonghua Kou Qiang Yi Xue Za Zhi. 25(29-31): 621990.(In
Chinese).
|
|
31
|
Wu Y, Zhu Y, Li S, Zeng M, Chu J, Hu P, Li
J, Guo Q, Lv XB and Huang G: Terrein performs antitumor functions
on esophageal cancer cells by inhibiting cell proliferation and
synergistic interaction with cisplatin. Oncol Lett. 13:2805–2810.
2017.PubMed/NCBI
|
|
32
|
Kou XX, Hao T, Meng Z, Zhou YH and Gan YH:
Acetylated Sp1 inhibits PTEN expression through binding to PTEN
core promoter and recruitment of HDAC1 and promotes cancer cell
migration and invasion. Carcinogenesis. 34:58–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Abdelrahim M and Safe S: Cyclooxygenase-2
inhibitors decrease vascular endothelial growth factor expression
in colon cancer cells by enhanced degradation of Sp1 and Sp4
proteins. Mol Pharmacol. 68:317–329. 2005.PubMed/NCBI
|