Introduction
Breast cancer is the second most common cause of
cancer-related deaths in women (1).
It is estimated that ~1.3 million females develop breast cancer
each year, with ~465,000 expected to succumb to the disease
(2,3). Not all patients derive therapeutic
benefit from treatment, and some breast cancer patients are
resistant to treatment.
Cisplatin, a chemotherapeutic agent, is used for
treating various malignancies, including lung, ovary and breast
cancer. Mechanistic studies indicated that cisplatin covalently
binds to the N-7 atoms of purines on DNA to form DNA adducts, which
distort DNA conformation and it also inhibits replication and
transcription, leading to cell cycle arrest and apoptosis through
the activation of multiple signaling pathways (4). Endoplasmic reticulum (ER) stress is
involved in cisplatin-induced apoptosis in human lung cancer cells
(5). However, cisplatin exhibits
limited therapeutic efficacy (6).
Therefore, more effective therapeutic options are required.
There is an established close association between
autophagy and cancer, where it has an important role in cell
survival and death (7,8). Autophagy is strongly activated by
various stress conditions, including cancer chemotherapy and
radiotherapy (9). Certain cytotoxic
drugs induce protective autophagy, impairing rather than enhancing
the action of the drug (10,11).
Recent studies have reported that cisplatin treatment activated
autophagy, which served as a survival factor to counter
cisplatin-induced apoptosis in other cancer cells (12). Mechanically, autophagy activation
was tightly regulated by mitogen-activated protein kinases (MAPKs),
including extracellular signal-related kinase 1/2 (ERK1/2), p38 and
c-jun N-terminal kinase (JNK) (13–15).
To the best of our knowledge, there has been no further
investigation of the underlying mechanisms between cisplatin and
autophagy in breast cancer cells.
The transcriptional co-activator, yes-associated
protein (YAP), is an important gene for cell proliferation,
metastasis, chemoresistance and other malignant properties
(16–18). In breast cancer, the relationship
between autophagy and YAP remains controversial. Recent studies,
using chemical inhibitors or gene silencing, have reported that
autophagy inhibition increased the phosphorylation rate of YAP,
promoting the proliferation and invasion of cancer cells (19,20).
Meanwhile, YAP enhanced autophagy in response to nutrient
deprivation (21). Connecting these
phenomena, in the present study, we focused on the communication
between YAP and cisplatin-induced autophagy.
Materials and methods
Cell lines and culture
The human breast cancer (BC) cell lines MCF-7 and
MDA-MB-231 cells were obtained from the American Type Culture
Collection (ATCC; Rockville, MD, USA). MCF-7 and MDA-MB-231 were
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA),
penicillin (100 U/ml), and streptomycin (100 µg/ml) at 37°C with 5%
CO2.
Drugs and reagents
Cisplatin and hydroxychloroquine (HCQ) were
purchased from Aladdin (Shanghai, China) and Selleck Chemicals
(Houston, TX, USA), respectively. The MAPK inhibitor PD98059 and
the JNK inhibitor SP600125 were purchased from Beyotime
Biotechnology (Shanghai, China). The p38 inhibitor PD169316 was
purchased from MedChem Express (Monmouth Junction, NJ, USA).
Hoechst 33342 was purchased from Beijing Solarbio Science &
Technology (Beijing, China). Cisplatin and HCQ were diluted in
ultrapure water. Antibodies against β-actin and YAP were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Antibodies against LC3, p62 and pJNK-Thr183/Tyr185 were purchased
from the Novus Biologicals (Littleton, CO, USA), Abcam (Cambridge,
MA, USA) and Bioworld Technology (St. Louis Park, MN, USA),
respectively. Antibodies against p-YAP, caspase 3, p-p38, p38,
pERK-Thr202/Tyr204, ERK and JNK1/2 were purchased from Cell
Signaling Technology (Danvers, MA, USA). The horseradish peroxidase
(HRP)-conjugated goat anti-mouse, anti-rabbit IgG and
FITC-conjugated anti-rabbit IgG secondary antibodies were purchased
from Beyotime Biotechnology.
Western blot assay
Human BC cells were lysed in RIPA lysis buffer
containing 1 mM phenylmethylsulfonyl fluoride (PMSF) (both from
Beyotime Biotechnology) as a protease inhibitor. Protein
concentrations were assessed using bicinchoninic acid (BCA) protein
assay reagent kit (Beyotime Biotechnology). A total of 40 µg of
cell lysate was separated by 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then
transferred onto polyvinylidene difluoride (PVDF) membranes. The
membranes were blocked with 5% non-fat dry milk in Tris-buffered
saline with 0.05% Tween-20 (TBST) for 1 h followed by incubation at
4°C overnight with the primary antibodies. HRP anti-mouse and
anti-rabbit IgG (Beyotime Biotechnology) were used as the secondary
antibodies. Immunoreactive bands were detected by ECL
chemiluminescent substrate (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The grey value of each band was calculated using
ImageJ 1.50i (National Institute of Health, Bethesda, MD, USA) and
normalized to the internal control β-actin.
Hoechst 33342 staining
Human BC cells were fixed for 10 min in 4%
paraformaldehyde after being treated. The fixed cells were
incubated in 0.25% TritonX-100 for 10 min, then incubated in
Hoechst 33342 for 10 min. After being washed three times with
phosphate-buffered saline (PBS), the cells were mounted with
antifade reagent. Apoptotic cells were identified by the
condensation and fragmentation of their nuclei, and were
photographed using an Eclipse 80i microscope (Nikon, Tokyo,
Japan).
Immunofluorescence microscopy
Human BC cells were fixed for 20 min in 4%
paraformaldehyde after being treated. The fixed cells were
incubated in 0.25% Triton X-100 for 10 min, and then blocked for 1
h in 5% goat serum. The cells were stained with anti-LC3 antibodies
(1:50) overnight at 4°C. After being washed three times with PBS,
the cells were stained with appropriate secondary antibodies
(1:200) for 1 h at room temperature. After being washed three times
with PBS, the cells were incubated with
4,6-diamidino-2-phenylindole (DAPI) for 10 min at room temperature.
Finally, after being washed another three times with PBS, the cells
were mounted with antifade reagent. Fluorescence images were
collected using an Eclipse 80i microscope.
Annexin V-FITC/PI double staining
BC cells were detached using EDTA-free trypsin and
re-suspended in growth medium. After centrifugation, the cell
precipitates were washed with cold PBS three times and resuspended
in 1 ml PBS. To determine the rate of apoptosis, the cells were
stained with 5 µl propidium iodide (PI) and 5 µl Annexin V-FITC for
15 min in the dark. Samples were then analyzed by flow cytometry on
a FACScan flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA).
Transmission electron microscopy
For observation under transmission electron
microscope (TEM), the cells were first washed with PBS and pelleted
by centrifugation before fixing the cell pellet in 2.5% glutaric
dialdehyde and 1% osmic acid for 2 h. After washing, the pellet was
dehydrated and embedded in epoxy resin. Ultrathin sections (45 nm)
were stained with lead acetate, and the stained sections were
observed using a transmission electron microscope (H-7500; Hitachi,
Tokyo, Japan).
Cell transfection and infection
MCF-7 and MDA-MB-231 cells were plated into 6-well
plates at 2×105 cells/well. Lentivirus-based short
hairpin RNA (shRNA) vector and lentivirus-based cDNA targeting YAP
were transfected into the cells according to the manufacturer's
protocol. Cells were selected with puromycin for 10 days at 37°C.
The effectiveness of transfection was ascertained by western
blotting. The lentivirus-based shRNA vector and lentivirus-based
cDNA targeting YAP were purchased from GenePharma (Shanghai,
China). The sequence for YAP shRNA was as follows: YAP,
5′-CCGGGCCACCAAGCTAGATAAAGAACTCGAGTTCTTTATCTAGCTTGGTGGCTTTTTG-3′.
The control shRNA was synthesized using a scrambled sequence.
Statistical analysis
Statistical analysis was performed using SPSS for
Windows (version 17.0) (SPSS, Inc., Chicago, IL, USA). Student's
t-test was used to compare groups. All quantitative results are
displayed as the average value ± the standard deviation. P-values
of <0.05, 0.01 or 0.001 were considered to indicate a
statistically significant result.
Results
Cisplatin induces apoptosis in BC
cells
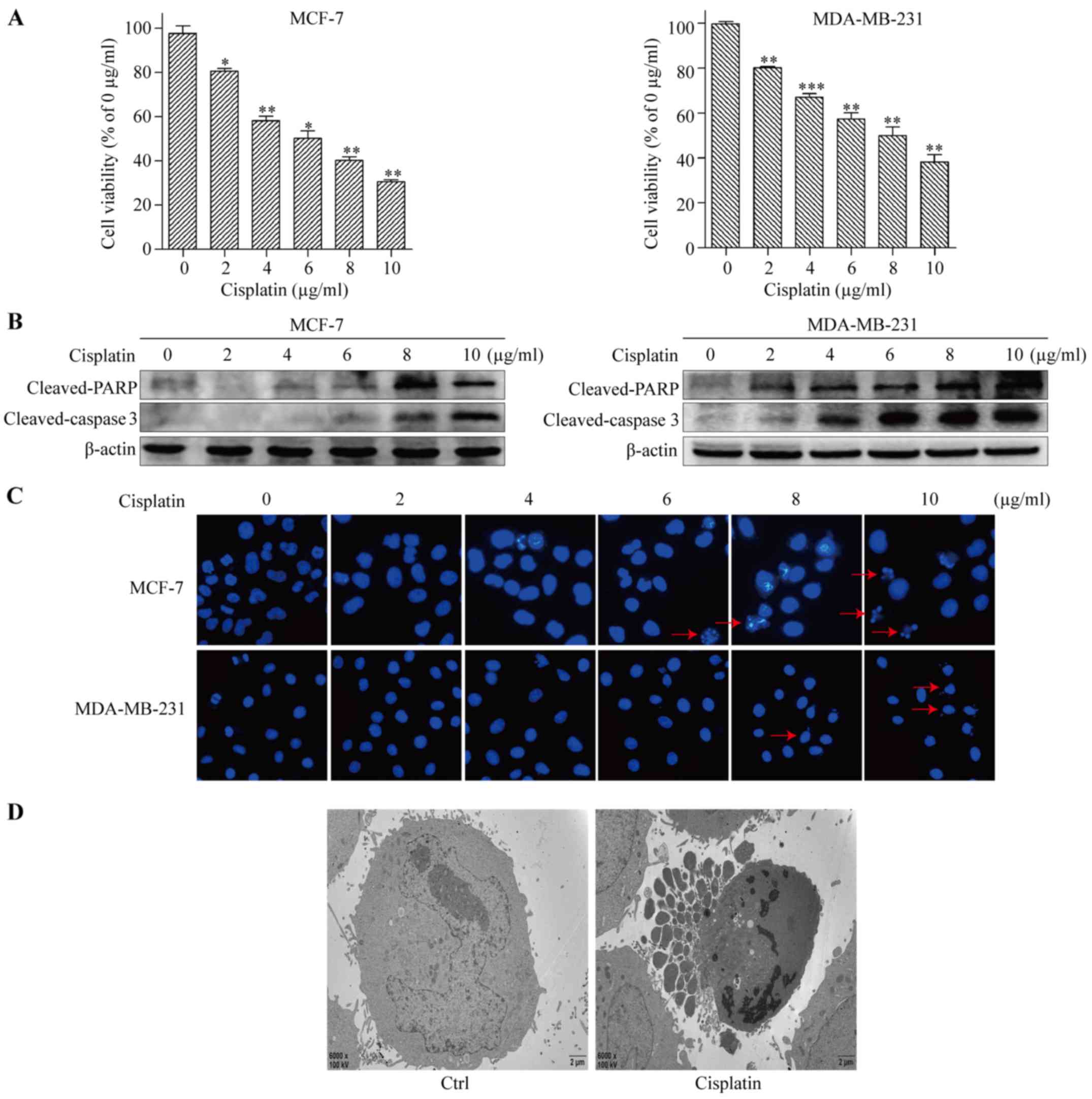
To test the cytotoxic effect of cisplatin on BC
cells, the human BC cell lines MCF-7 and MDA-MB-231 were treated
with 0, 2, 4, 6, 8 and 10 µg/ml of cisplatin for 48 h. Cell
viability was assessed by Cell Counting Kit-8 (CCK-8) cytotoxicity
assay. The results revealed that cisplatin-mediated cytotoxicity
was dose-dependent, and the half maximal inhibitory concentrations
(IC50) of cisplatin for MCF-7 and MDA-MB-231 were around
6 and 8 µg/ml, respectively (Fig.
1A). Therefore, cisplatin was applied at 6 and 8 µg/ml in
subsequent experiments in MCF-7 and MDA-MB-231 cells, respectively.
Next, apoptosis was detected by western blot analysis. The levels
of cleaved-caspase 3 and cleaved-PARP increased in a dose-dependent
manner (Fig. 1B). Compared with the
control, the cells exposed to cisplatin had increased
dose-dependent apoptosis, with the typical apoptosis morphological
changes of condensed nuclear chromatin detected by Hoechst 33342
staining (Fig. 1C). Apoptosis was
further evaluated using transmission electron microscopy. We
observed cytoplasmic vacuolation, mitochondrion swelling, chromatin
condensation, and apoptotic body formation in cisplatin-treated
MCF-7 cells (Fig. 1D). These
results clearly indicated that cisplatin significantly triggered
apoptosis in BC cells.
Cisplatin induces protective autophagy
in BC cells
Cisplatin is known to induce autophagy in cancer
cells (22). To investigate whether
cisplatin induced autophagy in BC cells, we examined autophagy
markers LC3 and p62 by immunoblotting. Compared with that of the
control, the expression levels of LC3-II increased, and the
expression levels of p62 decreased in a dose-dependent manner in
MCF-7 and MDA-MB-231 cells after 48 h of cisplatin treatment
(Fig. 2A). To further confirm the
role of cisplatin in inducing autophagy, fluorescence microscopy
was used to examine the localization of LC3 punctate. Compared with
that of the control, we observed increased specific punctate
distribution of endogenous LC3 as punctate dots of green
fluorescence after 48 h of cisplatin treatment in both BC cell
lines (Fig. 2B). In addition, we
examined autophagy using TEM. The control MCF-7 cells had obvious
nuclei, and numerous mitochondria. In the cisplatin-treated MCF-7
cells, we observed several clearly visible double-membrane
structures resembling autophagosomes, including non-degradable
organelles and macromolecules could be observed (Fig. 2C). Collectively, the aforementioned
results suggest that cisplatin can induce autophagy in BC cells. To
further clarify the function of autophagy as a pro-death or
pro-survival mechanism in cisplatin-induced apoptosis, we used the
autophagy inhibitor HCQ to suppress the autophagy induced by
cisplatin treatment. Immunoblotting assay was used to analyze the
activation of cleaved-caspase 3 during the induction of apoptosis.
When cells were exposed to cisplatin combined with HCQ, we observed
significantly enhanced apoptosis-related changes (Fig. 2D). We then used flow cytometric
analysis to confirm the role of autophagy induced by cisplatin
treatment in BC cells. Cisplatin combined with HCQ treatment
increased the apoptosis rate from 46.8 to 67.0%, and from 52.9 to
74.9% in MCF-7 and MDA-MB-231 cells, respectively (Fig. 2E). Collectively, these results
indicate that inhibition of autophagy enhanced cisplatin-induced
apoptosis in BC cells.
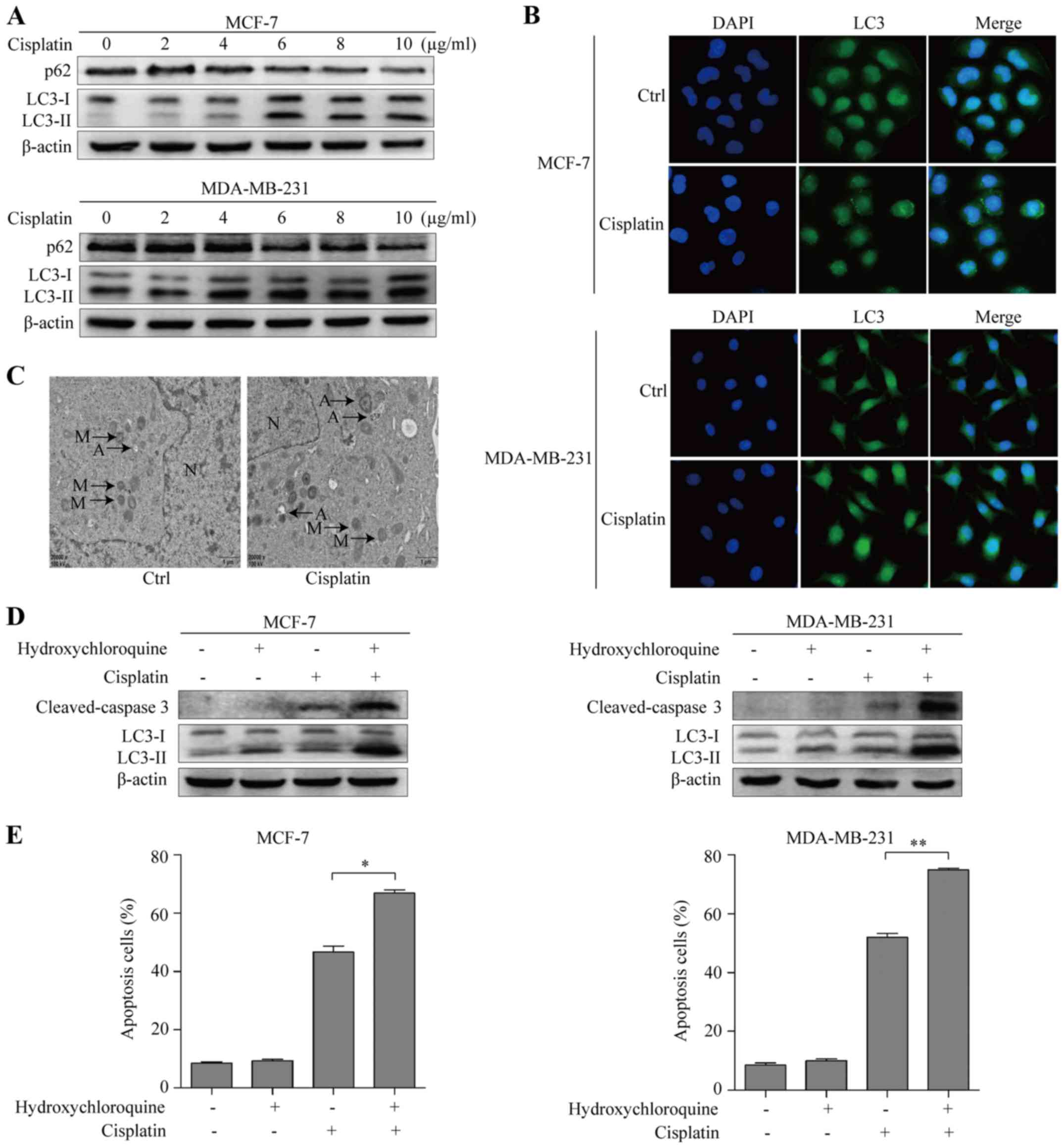 | Figure 2.Cisplatin induces protective autophagy
in breast cancer cells. (A) Western blotting examined autophagy
markers p62/SQSTM1, LC3-I and LC3-II with different concentrations
of cisplatin treatment in MCF-7 and MDA-MB-231 cells. (B)
Immunfluorescence assay detected LC3 puncta (green) of untreated
(control), and cisplatin-treated MCF-7 and MDA-MB-231 cells
(original magnification, ×400). (C) Transmission electron
microscopy was performed to observe autophagosome formation after
cisplatin treatment in MCF-7 cells. A, autophagosome; M,
mitochondria; N, nucleus. (D) Western blot assay examined apoptotic
marker cleaved-caspase 3 of untreated (control), treated with
hydroxychloroquine, cisplatin and both these agents in MCF-7 and
MDA-MB-231 cells. (E) Quantification of apoptotic cells in
untreated (control), treated with autophagy inhibitor
hydroxychloroquine, cisplatin and both these agents using Annexin
V-FITC/PI double staining in MCF-7 and MDA-MB-231 cells;
*P<0.05, **P<0.01. Data are presented as the mean ± SD of at
least three independent experiments. |
Cisplatin promotes autophagy through
activation of the ERK signaling pathway
Accumulating evidence indicates that a role for all
three MAPK subfamilies in autophagy (23). To investigate whether MAPKs
contributed to cisplatin-induced autophagy in BC cells, we assessed
the expression levels of phosphorylated ERK1/2, p38 and c-Jun
N-terminal kinases (JNK) by immunoblotting. MDA-MB-231 cells were
treated with 0, 2, 4, 6, 8 and 10 µg/ml of cisplatin for 48 h, and
the levels of p-ERK1/2, p-p38 and p-JNK increased in a
dose-dependent manner compared to that of the control, suggesting
that cisplatin activated all three MAPK pathways in BC cells
(Fig. 3A). To determine which MAPK
is involved in cisplatin-induced autophagy, ERK inhibitor PD98059
(10 µmol/ml), p38 inhibitor PD169316 (10 µmol/ml) and JNK inhibitor
SP600126 (10 µmol/ml) were used. MDA-MB-231 cells exposed to
cisplatin combined with the ERK inhibitor, but not the p38 or JNK
inhibitors, had decreased LC3-II when compared to cisplatin alone
(Fig. 3B-D), suggesting that the
ERK signaling pathway may be involved in cisplatin-induced
autophagy.
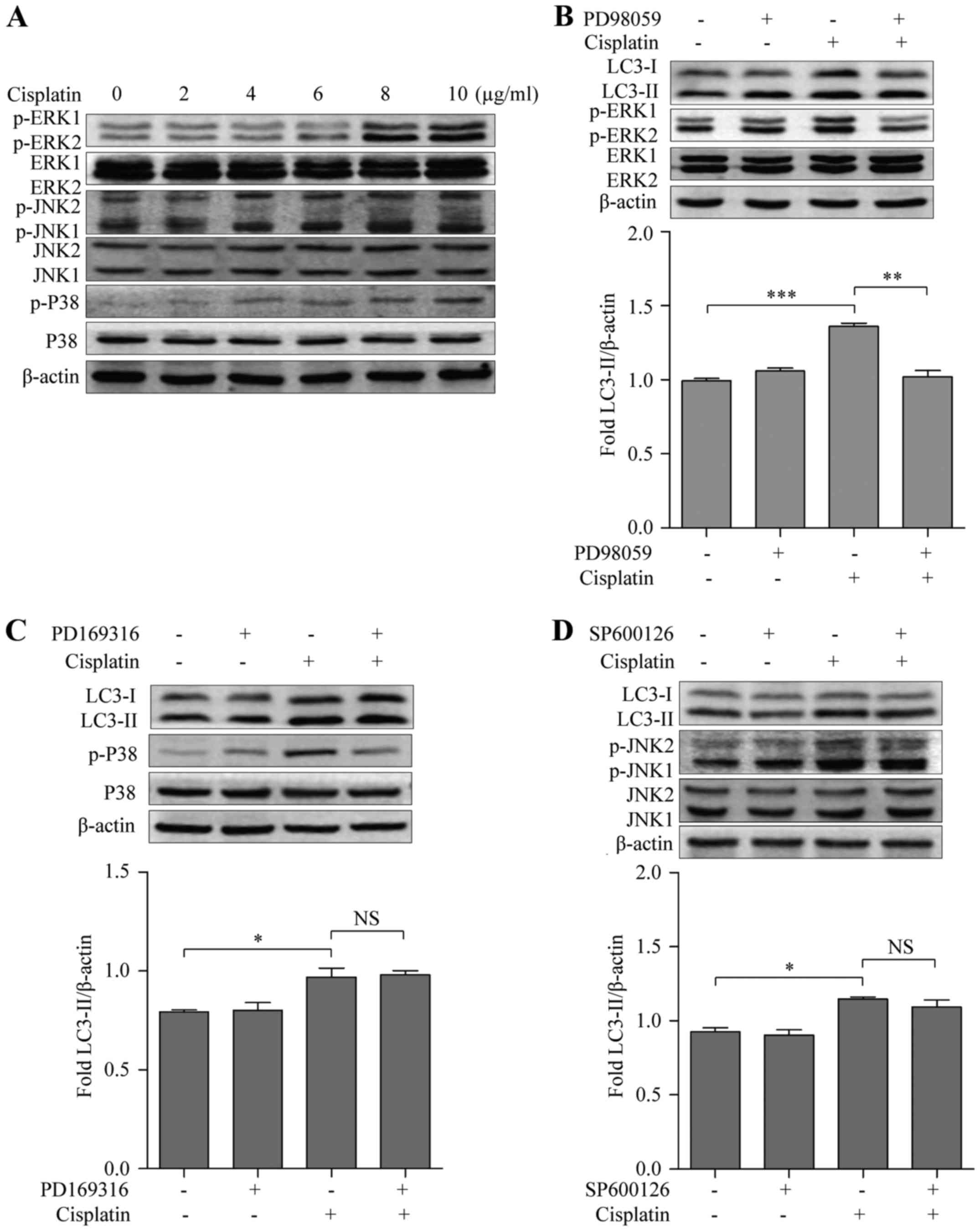 | Figure 3.Cisplatin induces autophagy in breast
cancer cells via the ERK signaling pathway. (A) Western blotting
examined the activation of the MAPK pathways including p-ERK1/2,
ERK1/2, p-P38, p38, p-JNK and JNK in MDA-MB-231 cells. (B) Western
blot analysis evaluated p-ERK1/2, ERK1/2, LC3 and quantification of
LC3-II in untreated (control), treated with the ERK inhibitor
PD98059, cisplatin and both these agents in MDA-MB-231 cells. (C)
Western blot analysis evaluated p-p38, p38, LC3 and quantification
of LC3-II in untreated (control), treated with the p38 inhibitor
PD169316, cisplatin, and both these agents in MDA-MB-231 cells. (D)
Western blot analysis evaluated p-JNK, JNK, LC3 and quantification
of LC3-II in untreated (control), treated with the JNK inhibitor
SP600126, cisplatin and both these agents in MDA-MB-231 cells;
*P<0.05, **P<0.01, ***P<0.001. Data are presented as the
mean ± SD of at least three independent experiments. |
Cisplatin-induced autophagy promotes
YAP activity
The transcription co-activator YAP is thought to be
involved in autophagy-dependent proliferation and invasion of BC
cells (19). However, the role of
YAP activation under cisplatin-induced autophagy remains unknown.
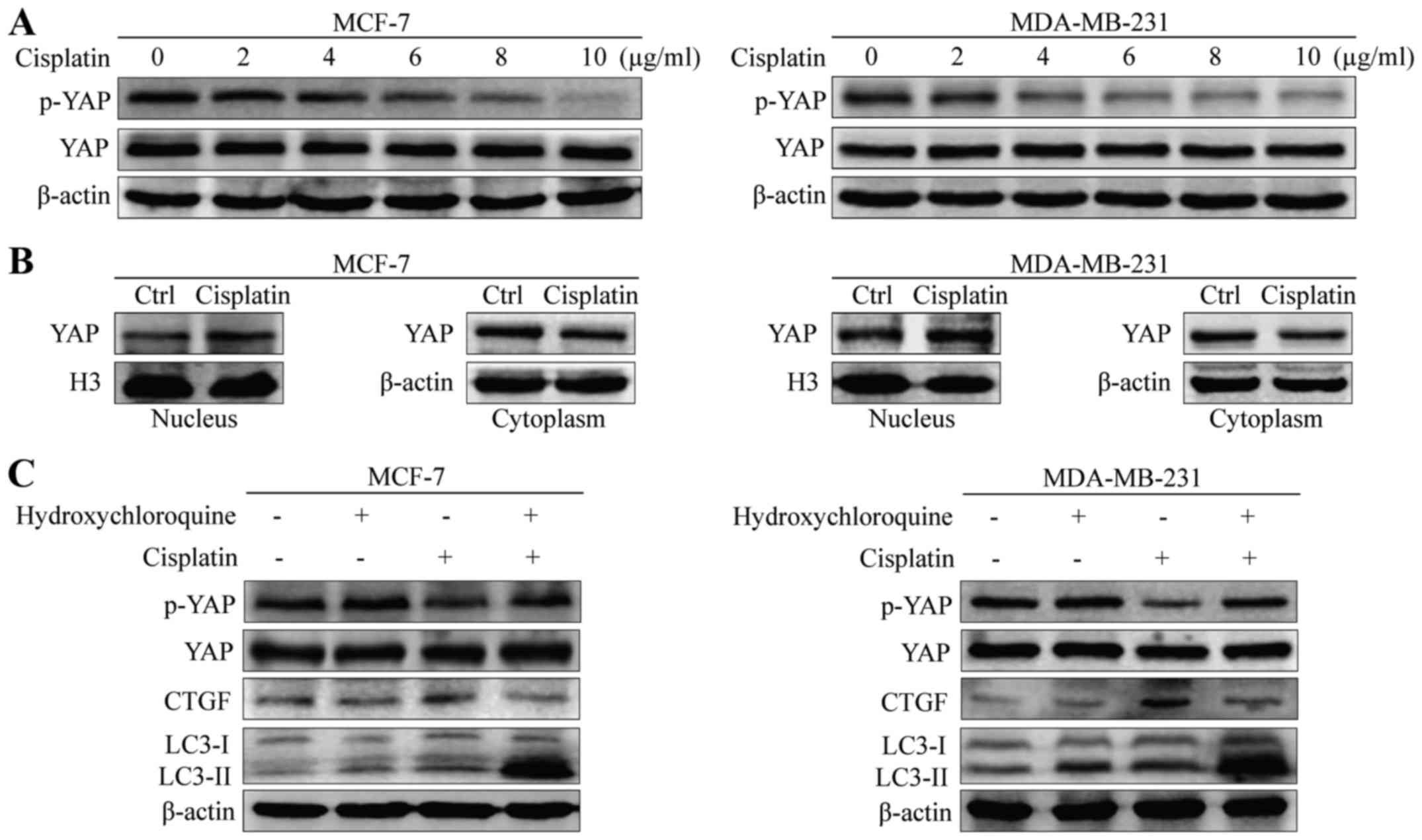
The rate of phosphorylated YAP decreased with cisplatin treatment
in a dose-dependent manner in MCF-7 and MDA-MB-231 cells (Fig. 4A). Consistent with this observation,
immunoblotting of the nuclear and cytoplasmic fractions revealed
increased accumulation of YAP protein in the nucleus of MCF-7 and
MDA-MB-231 cells with cisplatin treatment (Fig. 4B). As YAP could be regulated by
autophagy, we validated the impact of autophagy on YAP
phosphorylation using the autophagy inhibitor HCQ. Compared with
cisplatin alone, phosphorylation of YAP increased when treated with
cisplatin and HCQ in MCF-7 and MDA-MB-231 cells (Fig. 4C). In addition, we found that
cisplatin-induced autophagy increased the expression of CTGF, the
target gene of YAP. These results indicate that cisplatin-induced
autophagy had a significant impact on YAP activity.
Transcription co-activator YAP
contributes to cisplatin-induced apoptosis
Since cisplatin-induced autophagy has a significant
impact on YAP activity, we next investigated the role of YAP in the
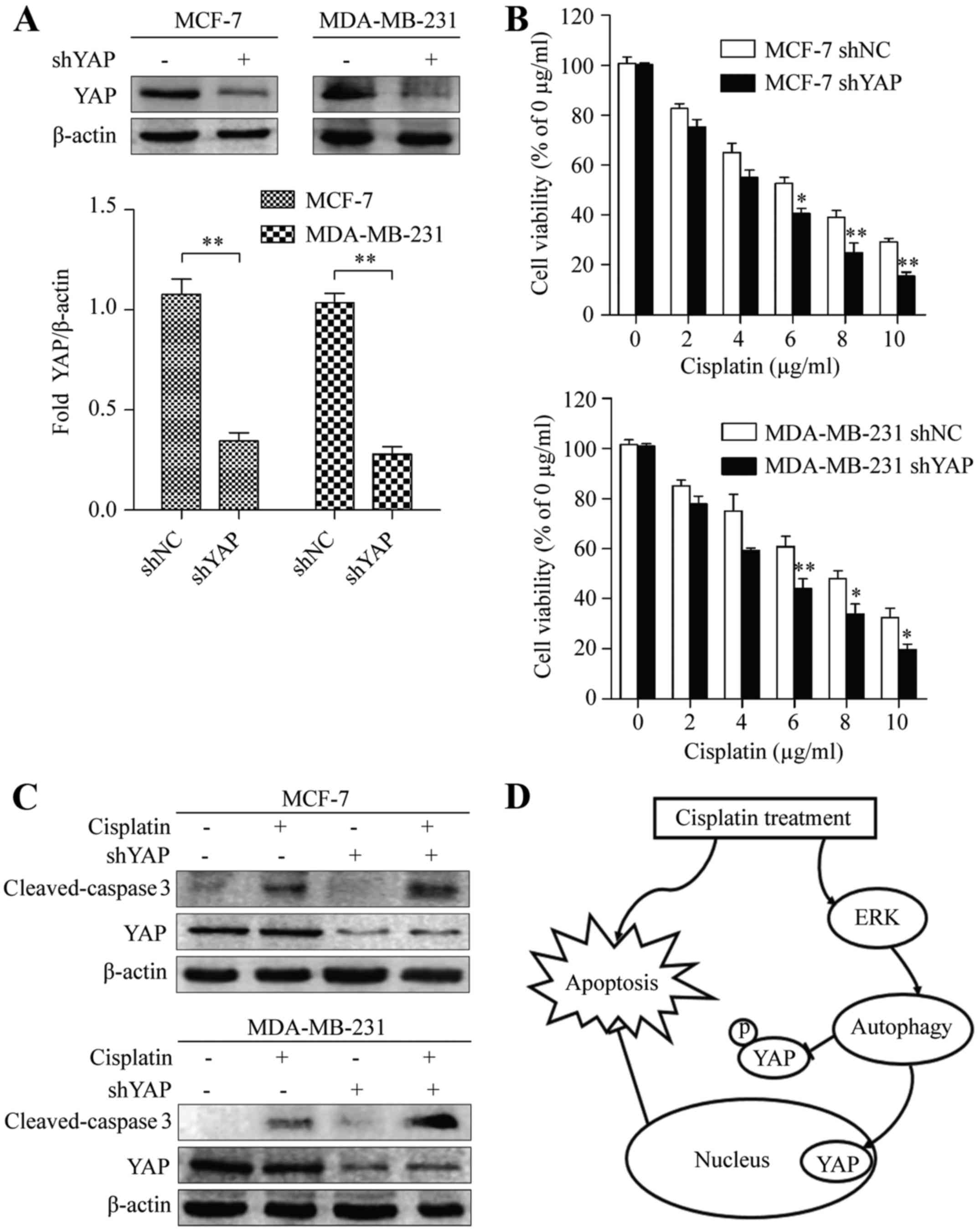
apoptosis of cisplatin-treated cells. MCF-7 and MDA-MB-231 cells
were transfected with either non-target (shNC) or YAP shRNAs
(shYAP), and the expression of YAP was examined by western
blotting. Compared with the control-transfected cells, the amount
of YAP in cells transfected with YAP shRNA decreased significantly
(Fig. 5A). Next, we used a CCK-8
assay to examine the cytotoxic effects of shNC and shYAP. Depletion
of YAP significantly increased the cytotoxicity of cisplatin in
MCF-7 and MDA-MB-231 cells (Fig.
5B). Consistent with this observation, we also found that the
apoptotic marker cleaved-caspase 3 was increased in YAP knockdown
cisplatin-treated cells, compared with that of their control
shRNA-expressing counterparts (Fig.
5C). This data indicated that YAP inactivation was essential
for the apoptotic effect of cisplatin in BC cells. Collectively,
these results revealed that cisplatin induced autophagy, which
functioned as a protective mechanism by regulating nuclear
translocation of YAP, and subsequently impairing the apoptosis of
BC cells (Fig. 5D).
Discussion
Cisplatin is a chemotherapeutic agent used for
treating various malignancies, and functions by causing lethal DNA
damage and inducing apoptosis (24). Although there are obvious benefits
of cisplatin-based treatment of breast cancer, accumulating
evidence indicates that the efficacy of cisplatin is still a
problem (25). Multiple mechanisms
contribute to the efficacy of cisplatin treatment in cancer cells.
For example, changes in cytokine and phosphorylation profiles of
human mesenchymal stromal cells led to increased chemoresistance
and stemness of breast cancer cells. MicroRNA-302b overexpression
enhanced cisplatin sensibility of breast cancer cells by inducing
apoptosis, but the mechanisms underlying this effect are not fully
understood (26). Investigation of
cisplatin signal transduction is of particular importance. Recent
studies suggest that the cytotoxic effects of drugs could by
mediated by autophagy (27).
Cisplatin has been reported to promote apoptosis and
inhibit proliferation in breast cancer cells (28). Consistently, in the present study we
demonstrated that cisplatin exhibited significant cytotoxic effects
in a dose-dependent manner in MCF-7 and MDA-MB-231 cells.
Furthermore, we found that cisplatin induced autophagy, which was
confirmed by evaluating the expression of several autophagy markers
and by electron microscopy. Autophagy plays an important role in
cancer treatment (29,30). Certain cytotoxic drugs can induce
protective autophagy in cancer cells, impairing the cytotoxic
effect of these drugs (31). We
speculated that the activation of autophagy weakens
cisplatin-induced apoptotic cell death. We used the lysosomal
inhibitor hydroxychloroquine (HCQ) to further investigate the
function of autophagy in cisplatin treatment. Inhibition of
autophagy enhanced cisplatin-induced apoptosis in both MCF-7 and
MDA-MB-231 cells, indicated that suppression of autophagy may be a
promising strategy for breast cancer therapy using cisplatin.
The mechanisms by which autophagy is regulated are
not fully understood. Emerging evidence has revealed that all three
MAPK subfamilies regulated autophagy in other cancer cells
(15,32,33).
For example, a recent study stated that ERK promoted autophagy in
human melanoma cells by CREB protein (34). The p38 MAPK blockade induced an
increase in autophagy through enhanced interactions between p38
interacting protein (p38IP) and autophagy protein 9 (ATG9) in an
mTOR-independent manner (35).
JNK1-mediated multisite phosphorylation of Bcl-2 stimulated
starvation-induced autophagy by disrupting the Bcl-2/Beclin 1
complex (32). In the present
study, we found that cisplatin treatment activated the MAPK
signaling pathway in breast cancer cells. We used an ERK, p38, and
JNK inhibitors to clarify the function of all three MAPKs in the
regulation of cisplatin-induced autophagy. We revealed that the p38
and JNK inhibitors did not block cisplatin-induced autophagy.
Therefore, we concluded that cisplatin treatment activated
autophagy via the ERK signaling pathway.
The transcription co-activator YAP is an important
regulator in cancer treatment (36). Notably, the phosphorylation level of
YAP decreased with cisplatin treatment, which was accompanied by
significant nuclear accumulation of YAP. The upstream kinase
molecule LATS was not altered (data not shown), indicating that YAP
was regulated by another process. A recent study revealed that YAP
was modulated by autophagy, and could control cell proliferation
and invasion (19). Consistent with
the present study, we found that YAP and the YAP-target gene CTGF
were regulated by cisplatin-induced autophagy using HCQ. Moreover,
YAP depletion potentiated the cytotoxic effects of cisplatin in
breast cancer cells. These observations strongly indicated that
cisplatin-induced autophagy promoted YAP activity, further
inhibiting apoptosis with cisplatin treatment in breast cancer
cells.
In conclusion, in the present study we demonstrated
that the inhibition of autophagy markedly enhanced
cisplatin-induced apoptosis in breast cancer cells, and that the
transcription co-activator YAP plays an important role in this
process. Therefore, the combination of cisplatin and an autophagy
inhibitor may provide an effective chemical therapy for breast
cancer treatment.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81272544) to
T.C.
References
|
1
|
Dey N, De P and Leyland-Jones B:
PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell
signaling to clinical trials. Pharmacol Ther. 175:91–106. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herranz M and Ruibal A: Optical imaging in
breast cancer diagnosis: The next evolution. J Oncol.
2012:8637472012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shah NR and Chen H: MicroRNAs in
pathogenesis of breast cancer: Implications in diagnosis and
treatment. World J Clin Oncol. 5:48–60. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller RP, Tadagavadi RK, Ramesh G and
Reeves WB: Mechanisms of cisplatin nephrotoxicity. Toxins.
2:2490–2518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shi S, Tan P, Yan B, Gao R, Zhao J, Wang
J, Guo J, Li N and Ma Z: ER stress and autophagy are involved in
the apoptosis induced by cisplatin in human lung cancer cells.
Oncol Rep. 35:2606–2614. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Czarnomysy R, Surażyński A, Popławska B,
Rysiak E, Pawłowska N, Czajkowska A, Bielawski K and Bielawska A:
Synergistic action of cisplatin and echistatin in MDA-MB-231 breast
cancer cells. Mol Cell Biochem. 427:13–22. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gewirtz DA: Cytoprotective and
nonprotective autophagy in cancer therapy. Autophagy. 9:1263–1265.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mah LY and Ryan KM: Autophagy and cancer.
Cold Spring Harb Perspect Biol. 4:a0088212012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liang DH, El-Zein R and Dave B: Autophagy
inhibition to increase radiosensitization in breast cancer. J Nucl
Med Radiat Ther. 6:2542015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aydinlik S, Erkisa M, Cevatemre B,
Sarimahmut M, Dere E, Ari F and Ulukaya E: Enhanced cytotoxic
activity of doxorubicin through the inhibition of autophagy in
triple negative breast cancer cell line. Biochim Biophys Acta.
1861:49–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wen J, Yeo S, Wang C, Chen S, Sun S, Haas
MA, Tu W, Jin F and Guan JL: Autophagy inhibition re-sensitizes
pulse stimulation-selected paclitaxel-resistant triple negative
breast cancer cells to chemotherapy-induced apoptosis. Breast
Cancer Res Treat. 149:619–629. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu L, Gu C, Zhong D, Shi L, Kong Y, Zhou Z
and Liu S: Induction of autophagy counteracts the anticancer effect
of cisplatin in human esophageal cancer cells with acquired drug
resistance. Cancer Lett. 355:34–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Basu S, Rajakaruna S, Reyes B, Van
Bockstaele E and Menko AS: Suppression of MAPK/JNK-MTORC1 signaling
leads to premature loss of organelles and nuclei by autophagy
during terminal differentiation of lens fiber cells. Autophagy.
10:1193–1211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng Y, Yang X, Wang J, Fan J, Kong Q and
Yu X: Aristolochic acid I induced autophagy extenuates cell
apoptosis via ERK 1/2 pathway in renal tubular epithelial cells.
PLoS One. 7:e303122012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang X, Wang J, Dai J, Shao J, Ma J, Chen
C, Ma S, He Q, Luo P and Yang B: Autophagy protects against
dasatinib-induced hepatotoxicity via p38 signaling. Oncotarget.
6:6203–6217. 2015.PubMed/NCBI
|
|
16
|
Zhi X, Zhao D, Zhou Z, Liu R and Chen C:
YAP promotes breast cell proliferation and survival partially
through stabilizing the KLF5 transcription factor. Am J Pathol.
180:2452–2461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sorrentino G, Ruggeri N, Zannini A,
Ingallina E, Bertolio R, Marotta C, Neri C, Cappuzzello E, Forcato
M, Rosato A, et al: Glucocorticoid receptor signalling activates
YAP in breast cancer. Nat Commun. 8:140732017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lamar JM, Stern P, Liu H, Schindler JW,
Jiang ZG and Hynes RO: The Hippo pathway target, YAP, promotes
metastasis through its TEAD-interaction domain. Proc Natl Acad Sci
USA. 109:E2441–E2450. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lefort S, Joffre C, Kieffer Y, Givel AM,
Bourachot B, Zago G, Bieche I, Dubois T, Meseure D, Vincent-Salomon
A, et al: Inhibition of autophagy as a new means of improving
chemotherapy efficiency in high-LC3B triple-negative breast
cancers. Autophagy. 10:2122–2142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kwon Y, Vinayagam A, Sun X, Dephoure N,
Gygi SP, Hong P and Perrimon N: The Hippo signaling pathway
interactome. Science. 342:737–740. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song Q, Mao B, Cheng J, Gao Y, Jiang K,
Chen J, Yuan Z and Meng S: YAP enhances autophagic flux to promote
breast cancer cell survival in response to nutrient deprivation.
PLoS One. 10:e01207902015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu
LZ and Jiang BH: Downregulation of ATG14 by EGR1-MIR152 sensitizes
ovarian cancer cells to cisplatin-induced apoptosis by inhibiting
cyto-protective autophagy. Autophagy. 11:373–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Skolekova S, Matuskova M, Bohac M, Toro L,
Durinikova E, Tyciakova S, Demkova L, Gursky J and Kucerova L:
Cisplatin-induced mesenchymal stromal cells-mediated mechanism
contributing to decreased antitumor effect in breast cancer cells.
Cell Commun Signal. 14:42016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cataldo A, Cheung DG, Balsari A, Tagliabue
E, Coppola V, Iorio MV, Palmieri D and Croce CM: miR-302b enhances
breast cancer cell sensitivity to cisplatin by regulating E2F1 and
the cellular DNA damage response. Oncotarget. 7:786–797. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carew JS, Nawrocki ST and Cleveland JL:
Modulating autophagy for therapeutic benefit. Autophagy. 3:464–467.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang S, Peng X, Li X, Yang P, Xie L, Li
Y, Du C and Zhang G: Silencing of CXCR4 sensitizes triple-negative
breast cancer cells to cisplatin. Oncotarget. 6:1020–1030. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ambjørn M, Ejlerskov P, Liu Y, Lees M,
Jäättelä M and Issazadeh-Navikas S: IFNB1/interferon-β-induced
autophagy in MCF-7 breast cancer cells counteracts its proapoptotic
function. Autophagy. 9:287–302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li D, Hu J, Wang T, Zhang X, Liu L, Wang
H, Wu Y, Xu D and Wen F: Silymarin attenuates cigarette smoke
extract-induced inflammation via simultaneous inhibition of
autophagy and ERK/p38 MAPK pathway in human bronchial epithelial
cells. Sci Rep. 6:377512016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu YL, Lai F, Wilmott JS, Yan XG, Liu XY,
Luan Q, Guo ST, Jiang CC, Tseng HY, Scolyer RA, et al: Noxa
upregulation by oncogenic activation of MEK/ERK through CREB
promotes autophagy in human melanoma cells. Oncotarget.
5:11237–11251. 2014.PubMed/NCBI
|
|
35
|
Henson SM, Lanna A, Riddell NE, Franzese
O, Macaulay R, Griffiths SJ, Puleston DJ, Watson AS, Simon AK,
Tooze SA, et al: p38 signaling inhibits mTORC1-independent
autophagy in senescent human CD8+ T cells. J Clin
Invest. 124:4004–4016. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ciamporcero E, Shen H, Ramakrishnan S, Ku
Yu S, Chintala S, Shen L, Adelaiye R, Miles KM, Ullio C, Pizzimenti
S, et al: YAP activation protects urothelial cell carcinoma from
treatment-induced DNA damage. Oncogene. 35:1541–1553. 2016.
View Article : Google Scholar : PubMed/NCBI
|